
Abstract
Recent developments in the field of genetics have accelerated our understanding of the aetiology of complex diseases. Type 2 diabetes mellitus and cancer are no exception, with large-scale genome-wide association studies (GWAS) facilitating exploration of the underlying pathology. Here, we discuss how genetics studies can be used to investigate the relationship between these complex diseases. Observational epidemiological studies consistently report that people with type 2 diabetes have a higher risk of several types of cancer. Indeed, type 2 diabetes and cancer share many common risk factors, such as obesity, ageing, poor diet and low levels of physical activity. However, questions remain regarding the biological mechanisms that link these two diseases. Large-scale GWAS of type 2 diabetes and cancer allow us to consider the evidence for shared genetic architecture. Several shared susceptibility genes have been identified, yet tissue specificity and direction of effect must be taken into account when considering common genetic aetiology. We also consider how GWAS, and associated techniques such as Mendelian randomisation, allow us to dissect the link between the two diseases and address questions such as ‘Does type 2 diabetes cause cancer or is the increased risk observed driven by higher adiposity or another associated metabolic feature?’

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.


Introduction
Of the 4.7 million people in the UK (>450 million worldwide) with diabetes, ~90% have type 2 diabetes mellitus [1]. Observational epidemiological studies have consistently reported that people with type 2 diabetes have a higher risk of certain types of cancer (not prostate cancer, where an inverse relationship has been reported) [2]. Although type 2 diabetes is typically diagnosed by elevated levels of circulating glucose, it presents as a collection of metabolic features, some of which may influence cancer development whereas others may not. To date, the biological mechanisms principally hypothesised to support associations between type 2 diabetes and cancer include hyperglycaemia, hyperinsulinaemia, sex hormone dysregulation and chronic low-grade inflammation [2]. However, questions remain as to the exact features of type 2 diabetes that drive the association and whether they are causal.
Here, we focus on the contribution to this field made by genetics studies and on the insights these studies have provided in elucidating the link between type 2 diabetes and certain types of cancer. First, we discuss whether shared genetic aetiology may explain the increased risk of cancer in people with type 2 diabetes (i.e. horizontal pleiotropy: genetic variants associated with type 2 diabetes are also independently [via different mechanisms] associated with cancer risk). Second, we consider the evidence for vertical pleiotropy (i.e. a causal path from type 2 diabetes [or a particular metabolic feature of type 2 diabetes] to cancer development) by appraising the evidence from Mendelian randomisation (MR) studies (Fig. 1 and Text box: Mendelian randomisation).

The relationship between type 2 diabetes and cancer. (a) The increased risk of cancer in people with type 2 diabetes could be due to shared genetic aetiology between the two diseases. In this scenario, genetic variants that predispose individuals to type 2 diabetes would also predispose individuals to cancer. The relationship between type 2 diabetes and cancer is therefore horizontal, as the effect of the genetic variant on each disease is independent and is exerted through different mechanisms. (b) The increased risk of cancer in people with type 2 diabetes may be due to a tumorigenic effect driven by either type 2 diabetes itself or an associated trait (such as adiposity). This is an example of vertical pleiotropy, as there is a linear pathway from type 2 diabetes (or associated trait) to cancer. MR uses genetic variants as a proxy for the exposure of interest to assess whether there is a vertical causal pathway from the exposure to the outcome. This figure is available as part of a downloadable slideset
Horizontal pleiotropy: do type 2 diabetes and cancer share common genetic aetiology?
Since the advent of genome-wide association studies (GWAS) in 2005, major progress has been made in our understanding of complex diseases including type 2 diabetes and cancer [3, 4]. Type 2 diabetes itself and the cancer types observationally associated with it are relatively common, yet high-impact variants that cause monogenic forms of diabetes or heritable cancer syndromes are exceedingly rare and tend to result in early-onset disease. GWAS have revealed that the inherited contribution to more prevalent, later-onset disease is comprised of many common variants, with each individual variant having a relatively small impact [3]. Here, we discuss whether common risk alleles predispose to both type 2 diabetes and particular cancers and whether this can explain the observed association between type 2 diabetes and particular cancer types. We discuss examples that provide the most convincing evidence for shared genetic aetiology between the two diseases, discuss shared susceptibility genes and highlight scenarios that may result in misinterpretation of common aetiology.
Evidence for type 2 diabetes risk alleles increasing risk of cancer
Several types of cancer, including liver, pancreatic, endometrial, breast, colorectal, bladder and kidney cancer and non-Hodgkin’s lymphoma, have been positively and observationally associated with type 2 diabetes [5]. However, evidence for shared genetic aetiology between type 2 diabetes and these cancers is scarce. Several type 2 diabetes susceptibility genes are associated with cancer development, yet this does not necessarily imply shared genetic aetiology. This would be demonstrated by the identification of the same genetic variant and risk allele (or those in linkage disequilibrium) that independently predisposes to both type 2 diabetes and cancer (Fig. 1a). Genetic variation at TCF7L2 (transcription factor 7 like 2) is perhaps the best and most extensively studied example of this.
The transcription factor encoded by TCF7L2 operates at the last stage of the canonical Wnt signalling transduction cascade [6]. Genetics studies have reported a positive association between type 2 diabetes predisposing alleles within TCF7L2 and cancer including colorectal [7,8,9], breast [10,11,12] hepatocellular [13] and aggressive prostate cancer [14]. A recent comprehensive meta-analysis found type 2 diabetes risk alleles at eight variants in TCF7L2 were associated with increased risk of breast, colorectal and lung cancer and glioma [15].
The association between type 2 diabetes risk alleles in TCF7L2 and higher risk of colorectal cancer has been replicated in several studies [7, 16, 17]. The Wnt pathway is a major driver of colorectal carcinogenesis and the TCF7L2 gene is frequently mutated in colorectal cancer [18]. The association with colorectal cancer has been shown to be independent of type 2 diabetes or obesity [7, 16, 17], suggesting the risk alleles are likely to have greatest impact specifically in colon tissue and not in the pancreatic islet (which is likely to mediate the impact of the variants on type 2 diabetes).
Studies investigating a number of different type 2 diabetes predisposing variants in relation to cancer risk have reported only a small number of weak associations and findings are often inconsistent. In one study, among 37 type 2 diabetes risk alleles, two (in FTO and MTNR1B) showed nominally positive associations with pancreatic cancer risk and one showed an inverse association (in BCL11A) [19]. In another, among 33 type 2 diabetes risk alleles, three (in FTO, TCF7L2 and PRC1) were found to be associated with breast cancer [20]. Inconsistently, two additional studies have shown null associations with breast cancer across all type 2 diabetes variants tested [21, 22].
