
Abstract
Aims/hypothesis
Here, we describe the characteristics of the Innovative Medicines Initiative (IMI) Diabetes Research on Patient Stratification (DIRECT) epidemiological cohorts at baseline and follow-up examinations (18, 36 and 48 months of follow-up).
Methods
From a sampling frame of 24,682 adults of European ancestry enrolled in population-based cohorts across Europe, participants at varying risk of glycaemic deterioration were identified using a risk prediction algorithm (based on age, BMI, waist circumference, use of antihypertensive medication, smoking status and parental history of type 2 diabetes) and enrolled into a prospective cohort study (n = 2127) (cohort 1, prediabetes risk). We also recruited people from clinical registries with type 2 diabetes diagnosed 6–24 months previously (n = 789) into a second cohort study (cohort 2, diabetes). Follow-up examinations took place at ~18 months (both cohorts) and at ~48 months (cohort 1) or ~36 months (cohort 2) after baseline examinations. The cohorts were studied in parallel using matched protocols across seven clinical centres in northern Europe.
Results
Using ADA 2011 glycaemic categories, 33% (n = 693) of cohort 1 (prediabetes risk) had normal glucose regulation and 67% (n = 1419) had impaired glucose regulation. Seventy-six per cent of participants in cohort 1 was male. Cohort 1 participants had the following characteristics (mean ± SD) at baseline: age 62 (6.2) years; BMI 27.9 (4.0) kg/m2; fasting glucose 5.7 (0.6) mmol/l; 2 h glucose 5.9 (1.6) mmol/l. At the final follow-up examination the participants’ clinical characteristics were as follows: fasting glucose 6.0 (0.6) mmol/l; 2 h OGTT glucose 6.5 (2.0) mmol/l. In cohort 2 (diabetes), 66% (n = 517) were treated by lifestyle modification and 34% (n = 272) were treated with metformin plus lifestyle modification at enrolment. Fifty-eight per cent of participants in cohort 2 was male. Cohort 2 participants had the following characteristics at baseline: age 62 (8.1) years; BMI 30.5 (5.0) kg/m2; fasting glucose 7.2 (1.4) mmol/l; 2 h glucose 8.6 (2.8) mmol/l. At the final follow-up examination, the participants’ clinical characteristics were as follows: fasting glucose 7.9 (2.0) mmol/l; 2 h mixed-meal tolerance test glucose 9.9 (3.4) mmol/l.
Conclusions/interpretation
The IMI DIRECT cohorts are intensely characterised, with a wide-variety of metabolically relevant measures assessed prospectively. We anticipate that the cohorts, made available through managed access, will provide a powerful resource for biomarker discovery, multivariate aetiological analyses and reclassification of patients for the prevention and treatment of type 2 diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction

The global prevalence of type 2 diabetes is burgeoning. There is no cure, nor are there treatments effective enough to halt the progression of the disease. The burden the disease conveys at a societal and personal level is enormous, with an estimated world prevalence in 2017 of around 425 million people with type 2 diabetes and a further 352 million at risk of developing the disease [1]. The global cost of diagnosing and treating the disease and its complications in 2017 was estimated to be around €730 billion [1]. This bleak picture emphasises the profound shortcomings in our understanding of type 2 diabetes aetiology and pathogenesis, and the inadequate tools available with which to combat the disease.
Like some other complex diseases, the clinical presentation and prognosis of type 2 diabetes is heterogeneous. The risk conveyed by established diabetogenic factors such as obesity, physical inactivity and certain dietary components varies widely from one person to the next, as does the response to interventions targeting these risk factors. This is also true for those in whom diabetes is manifest, with response to glucose-lowering therapies, occurrence of adverse events and rates of progression being variable and hard to predict.
The diagnosis of type 2 diabetes is relatively straightforward, relying primarily on evidence of chronically elevated blood glucose concentrations [2]. However, elevated blood glucose concentrations can be the consequence of multiple defects in energy metabolism occurring across several organs and tissues [3,4,5] caused by myriad acquired or inherited factors. Thus, type 2 diabetes as currently defined characterises a collection of underlying pathologies [6], each with the common feature of elevated blood glucose that may require tailored therapies. The stratification of type 2 diabetes into treatable subclasses might be possible if accessible biomarkers of the disease’s underlying pathologies were known.
Although improving the management of type 2 diabetes through subclassification may lead to more focused treatment, susceptibility to risk factors and response to treatments also vary. Thus, stratifying patient populations into subgroups defined using biomarkers quantifying susceptibility to risk factors and responsiveness to specific therapeutics would further enhance our ability to treat and ideally prevent the disease.
