
Abstract
Aims/hypothesis
Many individuals who develop type 2 diabetes also display increased glucagon levels (hyperglucagonaemia), which we have previously found to be associated with the metabolic syndrome. The concept of a liver–alpha cell axis provides a possible link between hyperglucagonaemia and elevated liver fat content, a typical finding in the metabolic syndrome. However, this association has only been studied in individuals with non-alcoholic fatty liver disease. Hence, we searched for a link between the liver and the alpha cells in individuals with non-steatotic levels of liver fat content. We hypothesised that the glucagon–alanine index, an indicator of the functional integrity of the liver–alpha cell axis, would associate with liver fat and insulin resistance in our cohort of women with low levels of liver fat.
Methods
We analysed data from 79 individuals participating in the Prediction, Prevention and Subclassification of Type 2 Diabetes (PPSDiab) study, a prospective observational study of young women at low to high risk for the development of type 2 diabetes. Liver fat content was determined by MRI. Insulin resistance was calculated as HOMA-IR. We conducted Spearman correlation analyses of liver fat content and HOMA-IR with the glucagon–alanine index (the product of fasting plasma levels of glucagon and alanine). The prediction of the glucagon–alanine index by liver fat or HOMA-IR was tested in multivariate linear regression analyses in the whole cohort as well as after stratification for liver fat content ≤0.5% (n = 39) or >0.5% (n = 40).
Results
The glucagon–alanine index significantly correlated with liver fat and HOMA-IR in the entire cohort (ρ = 0.484, p < 0.001 and ρ = 0.417, p < 0.001, respectively). These associations resulted from significant correlations in participants with a liver fat content >0.5% (liver fat, ρ = 0.550, p < 0.001; HOMA-IR, ρ = 0.429, p = 0.006). In linear regression analyses, the association of the glucagon–alanine index with liver fat remained significant after adjustment for age and HOMA-IR in all participants and in those with liver fat >0.5% (β = 0.246, p = 0.0.23 and β = 0.430, p = 0.007, respectively) but not in participants with liver fat ≤0.5% (β = −0.184, p = 0.286).
Conclusions/interpretation
We reproduced the previously reported association of liver fat content and HOMA-IR with the glucagon–alanine index in an independent study cohort of young women with low to high risk for type 2 diabetes. Furthermore, our data indicates an insulin-resistance-independent association of liver fat content with the glucagon–alanine index. In summary, our study supports the concept that even lower levels of liver fat (from 0.5%) are connected to relative hyperglucagonaemia, reflecting an imminent impairment of the liver–alpha cell axis.

Similar content being viewed by others
Avoid common mistakes on your manuscript.

Introduction
Increased plasma glucagon concentration (hyperglucagonaemia) has been suggested to play a crucial role in the development of type 2 diabetes. Indeed, inhibition of glucagon signalling by treatment with a glucagon receptor antagonist has shown favourable effects on glucose metabolism in individuals with type 2 diabetes [1, 2]. However, our previous finding that hyperglucagonaemia is not present in all individuals at high risk for type 2 diabetes suggests that hyperglucagonaemia might drive type 2 diabetes only in a subgroup of individuals [3]. Yet, the factors that drive hyperglucagonaemia in this subgroup have not been fully elucidated. Furthermore, many hyperglucagonaemic individuals exhibit characteristics of the metabolic syndrome [3], indicating that the metabolic syndrome might be closely related to hyperglucagonaemia.
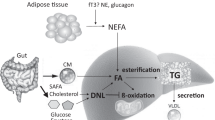
The concept of a liver–alpha cell axis provides a potential causal link between the metabolic syndrome and hyperglucagonaemia [4, 5]. According to this concept, fat deposition in the liver, a hallmark of the metabolic syndrome [6], might impair hepatic glucagon signalling. Impaired hepatic glucagon signalling fosters hyperaminoacidaemia because glucagon regulates ureagenesis from amino acids [7]. Hyperaminoacidaemia, in turn, increases glucagon secretion from the alpha cell, which causes hyperglucagonaemia [4]. Hyperglucagonaemia may compensate for the increasing glucagon resistance and thereby restore amino acid metabolism. However, the action of glucagon on hepatic glucose production might be preserved, leading to increased hepatic glucose production [8]. Importantly, hyperglucagonaemia may aggravate the glucagon resistance of hepatocytes. Similar to insulin resistance, a vicious cycle develops in which the steadily rising hepatic glucose production eventually evolves into type 2 diabetes [5].