Directionality of type 2 diabetes risk alleles on cancer risk
Observational epidemiology shows several cancers to be positively associated with type 2 diabetes; however, prostate cancer has been consistently found to be inversely associated. In agreement, several type 2 diabetes risk alleles have been found to associate with reduced risk of prostate cancer. Type 2 diabetes risk alleles in PPARG (encoding peroxisome proliferator-activated receptor γ [PPARγ]) [23, 24] and HNF1B (encoding hepatocyte nuclear factor 1 homeobox B) [25] lower the risk of prostate cancer. One study found that 10 out of 36 type 2 diabetes risk alleles were nominally inversely associated with prostate cancer, with only the HNF1B risk allele remaining significant following multiple testing correction [26]. Another study found that four of 13 type 2 diabetes predisposing alleles were nominally inversely associated [27]. Using a genetic risk score (GRS) consisting of 18 type 2 diabetes risk variants, it has been shown that people with higher genetic susceptibility to type 2 diabetes have a reduced risk of prostate cancer [28]. However, some studies have found no association between type 2 diabetes risk variants (either individually or in risk scores) and prostate cancer [29, 30].
Genetic variants in JAZF1 (encoding JAZF zinc finger 1) also have an inverse effect on the relationship between type 2 diabetes and prostate cancer. A recent GWAS found two genetic variants in JAZF1, one with a major allele predisposing to type 2 diabetes [31] and the other with a major allele protecting against prostate cancer [32]. As the genetic variants are not correlated (not in linkage disequilibrium), they may operate through different pathways to alter type 2 diabetes and prostate cancer risk, respectively. This example highlights how genetic variation in the same gene can affect the risk of two diseases without necessarily meaning that the diseases share genetic aetiology.
The direction of effect of type 2 diabetes risk alleles on cancer is often not clear-cut, with some type 2 diabetes predisposing alleles in the same gene increasing the risk of one cancer but protecting against another. For example, type 2 diabetes risk alleles in PPARG are associated with elevated risk of pancreatic cancer [33] and reduced risk of colorectal cancer [34]. This pattern of association suggests the role of PPARG likely depends on the context and should be interpreted accordingly. This finding is also consistent with the described multifaceted role of PPARγ in cancer, wherein it exhibits both tumour-suppressive and tumour-promoting properties [35]. Similar evidence comes from HNF1B, for which type 2 diabetes risk alleles are associated with ovarian cancer but the direction of effect varies by subtype (serous vs clear cell) [36, 37].
Shared susceptibility genes: directionality, tissue specificity and context
Several common susceptibility genes have been implicated in type 2 diabetes and particular cancers but there is currently little evidence for shared genetic aetiology for many of these. For example, common variants in KLF14 (encoding Kruppel-like factor 14, an imprinted transcription factor), associated with lower expression in adult adipose tissue, cause a defect of adipogenesis that is likely to reflect impaired glucose uptake and consequently higher risk of type 2 diabetes [38]. KLF14 expression is reduced in many types of human cancer, including breast, lymphatic, cervical, oral cavity, floor of mouth, pancreas and colorectal cancers [39]. Despite reduced expression having been associated with both type 2 diabetes and certain types of cancer, further studies are needed to investigate whether type 2 diabetes predisposing alleles resulting in reduced expression of KLF14 are also associated with risk of the implicated cancers.
GWAS of type 2 diabetes have consistently identified variants (or colocalising variants) in genes implicated in proliferation and cell cycle regulation [40, 41]. These hallmarks are traditionally associated with cancer; however, direction of effect and tissue specificity are key here. As an illustration, type 2 diabetes risk alleles identified at the CDKN2A/B locus [41] have been implicated in reduced beta cell function [42, 43]. Transgenic mice overexpressing CDKN2A/B display decreased islet proliferation [44], suggesting that the type 2 diabetes risk alleles influence pathology by increasing CDKN2A/B expression. Conversely, rare CDKN2A loss-of-function mutations cause familial melanoma and individuals carrying these mutations have improved beta cell function [45]. The direction of effect here is clearly opposing and even though the gene is implicated in both diseases, the context is vastly different and they would not be expected to share risk alleles at these loci. An important consideration when evaluating evidence for shared genetic aetiology is that many type 2 diabetes predisposing variants are in pancreatic islet enhancers and are therefore unlikely to be implicated in cancer development in other tissues.
Furthermore, type 2 diabetes predisposing variants in genes involved in the insulin signalling pathway are expected to have an opposing direction of effect on cancer. Variants in AKT2 and PTEN have been associated with type 2 diabetes risk; their presence reduces activity of the insulin signalling pathway, leading to reduced glucose uptake and insulin resistance in insulin-responsive tissues. Conversely, tumour cells would typically benefit from enhanced activity of this pathway (through pro-proliferative and anti-apoptotic signals). Indeed, PTEN (a negative regulator of the pathway) and AKT2 are well-characterised tumour suppressor and oncogenes, respectively. Indeed, loss-of-function mutations in PTEN cause a rare cancer-predisposition syndrome and protect against type 2 diabetes (through enhanced insulin sensitivity) [46].
Summary
Several type 2 diabetes susceptibility genes are known to play a role in cancer development (e.g. TCF7L2, CDKN2A/B, AKT2, PPARG, PTEN and HNF1B) but the evidence is relatively scarce for shared genetic aetiology between type 2 diabetes predisposing alleles and the observationally associated cancers. However, there are particular contexts wherein the evidence appears consistent and persuasive: the positive association between type 2 diabetes risk alleles in TCF7L2 and higher risk of breast and colorectal cancer; and the association between type 2 diabetes predisposing alleles and lower risk of prostate cancer.
What quickly becomes apparent when appraising the role of type 2 diabetes susceptibility genes in the development of a particular cancer is that the situation is very rarely clear-cut. Several genes are implicated in both diseases yet in most cases type 2 diabetes predisposing alleles have not been found to be associated with a cancer and often the biological impact of these alleles would be expected to protect against, rather than predispose to, cancer. When evaluating evidence from genetics studies, it is important to consider that in many of the studies the identity of the gene (or genes) through which the type 2 diabetes predisposing variants act has been assumed. The gene is easy to identify if the variant is in a coding region but most loci are in non-protein coding regions of the genome, making it difficult to determine how the variant impacts disease development. While some elegant work is being carried out in this area [47], further work is needed in the functional annotation of type 2 diabetes predisposing variants. Extrapolating this to determine how the gene may behave in the context of cancer is further complicated by the fact that cancer is very much not one disease. Cancers developing at different sites (and indeed different cancer subtypes developing at the same site) will be exposed to different environments, acquire different mutations and will invariably develop differently. Therefore, directionality, tissue specificity and context are key considerations when assessing evidence for shared genetic aetiology between type 2 diabetes and a particular cancer.