The Innovative Medicines Initiative (IMI) Diabetes Research on Patient Stratification (DIRECT) Consortium is a collaboration among investigators from some of Europe’s leading academic institutions and pharmaceutical companies [7]. The overarching objective of IMI DIRECT is to discover and validate biomarkers of glycaemic deterioration before and after the onset of type 2 diabetes. To this end, we established two new multicentre prospective cohort studies comprised of adults from northern Europe at risk of or with recently diagnosed type 2 diabetes. Within these cohorts, a comprehensive array of risk factors, intermediate phenotypes and metabolic outcomes were repeatedly assessed using cutting-edge technologies. The richly phenotyped IMI DIRECT cohorts will facilitate the discovery of biomarkers for glycaemic control in individuals at risk of or with type 2 diabetes.
Here we describe the characteristics of the two IMI DIRECT cohorts at baseline and at the two major follow-up visits up to 48 months later, to provide context for those subsequently analysing and reviewing studies based on these data. We also consider these results in the context of the implemented protocols and plans outlined at the beginning of the project, as described previously [7].
Methods
The rationale and design of the epidemiological cohorts within IMI DIRECT are reported elsewhere [7]; here we provide data and information about key variables and methods, respectively, that were not described in the rationale and design paper published previously.
Approval for the study protocol was obtained from each of the regional research ethics review boards separately and all participants provided written informed consent at enrolment. The research conformed to the ethical principles for medical research involving human participants outlined in the declaration of Helsinki.
Recruitment, enrolment and eligibility
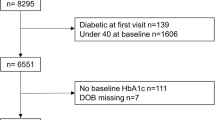
The derivations of cohort 1 and cohort 2 are shown in Fig. 1. The sampling frame for cohort 1 comprised four existing prospective cohort studies: Metabolic Syndrome in Men (METSIM, Finland) [8]; Relationship between Insulin Sensitivity and Cardiovascular disease (RISC) [9], Hoorn Meal Study (HMS) and New Hoorn Study (NHS) [10] (Netherlands); Health2010 [11], Health2006 [12], Danish Study of Functional Disorders (DanFunD) [13] and Gut, Grain and Greens (GGG) [14] studies (Denmark) and Malmö Diet and Cancer (MDC) study (Sweden) [15]. Participants for cohort 2 were identified through general practice and other registries, as described previously [7].

Participant flow of cohorts 1 and 2. DanFunD, Danish Functional Disability study; GGG, Gut, Grain and Greens study; HMS, Hoorn Meal Study; METSIM, Metabolic Syndrome in Men study; NHS, New Hoorn Study; RISC, Relationship between Insulin Sensitivity and Cardiovascular disease cohort
After excluding participants who did not meet the inclusion criteria or whose data failed quality control, a total of 2127 participants at risk of diabetes and 789 participants with type 2 diabetes were retained in cohort 1 and cohort 2, respectively. In cohort 1, emphasis was placed on recruiting participants deemed at high risk of type 2 diabetes according to ADA 2011 HbA1c criteria (HbA1c 40–48 mmol/mol [5.7–6.4%]) [2, 7]. As anticipated during the design phase, the sampling frame contained too few participants that fulfilled this criterion; thus, we proceeded to enrol participants with progressively lower HbA1c concentrations, who were also considered at highest risk of glycaemic deterioration according to the DIRECT-DETECT risk algorithm (based on age, BMI, waist circumference, use of antihypertensive medication, smoking status and parental history of type 2 diabetes) applied to the parent cohort sampling frame [7, 16].
In cohort 1, 1989 (93%) participants enrolled at baseline also attended the first major follow-up visit at a mean of 18.6 (SD 1.4) months and 1729 (81%) attended the final follow-up visit 30.8 (SD 1.3) months later. In cohort 2, 668 (85%) participants enrolled at baseline attended their first major follow-up visit at 18.2 (SD 0.6) months and 552 (70%) attended the final follow-up visit 18.2 (SD 1.0) months later.
Glycaemic biochemistry assays
Plasma glucose, insulin and C-peptide assays for cohort 1 were carried out centrally at the University of Eastern Finland (Kuopio, Finland), where plasma glucose was analysed using the enzymatic glucose hexokinase method and photometric measurement on Konelab 20 XT Clinical Chemistry analyser (Thermo Fisher Scientific, Vantaa, Finland). In cohort 2, plasma insulin and C-peptide were analysed using chemiluminometric immunoassay (CLIA) (Liaison Insulin [DiaSorin, Saluggia, Italy] and Liaison C-peptide [DiaSorin]). The instrument used was DiaSorin Liaison Analyser (DiaSorin Deutschland, Dietzenbach, Germany). Plasma glucose, insulin and C-peptide assessments for cohort 2 were carried out centrally at the University of Exeter (Exeter, UK). Assessments of HbA1c, blood lipids, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) for both cohorts were carried out centrally at the University of Exeter. Glucose was measured by the enzymatic colorimetric assay GOD-PAP using Roche MODULAR P analysers (Hoffmann-La Roche, Basel, Switzerland). Insulin was measured by electrochemiluminescence using Roche E170 Analysers (Hoffmann-La Roche). C-peptide concentrations in plasma and urine were measured by electrochemiluminescence using Roche E170 Analysers (Hoffmann-La Roche). HbA1c was measured by ion-exchange high-performance liquid chromatography using Tosoh G8 analysers (Tosoh Bioscience, San Francisco, CA, USA). Each biochemical assay was performed using validated standard methods. Reference samples were included in all procedures to control for inter-assay variation and laboratories regularly participated in international external quality assessment schemes. In addition, a subset of samples was assayed for C-peptide, insulin and glucose on both sites to assess inter-laboratory variation.