Evidence for this feedback cycle between the liver (amino acids) and alpha cells (glucagon) has previously been provided in participants of the ADDITION-PRO (‘Progression’ arm, nested within the Anglo–Danish–Dutch Study of Intensive Treatment In People with Screen Detected Diabetes in Primary Care study) study cohort [9]. In this context, the glucagon–alanine index was introduced as a surrogate marker for the functional status of the liver–alpha cell axis. The index is calculated as the product of fasting concentrations of plasma glucagon and plasma alanine and it was suggested that a higher index indicates functional impairments in the axis [9].
Hepatic steatosis might be causally involved in the impairment of the liver–alpha cell axis as individuals with non-alcoholic fatty liver disease (NAFLD) display elevated fasting glucagon and higher plasma levels of the sum of l-amino acids compared with individuals without the disease [10]. However, the possible contribution of lower levels of liver fat (i.e. lower than 5.5%) has not yet been studied.
In the present study, we examined the association of liver fat, at lower concentrations than those observed in NAFLD, and insulin resistance with the glucagon–alanine index. We hypothesised that liver fat content would associate with the glucagon–alanine index even at levels below the classification of NAFLD. This association would support the assumption that a functionally impaired liver–alpha cell axis is already present in the early pathogenesis of the metabolic syndrome and type 2 diabetes.
Methods
Cohort
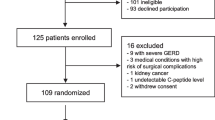
The present analyses were conducted on material obtained at the baseline visit of the Prediction, Prevention and Subclassification of Type 2 Diabetes (PPSDiab) study [11]. This study included 304 women 3–16 months after pregnancy. Participants were consecutively recruited from the diabetes centre and the obstetrics department of the University Hospital (Klinikum der Universität München) in Munich, Germany. Exclusion criteria for this study were alcohol or substance abuse, pre-pregnancy diabetes and chronic diseases requiring continuous medication (except for hypothyroidism [n = 52], bronchial asthma [n = 8], mild hypertension [n = 4], gastro-oesophageal reflux [n = 2] and history of pulmonary embolism resulting in rivaroxaban prophylaxis [n = 1]). Four women were excluded from the baseline visit of the PPSDiab study due to acute upper respiratory infection at the study visit (n = 1), overt hyperthyroidism (n = 2) or positive islet autoantibodies at baseline with diagnosis of type 1 diabetes during follow-up (n = 1) (Fig. 1).

Flow chart for the PPSDiab study cohort
For the present analyses, we further excluded women with type 2 diabetes diagnosis (n = 7) at the study visit to minimise possible bias from metabolic adaptions to overt diabetes. As previously described, measurement of plasma amino acids was only performed in the first half of the PPSDiab study cohort [12]. MRI for the determination of liver fat content was offered on a voluntary basis. Data on both plasma amino acids and liver fat content were available in 79 out of 293 (27%) participants without type 2 diabetes (Fig. 1). HOMA-IR and insulin sensitivity index (ISI) data were missing for one participant due to missing fasting insulin measurement.
All study participants provided written informed consent and the protocol was approved by the ethical review committee of the University Hospital in Munich (Ludwig-Maximilians-Universität, study ID 300-11). Detailed information on the study cohort is described in Rottenkolber et al [11].
OGTT
We conducted an OGTT with a five-point measurement of plasma glucose and serum insulin as previously described [11]. For the diagnosis of diabetes, we used ADA criteria (fasting glucose ≥ 7.0 mmol/l and/or 2 h post-load glucose ≥11.1 mmol/l).
Biochemical measurements and metabolomics
Glucagon and alanine levels were determined using frozen (−80°C) plasma samples collected in proteinase-stabilised tubes (BD P800; BD Biosciences, San Jose, CA, USA) after participants had fasted overnight. Plasma glucagon was measured by sandwich ELISA (catalogue no. 10-1271-01; Mercodia, Uppsala, Sweden). Alanine, as well as 20 other amino acids (arginine, asparagine, aspartate, citrulline, glutamine, glutamate, glycine, histidine, isoleucine, leucine, lysine, methionine, ornithine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine), were quantified by using targeted metabolomics (AbsoluteIDQ™ p180 Kit; Biocrates Life Sciences, Innsbruck, Austria) with LC-MS/MS. The measurement of plasma samples using this assay has been described in full detail previously [13]. Total amino acids represent the sum of all 21 l-amino acids, measured by the AbsoluteIDQ™ p180 Kit.