Vertical pleiotropy: using genetic variants to investigate a causal pathway to cancer through type 2 diabetes
Despite the lack of convincing evidence that shared genetic aetiology explains the observed positive association between type 2 diabetes and risk of a particular cancer, an observational correlation between diabetes and cancer quite clearly exists. To explore this further, we will appraise the evidence gained from genetics studies employing an MR strategy for a vertical (causal) pathway from type 2 diabetes (or a particular metabolic feature of type 2 diabetes) to cancer (Fig. 1b).
Determining whether there is a causal relationship between type 2 diabetes and cancer has been challenging. Observational epidemiological studies are limited due to certain biases including confounding, measurement error and reverse causation. Confounders are factors that may independently influence both the risk of type 2 diabetes and risk of cancer (e.g. alcohol consumption, adiposity and lower socioeconomic status). Reverse causation is also an important consideration, since the development of certain cancers can precede and cause the development of type 2 diabetes (e.g. pancreatic and liver cancer [problems with traditional epidemiological studies in this context are discussed further here [5, 48]]).
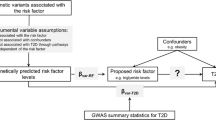
The gold standard study design for inferring causality is a randomised controlled trial (RCT), but it would be ethically unsound and methodologically unfeasible to apply this framework to the question of whether type 2 diabetes causes cancer. A safe, inexpensive alternative is to use a genetic approach: MR [49], essentially a genetic analogue of the RCT [3] (Fig. 2 and Text box: Mendelian randomisation). This powerful use of genetics allows researchers to consider how key environmental and lifestyle factors (e.g. type 2 diabetes) influence complex diseases (e.g. cancer). Certain assumptions must be satisfied in order to perform MR (Fig. 2) and there are particular limitations and considerations when performing MR with cancer as the outcome (comprehensively reviewed elsewhere [50]).

The assumptions of the MR approach. To perform MR, three assumptions (green arrows) need to be satisfied. First, the genetic variants used to proxy the risk factor need to be robustly associated with it. Second, these variants need to be independent of any confounding factors. Third, the variants should only be associated with the outcome through their effect on the risk factor. This figure is available as part of a downloadable slideset
Several studies have used MR to investigate whether there is a causal association between type 2 diabetes and cancer. These studies typically use a GRS composed of type 2 diabetes predisposing genetic variants weighted according to their effect on type 2 diabetes risk. These studies are in agreement in that they find no evidence for an association between the two diseases [51,52,53,54,55], consistent with the lack of evidence from studies assessing the association of single type 2 diabetes predisposing genetic variants and cancer.
Although MR studies do not support a role for type 2 diabetes in cancer development, a number of factors should be considered when interpreting these findings. First, power for an MR study is influenced by the statistical power of the GWAS used. Sample sizes are lower for cancer GWAS than for cardiometabolic disease GWAS, for instance, due to the rarer nature of the disease. Lack of power may limit the ability to detect smaller causal effects using MR. Second, some type 2 diabetes genetic variants have a paradoxical association with the classically defined metabolic features of type 2 diabetes (e.g. a variant in KCNQ1 is associated with both hyperglycaemia and hypoinsulinaemia [52]). These paradoxical associations could potentially cancel each other out. Finally, and most importantly, type 2 diabetes is a markedly complex and heterogeneous disease. It is characterised by a collection of metabolic events and each affected individual presents with a different contribution of metabolic features. Therefore, the observational association between type 2 diabetes and cancer may be driven by a particular metabolic trait of type 2 diabetes or by an associated condition, such as obesity. Below, we discuss the important metabolic traits thought to influence cancer risk in conjunction with evidence from MR studies.
Hyperglycaemia
The diagnostic hallmark of type 2 diabetes is hyperglycaemia. Several observational epidemiological studies have reported a positive correlation between fasting glucose levels and risk of and survival from particular cancers [56,57,58,59,60], even in people without diabetes [57, 59]. Tumour cells preferentially use glucose as their main source of energy and become ‘addicted’ to the pathways processing the sugar in the cell. However, there is little evidence to suggest that tumour cells benefit energetically from hyperglycaemia over and above normoglycaemic conditions [61]. Consistent with this, a meta-analysis of RCTs of intensified glycaemic control did not find evidence of any cancer risk reduction in people with type 2 diabetes [62].
Similarly, MR studies have been unable to support a role for hyperglycaemia in cancer development. Genetic variants inferring lifelong differences in fasting glucose were not associated with lung [63], endometrial [53] or pancreatic cancer [51] or renal cell carcinoma [54]. However, genetic variants inferring differences in 2 h (post-challenge) glucose were positively associated with breast cancer, despite there being no association with fasting glucose in the same study [64]. While this suggests that postprandial glucose levels may be pertinent for cancer development, further investigation is required.
Hyperinsulinaemia
Several observational epidemiological studies have demonstrated that high levels of endogenous insulin are associated with higher risk of cancer incidence and mortality, including risk of colorectal [65], endometrial [66, 67], prostate [68] and breast cancer [69, 70] and breast cancer mortality [71]. Unlike the findings with hyperglycaemia, MR studies support a causal association between higher levels of fasting insulin and risk of endometrial [53], breast [64], lung [63] and pancreatic cancer [51] and renal cell carcinoma [54]. With the exception of higher body fatness, this is by far the most consistent association between a type 2 diabetes-related trait and particular cancers in studies using an MR framework, although replication studies confirming the association found for particular cancers are lacking.
There are several explanations for the causal role of insulin, directly or indirectly, in cancer incidence and risk of mortality. Insulin and insulin-like growth factor 1 (IGF-1) may locally contribute to tumour cell proliferation [72]. Observational epidemiological studies have reported a positive link between circulating IGF-1 levels and increased risk of particular cancers, although associations vary between sites [73]. Evidence from MR studies implicate circulating IGFs in breast cancer [74] and prostate cancer risk and progression [75, 76]. Another purported mechanism is that hyperinsulinaemia inhibits the production of sex hormone binding globulin (SHBG) in the liver, resulting in an elevation of free hormones (including oestrogen and testosterone), which are pro-proliferative and anti-apoptotic [77]. Consistent with this, MR studies have reported an association between sex hormones and puberty timing and breast and endometrial cancer in women [78,79,80] and between puberty timing and prostate cancer in men [78].