Blood lipid and liver enzyme biochemistry assays
Triacylglycerol was measured by quantitative determination with glycerol blanking. HDL-cholesterol was measured directly using polyethylene glycol-modified enzymes and dextran sulphate. When cholesterol esterase and cholesterol oxidase enzymes are modified by polyethylene glycol, they show selective catalytic activity towards lipoprotein fractions, with reactivity increasing in the order LDL < VLDL ≈ chylomicrons < HDL. Total cholesterol was measured by an enzymatic, colorimetric method. LDL-cholesterol was calculated from the total cholesterol, HDL-cholesterol and triacylglycerol concentrations using the Friedewald formula: LDL = total cholesterol – HDL-cholesterol – (triacylglycerol/2.2). ALT and AST were measured by UV absorbance without pyridoxal phosphate activation. ALT, AST, cholesterol, glucose, triacylglycerol and HDL-cholesterol were measured using a Roche MODULAR P analyser (Roche Diagnostics, Indianapolis, IN, USA). Insulin and C-peptide were measured using a Roche E170 analyser (Roche Diagnostics).
Blood glucagon-like peptide-1 assays
Plasma concentrations of glucagon-like peptide-1 (GLP-1) were determined by drawing blood samples collected at two different time points (0 and 60 min) during the 75 g frequently sampled OGTT (fsOGTT)/mixed-meal tolerance test (MMTT) (baseline samples only). P800 tubes (Becton Dickinson, Wokingham, UK) were used to provide immediate protection from intrinsic proteolysis. Quantitative determination of active GLP-1 was achieved using MSD GLP-1 active kit (product code K150JWC; Meso Scale Diagnostics, Rockville, MD, USA). Total GLP-1 was assayed using MSD GLP-1 total kit (product code K150JVC; Meso Scale Diagnostics).
Abdominal MRI analyses
The volume of adipose tissue was measured in litres using MRI, as described elsewhere [17]. Total abdominal adipose tissue (TAAT) may be separated into intra-abdominal adipose tissue (IAAT), also known as ‘visceral’ fat, and abdominal subcutaneous adipose tissue (ASAT). IAAT is the volume of adipose tissue within the abdominal cavity. TAAT is the sum of IAAT and ASAT. Liver and pancreas fat and iron (T2*) were derived simultaneously using a multiecho MRI technique, as previously described [17, 18]. This method has the advantage over single voxel MR spectroscopy in that regional differences in ectopic fat distribution can be measured. Furthermore, it is often possible to obtain a single slice quantification of the liver and pancreas, allowing simultaneous measurement of fat and iron within two separate organs. A biexponential curve-fitting model was used to derive the relative signal contributions from fat and water from the many images normally obtained with the multiecho sequence. Briefly, tissue with no fat infiltration generates a very smooth decay curve, whereas tissue containing a higher level of fat is characterised by significant oscillations throughout the decay curve [18]. A further output from the multiecho technique is T2* tissue values; as changes in these are indicative of iron content, this provides a clinically relevant additional measurement. Tissue iron concentration (mg/g dry weight tissue) was estimated from T2* using a validated model [19].
Diet assessment
Self-reported dietary intake was assessed by a 24 h multi-pass method, using food habit and 24 h recall questionnaires. Analysis of these diet data was undertaken using Dietplan-6 (version 6.70.43, 2013; Forestfield Software, Horsham, UK). The specific analysis methods are described in detail elsewhere [7]. We also objectively assessed diet using discriminative metabolite signatures, an approach described in detail elsewhere [20]. Briefly, each participant’s serum metabolite profile was obtained using a targeted metabolomics assay (AbsoluteIDQ p180 Kit; BIOCRATES Life Sciences, Innsbruck, Austria), which simultaneously quantifies 188 metabolites. In a previously published diet intervention study [20], serum samples had been collected in 19 participants who had undergone a metabolic ward-based supervised diet intervention. We assayed these blood samples using the BIOCRATES AbsoluteIDQ p180 Kit and derived diet-discriminatory metabolomic signatures, using previously described methods [20]. These data were then used to predict the dietary characteristics of the IMI DIRECT study participants.