From the OGTT, plasma glucose was measured by a hexokinase method (Glucose HK Gen.3; Roche Diagnostics, Mannheim, Germany) and serum insulin by a chemiluminescent immunoassay (DiaSorin LIASON Systems, Saluggia, Italy). Fasting values of HDL-cholesterol and triacylglycerols were measured by enzymatic caloric test (Roche Diagnostics, Mannheim, Germany).
Anthropometrics
Height and waist circumference were measured to the nearest 1 cm. Body mass was determined by a bioelectrical impedance analysis scale (Tanita BC-418; Tanita Corporation, Tokyo, Japan).
MRI
Liver fat content was determined by MRI using an mDixon low-fat fraction map (3 Tesla System, Ingenia or Achieva; Philips Health Care, Hamburg, Germany). A detailed description of the MRI measurements is provided elsewhere [11].
Calculations
To measure hepatic insulin resistance, HOMA-IR was calculated as fasting glucose (mmol/l) × fasting insulin (mmol/l) / 22.5. To determine peripheral insulin resistance, the ISI was calculated from the OGTT according to Matsuda and DeFronzo [14].
For the primary confirmatory analysis, the glucagon–alanine index was calculated as fasting glucagon × fasting alanine, as has previously been suggested [9]. Analogously, for secondary exploratory analyses, the glucagon–glutamate index and the glucagon–total amino acid index were calculated as fasting glucagon × fasting glutamate and fasting glucagon × fasting total amino acids, respectively.
Statistical analysis
All metric and normally distributed variables are reported as mean ± SD; non-normally distributed variables are reported as median (first quartile–third quartile). Frequencies are presented as n (%). To address non-normality of the distribution of the glucagon–alanine index, liver fat content and HOMA-IR, all three variables were log-transformed for regression analyses. In addition, we compared liver fat quartiles using the Kruskal–Wallis test with Dwass, Steel, Critchlow-Fligner (DSCF) multiple comparison analysis [15]. The following analyses were conducted in all participants as well as stratified according to a liver fat content of ≤0.5% vs >0.5% (0.5% is used as proxy for the median of liver fat content in the cohort). Correlation analyses between glucagon, alanine, the glucagon–alanine index, liver fat and HOMA-IR were conducted using Spearman correlation coefficients (ρ). We also performed linear regression analyses with the glucagon–alanine index (loge-transformed) as the dependent variable and liver fat content or HOMA-IR (both loge-transformed) as independent variables adjusted for age and age plus HOMA-IR or liver fat, respectively. A p value <0.05 was considered statistically significant. All statistical calculations were performed using SAS statistical software package, version 9.4 (SAS Institute, Cary, NC, USA). Figures were created using Tableau 2020.3 (Tableau Software, Seattle, WA, USA).
Results
Table 1 shows the baseline characteristics of the study cohort.
First, we compared fasting glucagon, alanine and the glucagon–alanine index between liver fat quartiles. Alanine continuously increased from quartile 1 to quartile 4 (Fig. 2a and ESM Table 1) whereas fasting glucagon and the glucagon–alanine index increased from quartile 2 to quartile 4 (Fig. 2b, c and electronic supplementary material [ESM] Table 1).

Fasting plasma alanine levels (a), fasting plasma glucagon levels (b) and glucagon–alanine index (c) stratified by liver fat quartiles. Boxes represent first quartile to median (dark grey) and median to third quartile (light grey). Whiskers depict 1.5-times the IQR. Group comparison by Kruskal–Wallis test with Dwass, Steel, Critchlow-Fligner post hoc test for multiple comparisons. *p < 0.05 for difference between groups
Alanine, glucagon, and the glucagon–alanine index correlated with liver fat content (Table 2). The correlation between the glucagon–alanine index and liver fat content began at around 0.5% fat, which was the median fat content in our cohort (Fig. 3a, Table 2 and ESM Fig. 1). Below 0.5%, the correlation was absent. In addition, the glucagon–alanine index correlated positively with HOMA-IR, fasting glucose and 2 h plasma glucose (Table 2 and ESM Fig. 1). The correlation with HOMA-IR was linear throughout the study cohort (Fig. 3b). Exploratory correlation analyses of other amino acids with liver fat content, glucagon and HOMA-IR are displayed in ESM Table 2. Of all the amino acids, glutamate showed the highest correlation to both glucagon and liver fat content, followed by the branched-chain amino acids.