Adiposity
Among the risk factors shared by type 2 diabetes and cancer, the most dominant and the most likely to confound observational epidemiology is adiposity. Evidence from observational studies suggests there is a positive relationship between excess body fatness and the risk of 13 cancer types [81]. In support of this, observational studies of weight loss [82] and follow-up studies of patients undergoing bariatric surgery [83] show that weight reduction lowers cancer risk.
Several MR studies support a causal association between higher body fatness and risk of six obesity-related cancers [84], renal cell carcinoma [54], endometrial [85], ovarian [55, 86, 87], oesophageal [88], pancreatic [51, 89, 90] and colorectal cancer [87, 91] and reduced survival in oestrogen receptor-positive (ER+) breast cancer [92]. Higher body fatness has also been found to be causally associated with lung cancer [63, 87], a cancer not observationally associated with higher adiposity or type 2 diabetes. The same study also found a positive association between fasting insulin levels and lung cancer, which may mean we need to re-evaluate the cancers we think of as being associated with metabolic dysregulation.
The weight of evidence therefore suggests there is a causal path between higher adiposity and risk of particular cancers. Effort will now need to be directed into investigating which aspects of excess adiposity are important. Adiposity is a complex phenotype and is associated with considerable metabolic and endocrine abnormalities, therefore it may be metabolic dysfunction that increases the risk of cancer, rather than a higher level of body fat per se. In support of this, despite several metabolic features of type 2 diabetes being found to be positively associated with breast cancer, higher adiposity has been found to have a protective effect in MR studies of both pre- and postmenopausal women [64, 87, 93, 94]. This suggests that the association between metabolic features such as increased fasting insulin levels and breast cancer risk may be independent of higher adiposity. However, observational studies have consistently reported a positive association between high BMI and postmenopausal breast cancer, further emphasising that the relationship between BMI and breast cancer is not straightforward. How weight is gained and when across the life course may be important; there is strong evidence from a recent MR study of a protective effect for larger body size in early life with breast cancer risk, with little evidence for an association with adult body size [95].
MR provides a means to uncouple the impact of higher adiposity from its often associated metabolic dysfunction by using genetic variants associated with ‘favourable adiposity’. The adiposity-increasing alleles at these loci have a paradoxical effect on adiposity and risk of cardiometabolic disease [96,97,98] and are associated with a favourable metabolic profile. Studies using MRI scans of abdominal fat suggest that the underlying mechanism involves an ability to store excess fat in a safe place (subcutaneous adipose tissue), which protects against fat accumulation in ectopic organs like the liver [98]. Employing MR studies to consider the role of favourable vs unfavourable adiposity in cancer risk will indeed be intriguing.
Conclusions and future work
Here, we have considered the evidence from genetics studies that supports the observed association between type 2 diabetes and particular cancers. It is important to consider these studies in the context of the broader scientific literature. Thorough investigation of the link between type 2 diabetes and particular cancers requires the accumulation of several strands of evidence from studies across diverse methodologies including triangulating evidence across observational and genetic epidemiological studies and animal and laboratory-based studies. Indeed, observational and genetic epidemiological studies have different biases, strengths and weaknesses. Hence, if results are in agreement across different methodologies, they are more likely to be robust [99].
Based on appraisal of the available evidence from genetics studies it seems unlikely that the observational association between type 2 diabetes and cancer risk is driven by shared genetic aetiology. It seems more likely to be driven by a particular metabolic feature of type 2 diabetes itself, such as increased fasting insulin levels, or by an associated trait, such as higher adiposity, as demonstrated by MR studies (summarised in Fig. 3). However, further work is needed to fully define this complex relationship and to elucidate the underlying biological mechanisms. Recently, interest has grown in deconstructing or partitioning the type 2 diabetes GRS, grouping the genetic variants in relation to their biological role [100,101,102]. MR can then be employed to assess the causal association between the groups and various cancer types. This could help shed further light on what aspects of this heterogeneous disease are most influential in cancer development and help unpick the underlying mechanisms. In future studies it might also be possible to use MR to explore the potential of particular therapeutic strategies (see Text box: Translational relevance for genetics studies in type 2 diabetes and cancer). It is also likely that epigenetic alterations (driven by type 2 diabetes or associated metabolic features) play a role in promoting cancer development and genetics studies have the potential to help us explore this further [103].

Summary of evidence from MR studies supporting an association between type 2 diabetes (or associated metabolic traits) and cancer. Type 2 diabetes is a complex heterogeneous disease, characterised by a collection of metabolic events and each affected individual presents with a different contribution of metabolic features. MR allows us to consider the causal relationship between type 2 diabetes (when considered as a whole and also each metabolic trait in isolation) and cancer. This approach allows us to investigate whether a particular metabolic trait is responsible for the increased risk of cancer in people with type 2 diabetes. In the figure, the strength of evidence supporting a causal association is defined as follows: none, no evidence for a causal association between trait and cancer; weak, only one study reports a causal association between trait and cancer; moderate, more than one study reports a consistent causal association; strong, several studies report a consistent causal association but replication is limited; very strong, several studies report a consistent causal association and several studies have replicated analyses in the same and some in different cohort data sets. RCC, renal cell carcinoma; T2DM, type 2 diabetes mellitus. This figure is available as part of a downloadable slideset
There are several areas that need further exploration. In particular, genetics studies of type 2 diabetes and cancer progression are largely lacking. A recent analysis where this has been attempted used 143 type 2 diabetes predisposing variants and investigated their association with risk of mortality in ~800 people. Twelve SNPs were associated with risk of breast cancer, three with all-cause mortality and three with breast cancer specific mortality, yet none of the associations were significant after multiple testing corrections [104]. Until data from larger cohorts are available, power is likely to be an issue in such studies. Although a handful of MR studies have been undertaken on type 2 diabetes (or related traits) and cancer progression, methodological challenges remain, such as selection bias (discussed further here [50]).