Physical activity assessment
Objective measures of physical activity were derived from triaxial accelerometers (ActiGraph GT3X+/GT3X+w/GT3X+bt; ActiGraph Co., Pensacola, FL, USA) as described previously [7]. Raw data files (.gt3x) were converted to comma separated value (.csv) format storing rawest possible accelerations for each axis at a resolution of 30 Hz using ActiLife 6 (version 6.11.5; ActiGraph Co.). All inferred measures of physical activity were calculated using PAMPRO (version uploaded 21 Oct 2015; MRC Epidemiology unit, Cambridge, UK), custom open source software available under public license (https://github.com/Thomite/pampro). Data from each axis of acceleration was auto-calibrated to local gravity. Non-wear was inferred as a vector magnitude SD of less than 4 mg for a consecutive period greater than 60 min. All measures presented here have been adjusted for diurnal rhythm to account for bias from non-wear removal. However, due to the wear method (non-dominant wrist fastened using the manufacturer’s non-removable hospital band), intermittent non-wear time was rare. The main physical activity estimates presented here are high-pass-filtered vector magnitude (hpfVM), which infers intensity of participants’ movement in any direction at any given time (here, averaged during wear period). Time spent in established physical activity intensities by physical activity energy expenditure was estimated using calculated hpfVM cutpoints: sedentary (<48 mg hpfVM), light (48–154 mg hpfVM), moderate (154–389 mg hpfVM) and vigorous (>389 mg hpfVM). The methods used to infer these measures have been validated and described in detail elsewhere [21].
DNA extraction and genotyping
DNA extraction was carried out using Maxwell 16 Blood DNA purification kits and a Maxwell 16 semi-automated nucleic acid purification system (Promega, Southampton, UK). Genotyping was conducted using the Illumina HumanCore array (HCE24 v1.0) and genotypes were called using Illumina’s GenCall algorithm. Samples were excluded for any of the following reasons: call rate <97%; low or excess mean heterozygosity; sex discordance and monozygosity. Genotyping quality control was then performed to provide high-quality genotype data for downstream analyses using the following criteria: call rate <99%; deviation from Hardy–Weinberg equilibrium (exact p < 0.001); variants not mapped to human genome build GRCh37 and variants with duplicate chromosome positions (a total of 30,318 markers were excluded). A total of 3032 samples and 517,958 markers across the two studies passed quality control procedures. We took autosomal variants with MAF >1% that passed quality control and constructed axes of genetic variation using principal components analysis implemented in the GCTA (version 1.24.4, downloaded from https://cnsgenomics.com/software/gcta/#Download) software to identify ethnic outliers defined as non-European ancestry using the 1000 Genomes samples as reference. We identified six individuals as ethnic outliers.
Additional measures (not presented here)
Biomarker discovery analyses using these data also employ additional measures (including ‘omic’ measures), which are outside the scope of this cohort description. Additional measures that are not described here include transcriptomics (RNA sequencing from fasting whole blood), microbiomics (DNA isolation and deep sequencing in faecal samples), proteomics (targeted array in fasting plasma) and metabolomics (targeted and untargeted assays in fasting plasma). GAD/islet antigen-2 assessments from fasting serum samples were also undertaken. Data from the Recent Physical Activity Questionnaire (RPAQ) and sleep diaries were also collected in subcohorts.
Statistical power of study
A detailed section on sample size and power for the study is available in the previously published rationale and design paper [7]. Briefly, statistical power will vary depending on a number of factors specific to the analysis to be carried out, such as biomarker effect sizes, variance/frequency of outcome and biomarker, statistical modelling methods employed, number of tests (multiple testing adjustment) and of course sample size, available for the relevant variables included in the model. The dataset will therefore be well powered for some analyses while it may be underpowered for other analyses and will thus be covered in detail for the specific scenarios in subsequent analyses.
Statistical methods for descriptive data
Based on ADA 2011 diagnostic criteria, impaired fasting plasma glucose is 5.6–6.9 mmol/l (100–125 mg/dl), impaired glucose tolerance is 2 h 75 g OGTT plasma glucose 7.8–11.0 mmol/l (140–199 mg/dl) and prediabetes HbA1c is 40–48 mmol/mol (5.7–6.4%) [2]. Accordingly, below and above these cut-offs was considered ‘normal’ and ‘diabetic’ ranges, respectively. Cohort 1 was stratified into two categories of blood glucose level: normal glucose regulation (NGR) and impaired glucose regulation (IGR). NGR was defined as the HbA1c, fasting glucose and 2 h glucose values being within the normal ranges for each measure. IGR was defined as there being impaired values in at least one of HbA1c, fasting glucose or 2 h glucose. Cohort 2 was stratified into treatment categories: lifestyle advice only or metformin plus lifestyle advice. Descriptive statistics are presented as mean ± SD. Pairwise Pearson correlations were calculated for all key variables described here. For these analyses, continuous variables were first inverse normal transformed and then adjusted for age, sex and study centre by two-step residual regression. We present the same type of data for anthropometric and glycaemic variables for the main follow-up visits, as well as the differences for these variables between the baseline and the final follow-up visit measures (follow-upΔ = final follow-up value – baseline value). We also calculated pairwise Pearson correlations for the follow-upΔ values; for these analyses, continuous variables were first inverse normal transformed and then adjusted for age, sex, study centre and days since baseline visit by two-step residual regression. All statistics were computed using R software version 3.4.0 [22]. The IMI DIRECT data release version used for the analyses in this article was direct_03-29-2019.