Association of the glucagon–alanine index with liver fat content (a) and HOMA-IR (b). Trend lines of crude models are shown together with Spearman correlation coefficients (ρ) and p values. The association between liver fat content and the glucagon–alanine index is shown as separate models for participants with liver fat content ≤0.5% (light grey dots; n.s.: non-significant association) and >0.5% (dark grey dots)
In linear regression models, liver fat content still associated significantly with the glucagon–alanine index after an adjustment for age or age plus HOMA-IR, both in the whole study cohort as well as in the participants with a liver fat content of >0.5% (Table 3). Similarly, HOMA-IR associated with the glucagon–alanine index after adjustment for age or age plus liver fat content in all participants. However, in participants with a liver fat content of >0.5%, the association of HOMA-IR with the glucagon–alanine index was lost when liver fat content was added to the model (Table 3). No other glucagon–amino acid index associated as strongly as the glucagon–alanine index (ESM Tables 3, 4 show linear regression data for the glucagon–glutamate index and glucagon–total amino acid index, respectively).
Discussion
The present study provides evidence for a three-way association between liver fat content, the glucagon–alanine index and HOMA-IR. Thus, it supports the concept of a liver–alpha cell axis and suggests that alterations in this axis are already apparent at slightly increased amounts of liver fat content.
Our study substantiated the association between the liver–alpha cell axis and liver fat content. Studies in individuals with NAFLD and non-alcoholic steatohepatitis (NASH) illustrate that fat accumulation in the liver reduces the sensitivity of hepatocytes towards glucagon [16, 17]. One of glucagon’s actions on hepatocytes is the amplification of amino acid-induced urea synthesis [18]. Hence, a reduced glucagon sensitivity results in decreased urea formation from amino acids, leading to an increase in plasma amino acids [7, 19]. This rise in the levels of amino acids in turn stimulates glucagon secretion from the alpha cell, which promotes hyperglucagonaemia. Our analyses support this interrelation and further indicate that hepatic glucagon sensitivity might already be impaired at non-steatotic levels of liver fat, starting from 0.5%. Below this value, liver fat content and the glucagon–alanine index did not associate in our cohort. However, this may be related to the MRI measurement technique, which is the most sensitive among the non-invasive procedures but still not sufficiently sensitive for very low levels of fat [20].
Besides alanine, we also found glutamate to be involved in the liver–alpha cell axis (ESM Tables 2, 3). Glutamate activates AMPA/kainate receptors on the alpha cell, which increases glucagon secretion [21, 22]. Hence, a glucagon–glutamate index might also be suitable to represent the liver–alpha cell axis. In contrast to previous findings in individuals with and without NAFLD, total amino acids did not correlate with glucagon, liver fat or insulin resistance in our cohort of women with low levels of liver fat (ESM Table 2) and the association of a glucagon–total amino acid index was not superior to the glucagon–alanine index or the glucagon–glutamate index. Therefore, the determination of the glucagon–alanine index seems to be sufficient to characterise the status of the liver–alpha cell axis.
The extent to which the raised glucagon levels can compensate for the reduced glucagon sensitivity of hepatocytes regarding urea synthesis remains unknown. However, in the present study, the continuous association between liver fat content and the glucagon–alanine index points to an effective adaption of the liver–alpha cell axis to hepatic glucagon resistance in individuals without clinical steatosis (Fig. 3a).
Though adaption of the liver–alpha cell axis to hepatic glucagon resistance maintains amino acid turnover at a normal level, hyperglucagonaemia probably places a burden on glucose metabolism. In this context, our study revealed that insulin resistance and elevated plasma glucose levels associate with alterations in the liver–alpha cell axis (Table 2). Notably, the association of the glucagon–alanine index with insulin resistance was independent of liver fat content in the entire study cohort (Table 3). This independence was narrowly missing (p = 0.069) in the subgroup of participants with a liver fat content of >0.5% but we attribute this mainly to the smaller size of this subgroup. Previous research indicates that the action of glucagon on hepatic glucose production persists independent of a disturbance in hepatic glucagon sensitivity with regards to amino acid turnover [8]. In individuals with intact glucose metabolism, an increase in hepatic glucose production will be compensated for by enhanced insulin secretion. In the present study, the association of glucagon and the glucagon–alanine index with plasma glucose and insulin resistance (HOMA-IR) represented this compensation (Tables 2, 3). However, if insulin secretion and/or insulin sensitivity is not intact, hyperglucagonaemia is likely to aggravate the impaired glucose metabolism and finally lead to type 2 diabetes.