Abbreviations
- GRS:
-
Genetic risk score
- GWAS:
-
Genome-wide association studies
- HNF1B:
-
Hepatocyte nuclear factor 1 homeobox B
- IGF-1:
-
Insulin-like growth factor 1
- JAZF1:
-
JAZF zinc finger 1
- KLF14:
-
Kruppel-like factor 14
- MR:
-
Mendelian randomisation
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RCT:
-
Randomised controlled trial
- TCF7L2:
-
Transcription factor 7 like 2
References
Diabetes UK (2019) Number of people with diabetes reaches 4.7 million. Available from www.diabetes.org.uk/about_us/news/new-stats-people-living-with-diabetes. Accessed 23 Jun 2020
Giovannucci E, Harlan DM, Archer MC et al (2010) Diabetes and cancer: a consensus report. Diabetes Care 33(7):1674–1685. https://doi.org/10.2337/dc10-0666
Barroso I, McCarthy MI (2019) The genetic basis of metabolic disease. Cell 177(1):146–161. https://doi.org/10.1016/j.cell.2019.02.024
Sud A, Kinnersley B, Houlston RS (2017) Genome-wide association studies of cancer: current insights and future perspectives. Nat Rev Cancer 17(11):692–704. https://doi.org/10.1038/nrc.2017.82
Johnson JA, Carstensen B, Witte D, Bowker SL, Lipscombe L, Renehan AG (2012) Diabetes and cancer (1): evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 55(6):1607–1618. https://doi.org/10.1007/s00125-012-2525-1
Adams JD, Vella A (2018) What can diabetes-associated genetic variation in TCF7L2 teach us about the pathogenesis of type 2 diabetes? Metab Syndr Relat Disord 16(8):383–389. https://doi.org/10.1089/met.2018.0024
Folsom AR, Pankow JS, Peacock JM, Bielinski SJ, Heiss G, Boerwinkle E (2008) Variation in TCF7L2 and increased risk of colon cancer: The Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care 31(5):905–909. https://doi.org/10.2337/dc07-2131
Schumacher FR, Schmit SL, Jiao S et al (2015) Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun 6(1):7138. https://doi.org/10.1038/ncomms8138
Zhang B, Jia W-H, Matsuda K et al (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46(6):533–542. https://doi.org/10.1038/ng.2985
Burwinkel B, Shanmugam KS, Hemminki K et al (2006) Transcription factor 7-like 2 (TCF7L2) variant is associated with familial breast cancer risk: a case-control study. BMC Cancer 6(1):268. https://doi.org/10.1186/1471-2407-6-268
Michailidou K, Hall P, Gonzalez-Neira A et al (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45(4):353–361. https://doi.org/10.1038/ng.2563
Purrington KS, Slager S, Eccles D et al (2014) Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 35(5):1012–1019. https://doi.org/10.1093/carcin/bgt404
Ling Q, Dong F, Geng L et al (2013) Impacts of TCF7L2 gene polymorphisms on the susceptibility of hepatogenous diabetes and hepatocellular carcinoma in cirrhotic patients. Gene 522(2):214–218. https://doi.org/10.1016/j.gene.2013.03.089
Agalliu I, Suuriniemi M, Prokunina-Olsson L et al (2008) Evaluation of a variant in the transcription factor 7-like 2 (TCF7L2) gene and prostate cancer risk in a population-based study. Prostate 68(7):740–747. https://doi.org/10.1002/pros.20732
Zhang M, Tang M, Fang Y et al (2018) Cumulative evidence for relationships between multiple variants in the VTI1A and TCF7L2 genes and cancer incidence. Int J Cancer 142(3):498–513. https://doi.org/10.1002/ijc.31074
Shim H-J, Lee R, Shin M-H, Kim H-N, Kweon S-S (2016) Association between the TCF7L2 polymorphism and colorectal cancer does not differ by diabetes and obesity statuses. Cancer Epidemiol 45:108–111. https://doi.org/10.1016/j.canep.2016.10.012
Sainz J, Rudolph A, Hoffmeister M et al (2012) Effect of type 2 diabetes predisposing genetic variants on colorectal cancer risk. J Clin Endocrinol Metab 97(5):E845–E851. https://doi.org/10.1210/jc.2011-2565
Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487(7407):330–337. https://doi.org/10.1038/nature11252
Pierce BL, Austin MA, Ahsan H (2011) Association study of type 2 diabetes genetic susceptibility variants and risk of pancreatic cancer: an analysis of PanScan-I data. Cancer Causes Control 22(6):877–883. https://doi.org/10.1007/s10552-011-9760-5
Zhao Z, Wen W, Michailidou K et al (2016) Association of genetic susceptibility variants for type 2 diabetes with breast cancer risk in women of European ancestry. Cancer Causes Control 27(5):679–693. https://doi.org/10.1007/s10552-016-0741-6
Chen F, Wilkens LR, Monroe KR et al (2011) No association of risk variants for diabetes and obesity with breast cancer: the Multiethnic Cohort and PAGE studies. Cancer Epidemiol Biomark Prev 20(5):1039–1042. https://doi.org/10.1158/1055-9965.EPI-11-0135
Hou N, Zheng Y, Gamazon ER et al (2012) Genetic susceptibility to type 2 diabetes and breast cancer risk in women of European and African ancestry. Cancer Epidemiol Biomark Prev 21(3):552–556. https://doi.org/10.1158/1055-9965.EPI-11-0979
Paltoo D, Woodson K, Taylor P, Albanes D, Virtamo J, Tangrea J (2003) Pro12Ala polymorphism in the peroxisome proliferator-activated receptor-gamma (PPAR-γ) gene and risk of prostate cancer among men in a large cancer prevention study. Cancer Lett 191(1):67–74. https://doi.org/10.1016/s0304-3835(02)00617-1
Zmuda JM, Modugno F, Weissfeld JL et al (2006) Peroxisome proliferator-activated receptor-γ polymorphism, body mass and prostate cancer risk: evidence for gene-environment interaction. Oncology 70(3):185–189. https://doi.org/10.1159/000093805
Kasper JS, Giovannucci E (2006) A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomark Prev 15(11):2056–2062. https://doi.org/10.1158/1055-9965.EPI-06-0410
Machiela MJ, Lindström S, Allen NE et al (2012) Association of type 2 diabetes susceptibility variants with advanced prostate cancer risk in the Breast and Prostate Cancer Cohort Consortium. Am J Epidemiol 176(12):1121–1129. https://doi.org/10.1093/aje/kws191
Meyer TE, Boerwinkle E, Morrison AC et al (2010) Diabetes genes and prostate cancer in the Atherosclerosis Risk in Communities study. Cancer Epidemiol Biomark Prev 19(2):558–565. https://doi.org/10.1158/1055-9965.EPI-09-0902