Glycaemic trait modelling
Glycaemic traits were derived from the 75 g fsOGTT (sampling at 0, 15, 30, 45, 60, 90, 120 min) and MMTT (sampling at 0, 30, 60, 90, 120 min) for cohort 1 and cohort 2, respectively. Analyses used a mathematical model that describes the relationship between insulin secretion and glucose concentration [23, 24]. The model expresses insulin secretion as the sum of two components, the first of which represents the dependence of insulin secretion on absolute glucose concentration at any time during the fsOGTT/MMTT, through a dose–response function. Characteristic parameters of the dose–response relationship are the mean slope over the observed glucose range, denoted as glucose sensitivity. The dose–response relationship is modulated by a potentiation factor, which accounts for the fact that during acute stimulation, insulin secretion is higher on the descending phase of hyperglycaemia than at the same glucose concentration on the ascending phase. In participants with NGR and normal insulin secretion, the potentiation factor typically increases from baseline to the end of a 2 h OGTT [25]. To quantify this excursion, the ratio between the 2 h and the baseline value was calculated. This ratio is denoted as potentiation ratio and reflects late insulin release. The second insulin secretion component represents the dependence of insulin secretion on the rate of change of glucose concentration. This component is termed derivative component and is determined by a single parameter, denoted as rate sensitivity. Rate sensitivity reflects early insulin release [25].
The model parameters were estimated from glucose and C-peptide concentrations by regularised least-squares, as previously described [23]. Regularisation involves the choice of smoothing factors, which were selected to obtain glucose and C-peptide model residuals with SDs close to expected measurement error (~1% for glucose and ~4% for C-peptide). Insulin secretion rates were calculated from the model every 5 min. The integral of insulin secretion during the fsOGTT was denoted as total insulin output.
The validity of the fsOGTT and MMTT for the assessment of insulin sensitivity has been shown in the original publications presenting the indices [26,27,28]. In the studies, the OGTT/MMTT indices are compared with values obtained by euglycaemic glucose clamp. The validity of the beta cell function model is supported by numerous studies [25]. For beta cell function, it is not possible to validate an OGTT/MMTT method against the classical tests with glucose intravenous infusion, due to the presence of the incretin effect. However, it has been shown that the estimated beta cell dose–response is consistent with the graded glucose infusion test across the spectrum of glucose tolerance [29, 30].
Results
Cohort 1 (prediabetes)
Of 2235 enrolled participants in cohort 1, 2127 passed all inclusion, exclusion and quality control criteria. Of these, 1419 (67%) had IGR according to at least one ADA category for HbA1c, fasting glucose or 2 h glucose [2] and were thus within the target ‘prediabetes’ range. A total of 693 participants (33% of cohort 1) displayed NGR according to all three glycaemic measures. Participants with prevalent type 2 diabetes (n = 105) or who withdrew from the study (n = 3) were excluded from further analyses.
The number of participants enrolled into cohort 1 varied between centres, with the Finnish subcohort being the largest (providing 58% [n = 1240]) of the total cohort 1 baseline sample. The other centres in the Netherlands, Denmark and Sweden enrolled 22% (n = 473), 13% (n = 275) and 7% (n = 139) of the total cohort, respectively.
The ratio of men to women varied in each subcohort, with all participants at the Finnish centre being male, and 43%, 45% and 29% being male in the subcohorts from the Netherlands, Denmark and Sweden, respectively.
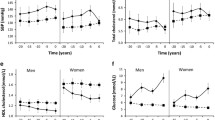
Detailed participant baseline characteristics for cohort 1 are shown in Table 1 (and stratified by glycaemic category in electronic supplementary material [ESM] Table 1). Figure 2 shows the pairwise correlations between a selection of key phenotypic variables at baseline. Participant characteristics at the follow-up visits and the difference (Δ) between baseline and final follow-up for cohort 1 are shown in Table 2. The pairwise correlations between the baseline to final follow-up difference for anthropometric and glucose-control variables are shown in Fig. 3.