The present study supports the potential link between hepatic fat accumulation and insulin resistance via glucagon signalling despite the lack of a proven cause–effect relationship. Recent data in mice indicates that liver fat reduces the sensitivity of hepatocytes towards glucagon at both a transcriptional and a non-transcriptional level [23]. Conversely, pharmacological studies reveal that treatment with the glucagon receptor antagonist LY2409021 increased hepatic fat content in individuals with type 2 diabetes [2]. This might result from blockage of glucagon’s action on β-oxidation in the hepatocytes [24]. Hence, an increase in liver fat content might not only be the cause but also the result of glucagon resistance in hepatocytes. In any case, liver fat content seems to govern the sensitivity of hepatocytes towards glucagon. Thus, our results strongly support the concept of a liver–alpha cell axis.
Previous studies indicate that amino acids other than alanine may also be involved in the liver–alpha cell axis [7, 19]. The close associations of glutamate and the branched-chain amino acids with plasma glucagon that we observed support these previous results (ESM Table 2). However, no other glucagon–amino acid index performed better than the glucagon–alanine index in its associations with liver fat content and insulin resistance.
The strengths of our study include its cohort of young women at varying risk for type 2 diabetes who otherwise constitute an extremely homogeneous sample. By including only women with normoglycaemia, impaired fasting glucose or impaired glucose tolerance, we prevented possible bias resulting from sex differences and secondary changes in metabolism due to overt diabetes. Using MRI, we could quantify even low amounts of hepatic fat. The main weakness of our study is its cross-sectional design, which precludes the determination of a cause–effect relationship. Further, the homogeneity of the cohort regarding sex and age and the predominantly white ethnicity limits the transferability of our findings to the general public.
In conclusion, we reproduced the suggested association between liver fat, the glucagon–alanine index and insulin resistance in an independent study cohort of young women with low to high risk for type 2 diabetes. Our study supports the concept that even low levels of liver fat (up from 0.5%) are associated with the integrity of the liver–alpha cell axis, which probably affects glucose metabolism. To fully establish this concept, further mechanistic studies to examine the signalling at a molecular level are necessary. In addition, larger cohorts including men and more participants with an ethnicity other than white are required.
Data availability
The data that support the findings of this study are available from the corresponding author, AL, upon reasonable request.
Abbreviations
- ISI:
-
Insulin sensitivity index
- NAFLD:
-
Non-alcoholic fatty liver disease
- PPSDiab:
-
Prediction, Prevention and Subclassification of Type 2 Diabetes
References
Unger RH, Cherrington AD (2012) Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122(1):4–12. https://doi.org/10.1172/jci60016
Guzman CB, Zhang XM, Liu R et al (2017) Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes Metab. https://doi.org/10.1111/dom.12958
Gar C, Rottenkolber M, Sacco V et al (2018) Patterns of plasma glucagon dynamics do not match metabolic phenotypes in young women. J Clin Endocrinol Metab 103(3):972–982. https://doi.org/10.1210/jc.2017-02014
Holst JJ, Wewer Albrechtsen NJ, Pedersen J, Knop FK (2017) Glucagon and amino acids are linked in a mutual feedback cycle: the liver-α-cell axis. Diabetes 66(2):235–240. https://doi.org/10.2337/db16-0994
Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD et al (2019) The liver-α-cell axis and type 2 diabetes. Endocr Rev 40(5):1353–1366. https://doi.org/10.1210/er.2018-00251
Asrih M, Jornayvaz FR (2015) Metabolic syndrome and nonalcoholic fatty liver disease: is insulin resistance the link? Mol Cell Endocrinol 418(Pt 1):55–65. https://doi.org/10.1016/j.mce.2015.02.018
Galsgaard KD, Winther-Sørensen M, Ørskov C et al (2018) Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am J Phys Endocrinol Metab 314(1):E93–e103. https://doi.org/10.1152/ajpendo.00198.2017
Suppli MP, Bagger JI, Lund A et al (2020) Glucagon resistance at the level of amino acid turnover in obese subjects with hepatic steatosis. Diabetes 69(6):1090–1099. https://doi.org/10.2337/db19-0715