Pierce BL, Ahsan H (2010) Genetic susceptibility to type 2 diabetes is associated with reduced prostate cancer risk. Hum Hered 69(3):193–201. https://doi.org/10.1159/000289594
Waters KM, Wilkens LR, Monroe KR et al (2011) No association of type 2 diabetes risk variants and prostate cancer risk: the multiethnic cohort and PAGE. Cancer Epidemiol Biomark Prev 20(9):1979–1981. https://doi.org/10.1158/1055-9965.EPI-11-0019
Yu OHY, Foulkes WD, Dastani Z et al (2013) An assessment of the shared allelic architecture between type II diabetes and prostate cancer. Cancer Epidemiol Biomark Prev 22(8):1473–1475. https://doi.org/10.1158/1055-9965.EPI-13-0476
Zeggini E, Scott LJ, Saxena R et al (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40(5):638–645. https://doi.org/10.1038/ng.120
Thomas G, Jacobs KB, Yeager M et al (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40(3):310–315. https://doi.org/10.1038/ng.91
Tang H, Dong X, Hassan M, Abbruzzese JL, Li D (2011) Body mass index and obesity- and diabetes-associated genotypes and risk for pancreatic cancer. Cancer Epidemiol Biomark Prev 20(5):779–792. https://doi.org/10.1158/1055-9965.EPI-10-0845
Theodoropoulos G, Papaconstantinou I, Felekouras E et al (2006) Relation between common polymorphisms in genes related to inflammatory response and colorectal cancer. World J Gastroenterol 12(31):5037–5043. https://doi.org/10.3748/wjg.v12.i31.5037
Krishnan A, Nair S, Pillai M (2007) Biology of PPARγ in cancer: a critical review on existing lacunae. Curr Mol Med 7(6):532–540. https://doi.org/10.2174/156652407781695765
Shen H, Fridley BL, Song H et al (2013) Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun 4(1):1628. https://doi.org/10.1038/ncomms2629
Pharoah PDP, Tsai Y-Y, Ramus SJ et al (2013) GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet 45(4):362–370. https://doi.org/10.1038/ng.2564
Small KS, Todorčević M, Civelek M et al (2018) Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat Genet 50(4):572–580. https://doi.org/10.1038/s41588-018-0088-x
Fan G, Sun L, Shan P et al (2015) Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun 6(1):8450. https://doi.org/10.1038/ncomms9450
Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42(2):105–116. https://doi.org/10.1038/ng.520
Morris AP, Voight BF, Teslovich TM et al (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44(9):981–990. https://doi.org/10.1038/ng.2383
Dimas AS, Lagou V, Barker A et al (2014) Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 63(6):2158–2171. https://doi.org/10.2337/db13-0949
Grarup N, Rose CS, Andersson EA et al (2007) Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes 56(12):3105–3111. https://doi.org/10.2337/db07-0856
Krishnamurthy J, Ramsey MR, Ligon KL et al (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443(7110):453–457. https://doi.org/10.1038/nature05092
Pal A, Potjer TP, Thomsen SK et al (2016) Loss-of-function mutations in the cell-cycle control gene CDKN2A impact on glucose homeostasis in humans. Diabetes 65(2):527–533. https://doi.org/10.2337/db15-0602
Pal A, Barber TM, Van de Bunt M et al (2012) PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med 367(11):1002–1011. https://doi.org/10.1056/NEJMoa1113966
Mattis KK, Gloyn AL (2020) From genetic association to molecular mechanisms for islet-cell dysfunction in type 2 diabetes. J Mol Biol 432(5):1551–1578. https://doi.org/10.1016/j.jmb.2019.12.045
Renehan AG, Yeh H-C, Johnson JA, Wild SH, Gale EAM, Møller H (2012) Diabetes and cancer (2): evaluating the impact of diabetes on mortality in patients with cancer. Diabetologia 55(6):1619–1632. https://doi.org/10.1007/s00125-012-2526-0
Davey Smith G, Hemani G (2014) Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23(R1):R89–R98. https://doi.org/10.1093/hmg/ddu328
Yarmolinsky J, Wade KH, Richmond RC et al (2018) Causal inference in cancer epidemiology: what is the role of Mendelian randomization? Cancer Epidemiol Biomark Prev 27(9):995–1010. https://doi.org/10.1158/1055-9965.EPI-17-1177
Carreras-Torres R, Johansson M, Gaborieau V et al (2017) The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a Mendelian randomization study. J Natl Cancer Inst 109(9). https://doi.org/10.1093/jnci/djx012
Goto A, Yamaji T, Sawada N et al (2019) Diabetes and cancer risk: a Mendelian randomization study. Int J Cancer 146:712–719. https://doi.org/10.1002/ijc.32310
Nead KT, Sharp SJ, Thompson DJ et al (2015) Evidence of a causal association between insulinemia and endometrial cancer: a Mendelian randomization analysis. J Natl Cancer Inst 107(9). https://doi.org/10.1093/jnci/djv178
Johansson M, Carreras-Torres R, Scelo G et al (2019) The influence of obesity-related factors in the etiology of renal cell carcinoma—a mendelian randomization study. PLoS Med 16(1):e1002724. https://doi.org/10.1371/journal.pmed.1002724
Yarmolinsky J, Relton CL, Lophatananon A et al (2019) Appraising the role of previously reported risk factors in epithelial ovarian cancer risk: a Mendelian randomization analysis. PLoS Med 16(8):e1002893. https://doi.org/10.1371/journal.pmed.1002893
Rao Kondapally Seshasai S, Kaptoge S, Thompson A et al (2011) Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med 364(9):829–841. https://doi.org/10.1056/NEJMoa1008862
Stocks T, Rapp K, Bjørge T et al (2009) Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): analysis of six prospective cohorts. PLoS Med 6(12):e1000201. https://doi.org/10.1371/journal.pmed.1000201
Park SM, Lim MK, Shin SA, Yun YH (2006) Impact of prediagnosis smoking, alcohol, obesity, and insulin resistance on survival in male cancer patients: National Health Insurance Corporation Study. J Clin Oncol 24(31):5017–5024. https://doi.org/10.1200/JCO.2006.07.0243
Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, Samet JM (2005) Fasting serum glucose level and cancer risk in Korean men and women. JAMA 293(2):194–202. https://doi.org/10.1001/jama.293.2.194
Pisani P (2008) Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem 114(1):63–70. https://doi.org/10.1080/13813450801954451