Pairwise correlation matrix. Fill colour indicates Pearson correlation coefficient (r), where positive is denoted by red fill, inverse by blue fill and magnitude by intensity. Cohorts 1 and 2 are separate, above and below the diagonal, respectively. All continuous variables were normally transformed and adjusted for age, sex and study centre. ActGLP1min0, fasting intact GLP-1 concentration; BasalISR, fasting insulin secretion; CHOI, carbohydrate intake; Chol, total cholesterol; DBP, diastolic blood pressure; FatI, fat intake; FibreI, fibre intake; Glucagonmin0, fasting glucagon; Glucose, fasting glucose; GlucoseSens, glucose sensitivity; HDL, fasting HDL-cholesterol; IncGLP1min60, 1 h GLP-1 increment; IncGlucagonmin60, 1 h glucagon increment; Insulin, fasting insulin; LDL, fasting LDL-cholesterol; LiverFat, liver fat; LiverIron, liver iron content; LPA, light physical activity (% of time); Matsuda, Matsuda insulin sensitivity index; MeanGlucose, mean 2 h glucose; MeanInsulin, mean 2 h insulin; MPA, moderate physical activity (% of time); MUFatI, monounsaturated fat intake; OGIS, 2 h insulin sensitivity; PA, mean physical activity intensity; hpfVM; PancFat, pancreatic fat; PancIron, pancreatic iron content; PFR, potentiation factor ratio; ProInsmin60, 1 h intact proinsulin; ProteinI, protein intake; PUFatI, polyunsaturated fat intake; RateSens, rate sensitivity; SatFatI, saturated fat intake; SBP, systolic blood pressure; SPA, sedentary (% of time); Stumvoll, Stumvoll insulin sensitivity index; SugarI, sugar intake; TEI, total energy intake; TG, fasting triacylglycerol; TotalISR, integral of total insulin secretion; TotGLP1min0, fasting total GLP-1 concentration; TwoGlucose, 2 h glucose; TwoInsulin, 2 h insulin; VPA, vigorous physical activity (% of time)

Pairwise correlation matrix of follow-up Δ (difference between 48 month follow-up assessment and baseline visit). Fill colour indicates Pearson correlation coefficient (r), where positive is denoted by red fill, inverse by blue fill and magnitude by intensity. Cohorts 1 and 2 are separate, above and below diagonal, respectively. All continuous variables were normally transformed and adjusted for age, sex and study centre. BasalISR, fasting insulin secretion; DBP, diastolic blood pressure; Glucose, fasting glucose; GlucoseSens, glucose sensitivity; Insulin, fasting insulin; Matsuda, Matsuda insulin sensitivity index; MeanGlucose, mean 2 h glucose; MeanInsulin, mean 2 h insulin; OGIS, insulin sensitivity (2 h OGIS); PFR, potentiation factor ratio; RateSens, rate sensitivity; SBP, systolic blood pressure; Stumvoll, Stumvoll insulin sensitivity index; TotalISR, integral of total insulin secretion; TwoGlucose, 2 h glucose; TwoInsulin, 2 h insulin.
Briefly, at baseline, participants had a mean (SD) age of 62 (6.2) years, BMI 27.9 (4.0) kg/m2, HbA1c 37 (2.9) mmol/mol [5.5 (0.27)%], fasting glucose 5.7 (0.6) mmol/l, 2 h glucose 5.9 (1.6) mmol/l, fasting insulin 10.9 (7.6) pmol/l, glucose sensitivity 113 (55) pmol min−1 m−2 (mmol/l)−1 and insulin sensitivity (2 h oral glucose insulin sensitivity [OGIS]) 381 (59) ml min−1 m−2. Participants had a 0–48 month mean (SD) difference in fasting plasma glucose levels of 0.3 (0.2) mmol/l.
Cohort 2 (diabetes)
Of 830 individuals in cohort 2 enrolled to attend the screening visit, 789 passed all inclusion, exclusion and quality control criteria. Of these, 272 were treated with lifestyle modification plus metformin and 517 were treated with lifestyle intervention only. Participants who withdrew consent, who were receiving any other oral hypoglycaemic agent or who reported ever receiving insulin treatment were excluded (n = 41).
Of the participants in cohort 2 at baseline, the UK (Dundee, Exeter, Newcastle), Dutch (Amsterdam), Swedish (Lund) and Danish (Copenhagen) study centres enrolled 21% (n = 167), 18% (n = 141), 21% (n = 166), 21% (n = 167), 12% (n = 96) and 7% (n = 52) of the total cohort, respectively; 52–63% of the subcohort participants were male.
Detailed participant characteristics and key variables for cohort 2 at baseline are shown in Table 1 (and stratified by treatment category in ESM Table 1). Figure 2 shows the pairwise correlation matrix for key variables at baseline adjusted for age, sex and study centre. Participant characteristics for follow-up visits and the difference between the baseline and final follow-up visit (Δ) for cohort 2 are shown in Table 2. A pairwise correlation matrix for the anthropometric and glucose-control Δ variables are shown in Fig. 3.
Briefly, at baseline, participants had a mean (SD) age 62 (8.1) years, BMI 30.5 (5.0) kg/m2, HbA1c 46.5 (5.8) mmol/mol [6.4 (0.53)%], fasting glucose 7.2 (1.4) mmol/l, 2 h glucose 8.6 (2.8) mmol/l, fasting insulin 107 (71) pmol/l, glucose sensitivity 83 (55) pmol min−1 m−2 (mmol/l)−1 and insulin sensitivity (2 h OGIS) 298 (69) ml min−1 m−2. Participants had a 0 months to 18 months mean difference in fasting plasma glucose levels of 0.8 (1.9) mmol/l.