Wewer Albrechtsen NJ, Færch K, Jensen TM et al (2018) Evidence of a liver-alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 61(3):671–680. https://doi.org/10.1007/s00125-017-4535-5
Wewer Albrechtsen NJ, Junker AE, Christensen M et al (2018) Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am J Physiol Gastrointest Liver Physiol 314(1):G91–g96. https://doi.org/10.1152/ajpgi.00216.2017
Rottenkolber M, Ferrari U, Holland L et al (2015) The diabetes risk phenotype of young women with recent gestational diabetes. J Clin Endocrinol Metab 100(6):E910–E918. https://doi.org/10.1210/jc.2014-3898
Gar C, Rottenkolber M, Prehn C, Adamski J, Seissler J, Lechner A (2018) Serum and plasma amino acids as markers of prediabetes, insulin resistance, and incident diabetes. Crit Rev Clin Lab Sci 55(1):21–32. https://doi.org/10.1080/10408363.2017.1414143
Zukunft S, Sorgenfrei M, Prehn C, Möller G, Adamski J (2013) Targeted metabolomics of dried blood spot extracts. Chromatographia 76(19):1295–1305. https://doi.org/10.1007/s10337-013-2429-3
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22(9):1462–1470. https://doi.org/10.2337/diacare.22.9.1462
Douglas CE, Michael FA (1991) On distribution-free multiple comparisons in the one-way analysis of variance. Communications in Statistics - Theory and Methods 20(1):127–139. https://doi.org/10.1080/03610929108830487
De Chiara F, Heebøll S, Marrone G et al (2018) Urea cycle dysregulation in non-alcoholic fatty liver disease. J Hepatol 69(4):905–915. https://doi.org/10.1016/j.jhep.2018.06.023
Eriksen PL, Vilstrup H, Rigbolt K et al (2019) Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int 39(11):2094–2101. https://doi.org/10.1111/liv.14205
Hamberg O, Vilstrup H (1994) Regulation of urea synthesis by glucose and glucagon in normal man. Clin Nutr 13(3):183–191. https://doi.org/10.1016/0261-5614(94)90099-x
Dean ED, Li M, Prasad N et al (2017) Interrupted glucagon signaling reveals hepatic alpha cell axis and role for L-glutamine in alpha cell proliferation. Cell Metab 25(6):1362–1373 e1365. https://doi.org/10.1016/j.cmet.2017.05.011
West J, Romu T, Thorell S et al (2018) Precision of MRI-based body composition measurements of postmenopausal women. PLoS One 13(2):e0192495. https://doi.org/10.1371/journal.pone.0192495
Bertrand G, Gross R, Puech R, Loubatieres-Mariani MM, Bockaert J (1993) Glutamate stimulates glucagon secretion via an excitatory amino acid receptor of the AMPA subtype in rat pancreas. Eur J Pharmacol 237(1):45–50. https://doi.org/10.1016/0014-2999(93)90091-U
Otter S, Lammert E (2016) Exciting times for pancreatic islets: glutamate signaling in endocrine cells. Trends Endocrinol Metab 27(3):177–188. https://doi.org/10.1016/j.tem.2015.12.004
Winther-Sørensen M, Galsgaard KD, Santos A et al (2020) Glucagon acutely regulates hepatic amino acid catabolism and the effect may be disturbed by steatosis. Mol Metab 42:101080. https://doi.org/10.1016/j.molmet.2020.101080
Janah L, Kjeldsen S, Galsgaard KD et al (2019) Glucagon receptor signaling and glucagon resistance. Int J Mol Sci 20(13). https://doi.org/10.3390/ijms20133314
Acknowledgements
We thank M. Scholz (Targeting Communications, Munich, Germany) for proofreading the manuscript and thank the women in the PPSDiab study for their participation.
Relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Helmholtz Zentrum München, Klinikum der Universität München, and the German Center for Diabetes Research (DZD) (to AL).
Author information
Authors and Affiliations
Contributions
CG, SJH, SKM, BR, JS, NJWA, JJH and AL substantially contributed to the conception or design of the work. CG, SJH, SKM, BR, CP and JA contributed to the acquisition of the data. CG, SJH, SKM, BR, JS, NJWA, JJH, and AL analysed and interpreted the work. CG drafted the article. All authors revised, reviewed and approved the final version of the manuscript. AL is the guarantor of this work.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 297 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gar, C., Haschka, S.J., Kern-Matschilles, S. et al. The liver–alpha cell axis associates with liver fat and insulin resistance: a validation study in women with non-steatotic liver fat levels. Diabetologia 64, 512–520 (2021). https://doi.org/10.1007/s00125-020-05334-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05334-x