Pollak M (2009) Do cancer cells care if their host is hungry? Cell Metab 9(5):401–403. https://doi.org/10.1016/J.CMET.2009.04.006
Johnson JA, Bowker SL (2011) Intensive glycaemic control and cancer risk in type 2 diabetes: a meta-analysis of major trials. Diabetologia 54(1):25–31. https://doi.org/10.1007/s00125-010-1933-3
Carreras-Torres R, Johansson M, Haycock PC et al (2017) Obesity, metabolic factors and risk of different histological types of lung cancer: a Mendelian randomization study. PLoS One 12(6):e0177875. https://doi.org/10.1371/journal.pone.0177875
Shu X, Wu L, Khankari NK et al (2019) Associations of obesity and circulating insulin and glucose with breast cancer risk: a Mendelian randomization analysis. Int J Epidemiol 48(3):795–806. https://doi.org/10.1093/ije/dyy201
Jenab M, Riboli E, Cleveland RJ et al (2007) Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer 121(2):368–376. https://doi.org/10.1002/ijc.22697
Lukanova A, Zeleniuch-Jacquotte A, Lundin E et al (2004) Prediagnostic levels of C-peptide, IGF-I, IGFBP -1, -2 and -3 and risk of endometrial cancer. Int J Cancer 108(2):262–268. https://doi.org/10.1002/ijc.11544
Gunter MJ, Hoover DR, Yu H et al (2008) A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol Biomark Prev 17(4):921–929. https://doi.org/10.1158/1055-9965.EPI-07-2686
Di Sebastiano KM, Pinthus JH, Duivenvoorden WCM, Mourtzakis M (2018) Glucose impairments and insulin resistance in prostate cancer: the role of obesity, nutrition and exercise. Obes Rev 19(7):1008–1016. https://doi.org/10.1111/obr.12674
Ahern TP, Hankinson SE, Willett WC, Pollak MN, Eliassen AH, Tamimi RM (2013) Plasma C-peptide, mammographic breast density, and risk of invasive breast cancer. Cancer Epidemiol Biomark Prev 22(10):1786–1796. https://doi.org/10.1158/1055-9965.EPI-13-0375
Verheus M, Peeters PHM, Rinaldi S et al (2006) Serum C-peptide levels and breast cancer risk: results from the European Prospective Investigation into Cancer and Nutrition (EPIC). Int J Cancer 119(3):659–667. https://doi.org/10.1002/ijc.21861
Irwin ML, Duggan C, Wang C-Y et al (2011) Fasting C-peptide levels and death resulting from all causes and breast cancer: the health, eating, activity, and lifestyle study. J Clin Oncol 29(1):47–53. https://doi.org/10.1200/JCO.2010.28.4752
Ordener C, Cypriani B, Vuillermoz C, Adessi GL (1993) Epidermal growth factor and insulin induce the proliferation of guinea pig endometrial stromal cells in serum-free culture, whereas estradiol and progesterone do not. Biol Reprod 49(5):1032–1044. https://doi.org/10.1095/biolreprod49.5.1032
Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M (2004) Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet 363(9418):1346–1353. https://doi.org/10.1016/S0140-6736(04)16044-3
Murphy N, Knuppel A, Papadimitriou N et al (2020) Insulin-like growth factor-1, insulin-like growth factor-binding protein-3, and breast cancer risk: observational and Mendelian randomization analyses with ∼430 000 women. Ann Oncol 31(5):641–649. https://doi.org/10.1016/j.annonc.2020.01.066
Bonilla C, Lewis SJ, Rowlands M-A et al (2016) Assessing the role of insulin-like growth factors and binding proteins in prostate cancer using Mendelian randomization: genetic variants as instruments for circulating levels. Int J Cancer 139(7):1520–1533. https://doi.org/10.1002/ijc.30206
Tan VY, Biernacka KM, Dudding T et al (2018) Reassessing the association between circulating vitamin d and igfbp-3: observational and Mendelian randomization estimates from independent sources. Cancer Epidemiol Biomark Prev 27(12):1462–1471. https://doi.org/10.1158/1055-9965.EPI-18-0113
Nestler JE, Powers LP, Matt DW et al (1991) A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab 72(1):83–89. https://doi.org/10.1210/jcem-72-1-83
Day FR, Thompson DJ, Helgason H et al (2017) Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet 49(6):834–841. https://doi.org/10.1038/ng.3841
Thompson DJ, O’Mara TA, Glubb DM et al (2016) CYP19A1 fine-mapping and Mendelian randomization: estradiol is causal for endometrial cancer. Endocr Relat Cancer 23(2):77–91. https://doi.org/10.1530/ERC-15-0386
Dimou NL, Papadimitriou N, Gill D et al (2019) Sex hormone binding globulin and risk of breast cancer: a Mendelian randomization study. Int J Epidemiol 48(3):807–816. https://doi.org/10.1093/ije/dyz107
Lauby-Secretan B, Scoccianti C, Loomis D et al (2016) Body fatness and cancer--viewpoint of the IARC Working Group. N Engl J Med 375(8):794–798. https://doi.org/10.1056/NEJMsr1606602
Eliassen AH, Colditz GA, Rosner B, Willett WC, Hankinson SE (2006) Adult weight change and risk of postmenopausal breast cancer. JAMA 296(2):193–201. https://doi.org/10.1001/jama.296.2.193
Tee MC, Cao Y, Warnock GL, Hu FB, Chavarro JE (2013) Effect of bariatric surgery on oncologic outcomes: a systematic review and meta-analysis. Surg Endosc 27(12):4449–4456. https://doi.org/10.1007/s00464-013-3127-9
Mariosa D, Carreras-Torres R, Martin RM, Johansson M, Brennan P (2019) Commentary: what can Mendelian randomization tell us about causes of cancer? Int J Epidemiol 48(3):816–821. https://doi.org/10.1093/ije/dyz151
Painter JN, O’Mara TA, Marquart L et al (2016) Genetic risk score Mendelian randomization shows that obesity measured as body mass index, but not waist:hip ratio, is causal for endometrial cancer. Cancer Epidemiol Biomark Prev 25(11):1503–1510. https://doi.org/10.1158/1055-9965.EPI-16-0147
Dixon SC, Nagle CM, Thrift AP et al (2016) Adult body mass index and risk of ovarian cancer by subtype: a Mendelian randomization study. Int J Epidemiol 45(3):884–895. https://doi.org/10.1093/ije/dyw158
Gao C, Patel CJ, Michailidou K et al (2016) Mendelian randomization study of adiposity-related traits and risk of breast, ovarian, prostate, lung and colorectal cancer. Int J Epidemiol 45(3):896–908. https://doi.org/10.1093/ije/dyw129
Thrift AP, Shaheen NJ, Gammon MD et al (2014) Obesity and risk of esophageal adenocarcinoma and Barrett’s esophagus: a Mendelian randomization study. J Natl Cancer Inst 106(11):dju252. https://doi.org/10.1093/jnci/dju252