Genetic population substructure
As some study centres enrolled participants into both cohorts, we elected to characterise the genetic population substructure across the cohorts by study centre (i.e. pooling both cohorts at a given centre where possible). Genetic substructures closely map to the geographic location of the populations [31], indicating ethnic homogeneity within regions from which the cohorts were recruited, whereas there is far greater heterogeneity between centres, the latter driven mainly by the inclusion of Finnish participants. This is illustrated in Fig. 4 where Finnish participants form a distinct cluster (to the north east) compared with the population from the other cohorts.

Genetic population structure within the cohorts. A statistical summary of genetic data from cohorts 1 and 2 (combined) based on principal component axis one (PC1) and axis two (PC2). Points are coloured according to recruitment centre
Discussion
Here, we report the characteristics of the IMI DIRECT cohorts at baseline, at 18 months follow-up, and at 48 or 36 months follow-up (for cohorts 1 and 2 respectively) for glycaemic deterioration and consider these results in the context of the implemented protocols and the plans outlined in the design and rationale paper published previously [7]. The descriptive statistics, pairwise correlations and genetic substructures presented in this article are not intended for aetiological inference; instead, the purpose is to provide context and details for subsequent IMI DIRECT papers, as well as to inform scientists outside the Consortium who might in the future consider using the IMI DIRECT data in their research.
Major advances in technologies and methods over the past decade make high-resolution quantification of disease phenotypes and processes possible in large sample collections. Applying modern assays to historical biosamples is particularly useful when studying processes that take decades to unfold. However, biosamples often degrade during long-term storage and many older studies did not deploy the advanced phenotyping methods available today. Recognising these limitations, we designed and initiated two state-of-the-art prospective cohort studies as part of the IMI DIRECT Consortium. Designed for biomarker discovery in glycaemic deterioration and diabetes progression, the IMI DIRECT cohorts include conventional and cutting-edge phenotyping techniques and technologies that are repeated on multiple occasions during a follow-up period of up to 48 months (currently ongoing). We note that the subsequent biomarker discovery analyses using these cohorts will combine the clinical phenotypic data described in this paper with omic measures such as transcriptomics (RNA sequencing from fasting whole blood), microbiomics (DNA isolation and deep sequencing in faecal samples), proteomics (targeted array in fasting plasma) and metabolomics (targeted and untargeted assays in fasting plasma).
The recruitment strategies for the two IMI DIRECT cohorts differed in that cohort 1 focused on recruiting participants from an existing large sample frame (N = 24,682) derived from established prospective cohort studies, whereas cohort 2 used clinical registries to identify eligible participants. The strategy for recruiting participants from existing prospective studies for cohort 1 facilitated access to data that were used to predict risk of rapid glycaemic deterioration. However, despite the relatively large sampling frame, it was necessary to enrol lower-risk participants in order to achieve the target sample size; in doing so, we recognised that this would likely reduce the overall rate of glycaemic deterioration in the cohort, although the generalisability of the study’s findings will be greater. In cohort 2, we fell slightly short of the target sample size of 1000 participants (N = 789 with complete and high-quality data eventually enrolled). This reflects the difficulties in engaging some general practices, which was necessary to access diabetes registries in some regions.
We stratified cohorts 1 and 2 by broad glucose-control category (overtly normoglycaemic or impaired glycaemic regulation in any ADA category) or treatment category (lifestyle only vs lifestyle plus metformin), respectively, to reflect the basic stages of progression at baseline for descriptive purposes.
The two IMI DIRECT cohorts are not identical. However, they share many methodology parallels that permit complimentary analyses to be performed, such as determining whether biomarkers for glycaemic deterioration are conditional on disease state. Nevertheless, several key differences in the protocols (e.g. fsOGTT vs MMTT) should be considered when interpreting results. We also note a difference in missing accelerometry (physical activity) data between cohort 1 (19%) and cohort 2 (8.5%); we were unable to definitively explain this discrepancy. Partitioning change from error is very challenging when variables are assessed at only two time points owing to regression to the mean. Notwithstanding this, we note a modest 0 month to 48 month difference in mean (SD) fasting plasma glucose levels, 0.3 (0.5) mmol/l and 0.8 (1.9) mmol/l in cohort 1 and cohort 2, respectively (Table 2), which likely reflects the relatively brief between-visit interval. Furthermore, we note that the 0.5 mmol/l and 1.9 mmol/l SDs in these differences, for cohort 1 and cohort 2, respectively, suggest the potential heterogeneity in changes in glycaemic control within each cohort. With this in mind, the IMI DIRECT cohorts are being followed further with record-linkage through 2026. It should also be noted that the IMI DIRECT cohorts are predominantly of European ancestry; therefore results from subsequent analyses on these cohorts will need to be replicated in other cohorts of relevant ancestry before generalising findings to other ethnicities. Finally, we note that the results presented here reflect the data available at publication and as long-term follow-up progresses additional data will accrue.