Langdon RJ, Richmond RC, Hemani G et al (2019) A phenome-wide Mendelian randomization study of pancreatic cancer using summary genetic data. Cancer Epidemiol Biomarkers Prev cebp.0036.2019. https://doi.org/10.1158/1055-9965.EPI-19-0036
Lu Y, Gentiluomo M, Lorenzo-Bermejo J et al (2020) Mendelian randomisation study of the effects of known and putative risk factors on pancreatic cancer. J Med Genet. https://doi.org/10.1136/jmedgenet-2019-106200
Thrift AP, Gong J, Peters U et al (2015) Mendelian randomization study of body mass index and colorectal cancer risk. Cancer Epidemiol Biomark Prev 24(7):1024–1031. https://doi.org/10.1158/1055-9965.EPI-14-1309
Guo Q, Burgess S, Turman C et al (2017) Body mass index and breast cancer survival: a Mendelian randomization analysis. Int J Epidemiol 46(6):1814–1822. https://doi.org/10.1093/ije/dyx131
Guo Y, Warren Andersen S, Shu X-O et al (2016) Genetically predicted body mass index and breast cancer risk: Mendelian randomization analyses of data from 145,000 women of European descent. PLoS Med 13(8):e1002105. https://doi.org/10.1371/journal.pmed.1002105
Ooi BNS, Loh H, Ho PJ et al (2019) The genetic interplay between body mass index, breast size and breast cancer risk: a Mendelian randomization analysis. Int J Epidemiol 48(3):781–794. https://doi.org/10.1093/ije/dyz124
Richardson TG, Sanderson E, Elsworth B, Tilling K, Davey Smith G (2020) Use of genetic variation to separate the effects of early and later life adiposity on disease risk: Mendelian randomisation study. BMJ 369:m1203. https://doi.org/10.1136/bmj.m1203
Yaghootkar H, Scott RA, White CC et al (2014) Genetic evidence for a normal-weight “metabolically” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63(12):4369–4377. https://doi.org/10.2337/db14-0318
Yaghootkar H, Lotta LA, Tyrrell J et al (2016) Genetic evidence for a link between favorable adiposity and lower risk of type 2 diabetes, hypertension, and heart disease. Diabetes 65(8):2448–2460. https://doi.org/10.2337/db15-1671
Ji Y, Yiorkas AM, Frau F et al (2019) Genome-wide and abdominal MRI data provide evidence that a genetically determined favorable adiposity phenotype is characterized by lower ectopic liver fat and lower risk of type 2 diabetes, heart disease, and hypertension. Diabetes 68(1):207–219. https://doi.org/10.2337/db18-0708
Munafò MR, Davey Smith G (2018) Robust research needs many lines of evidence. Nature 553(7689):399–401. https://doi.org/10.1038/d41586-018-01023-3
Mahajan A, Wessel J, Willems SM et al (2018) Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet 50(4):559–571. https://doi.org/10.1038/s41588-018-0084-1
Udler MS, Kim J, von Grotthuss M et al (2018) Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med 15(9):e1002654. https://doi.org/10.1371/journal.pmed.1002654
Udler MS, McCarthy MI, Florez JC, Mahajan A (2019) Genetic risk scores for diabetes diagnosis and precision medicine. Endocr Rev 40(6):1500–1520. https://doi.org/10.1210/er.2019-00088
Izquierdo AG, Crujeiras AB (2019) Role of epigenomic mechanisms in the onset and management of insulin resistance. Rev Endocr Metab Disord 20(1):89–102. https://doi.org/10.1007/s11154-019-09485-0
Parada H, Cleveland RJ, North KE et al (2019) Genetic polymorphisms of diabetes-related genes, their interaction with diabetes status, and breast cancer incidence and mortality: the Long Island Breast Cancer Study Project. Mol Carcinog 58(3):436–446. https://doi.org/10.1002/mc.22940
Timpson NJ, Wade KH, Smith GD (2012) Mendelian randomization: application to cardiovascular disease. Curr Hypertens Rep 14(1):29–37. https://doi.org/10.1007/s11906-011-0242-7
Au Yeung SL, Schooling CM (2019) Impact of glycemic traits, type 2 diabetes and metformin use on breast and prostate cancer risk: a Mendelian randomization study. BMJ Open Diabetes Res Care 7(1):e000872. https://doi.org/10.1136/bmjdrc-2019-000872
Au Yeung SL, Luo S, Schooling CM (2019) The impact of GDF-15, a biomarker for metformin, on the risk of coronary artery disease, breast and colorectal cancer, and type 2 diabetes and metabolic traits: a Mendelian randomisation study. Diabetologia 62(9):1638–1646. https://doi.org/10.1007/s00125-019-4913-2
Yarmolinsky J, Bull C, Walker V, Nounu A, Davey Smith G (2020) Mendelian randomization applied to pharmaceutical use: the case of metformin and lung cancer. Int J Epidemiol. https://doi.org/10.1093/ije/dyaa059
Acknowledgements
We would like to acknowledge all researchers that have contributed to this field. We also thank C. Bull (Cellular and Molecular Medicine, University of Bristol, UK), V. Tan (Population Health Science, University of Bristol, UK) and M. Gormley (Population Health Science, University of Bristol, UK) for critical feedback on the manuscript. We apologise to scientists whose work could not be highlighted due to space and reference limitations.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
EEV acknowledges funding by grants from a Diabetes UK RD Lawrence Fellowship (17/0005587), the World Cancer Research Fund (WCRF UK), as part of the World Cancer Research Fund International grant programme (IIG_2019_2009) and Cancer Research UK (C18281/A19169). HY acknowledges support by grants from a Diabetes UK RD Lawrence fellowship (17/0005594).
Author information
Authors and Affiliations
Contributions
Both authors were responsible for drafting the article and revising it critically for important intellectual content. Both authors approved the version to be published.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Slideset of figures
(PPTX 431 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vincent, E.E., Yaghootkar, H. Using genetics to decipher the link between type 2 diabetes and cancer: shared aetiology or downstream consequence?. Diabetologia 63, 1706–1717 (2020). https://doi.org/10.1007/s00125-020-05228-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05228-y