Conclusion
The study described here is being used to unravel the heterogeneous nature of glycaemic deterioration in individuals at risk of diabetes and in those with diabetes, and to discover biomarkers that might prove useful for patient stratification and therapeutic optimisation. As more prospective data are accrued, the IMI DIRECT cohorts will grow in value. In the long term, the IMI DIRECT Consortium intends to make these data available to other researchers through a managed-access repository.
Data availability
Requests for access to IMI DIRECT data, including data presented here, can made to DIRECTdataaccess@Dundee.ac.uk.
Abbreviations
- ALT:
-
Alanine aminotransferase
- ASAT:
-
Abdominal subcutaneous adipose tissue
- AST:
-
Aspartate aminotransferase
- DIRECT:
-
Diabetes Research on Patient Stratification
- fsOGTT:
-
Frequently sampled OGTT
- GLP-1:
-
Glucagon-like peptide-1
- hpfVM:
-
High-pass-filtered vector magnitude
- IAAT:
-
Intra-abdominal adipose tissue
- IGR:
-
Impaired glucose regulation
- IMI:
-
Innovative Medicines Initiative
- MMTT:
-
Mixed-meal tolerance test
- NGR:
-
Normal glucose regulation
- OGIS:
-
Oral glucose insulin sensitivity
- TAAT:
-
Total abdominal adipose tissue
References
Cho NH, Shaw JE, Karuranga S et al (2018) IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138:271–281. https://doi.org/10.1016/j.diabres.2018.02.023
American Diabetes Association (2011) Standards of medical care in diabetes--2011. Diabetes Care 34(Suppl 1):S11–S61. https://doi.org/10.2337/dc11-S011
Weyer C, Bogardus C, Mott DM, Pratley RE (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104(6):787–794. https://doi.org/10.1172/JCI7231
Ringborg A, Lindgren P, Yin DD, Martinell M, Stalhammar J (2010) Time to insulin treatment and factors associated with insulin prescription in Swedish patients with type 2 diabetes. Diabetes Metab 36(3):198–203. https://doi.org/10.1016/j.diabet.2009.11.006
Rathmann W, Strassburger K, Heier M et al (2009) Incidence of type 2 diabetes in the elderly German population and the effect of clinical and lifestyle risk factors: KORA S4/F4 cohort study. Diabet Med 26(12):1212–1219. https://doi.org/10.1111/j.1464-5491.2009.02863.x
Ahlqvist E, Storm P, Karajamaki A et al (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6(5):361–369. https://doi.org/10.1016/S2213-8587(18)30051-2
Koivula RW, Heggie A, Barnett A et al (2014) Discovery of biomarkers for glycaemic deterioration before and after the onset of type 2 diabetes: rationale and design of the epidemiological studies within the IMI DIRECT Consortium. Diabetologia 57(6):1132–1142. https://doi.org/10.1007/s00125-014-3216-x
Stancakova A, Javorsky M, Kuulasmaa T, Haffner SM, Kuusisto J, Laakso M (2009) Changes in insulin sensitivity and insulin release in relation to glycemia and glucose tolerance in 6,414 Finnish men. Diabetes 58(5):1212–1221. https://doi.org/10.2337/db08-1607
Hills SA, Balkau B, Coppack SW et al (2004) The EGIR-RISC STUDY (the European group for the study of insulin resistance: relationship between insulin sensitivity and cardiovascular disease risk): I. Methodology and objectives. Diabetologia 47(3):566–570. https://doi.org/10.1007/s00125-004-1335-5
Rutters F, Nijpels G, Elders P et al (2018) Cohort profile: the Hoorn studies. Int J Epidemiol 47(2):396–396j. https://doi.org/10.1093/ije/dyx227
Christensen AI, Ekholm O, Glumer C et al (2012) The Danish National Health Survey 2010. Study design and respondent characteristics. Scand J Public Health 40(4):391–397. https://doi.org/10.1177/1403494812451412
Thuesen BH, Cerqueira C, Aadahl M et al (2014) Cohort profile: the Health2006 cohort, research centre for prevention and health. Int J Epidemiol 43(2):568–575. https://doi.org/10.1093/ije/dyt009
Region Hovedstaden (2016) DanFunD: Danish study of functional disorders. Available from https://www.regionh.dk/fcfs/befolkningsbaseret-epidemiologi/befolkningsunders%C3%B8gelser/Sider/DanFund.aspx. Accessed 5 Mar 2018 2018 [in Danish]
Roager HM, Vogt JK, Kristensen M et al (2019) Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: a randomised cross-over trial. Gut 68(1):83–93
Berglund G, Elmstahl S, Janzon L, Larsson SA (1993) The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med 233(1):45–51. https://doi.org/10.1111/j.1365-2796.1993.tb00647.x
Rauh SP, Heymans MW, Koopman AD et al (2017) Predicting glycated hemoglobin levels in the non-diabetic general population: development and validation of the DIRECT-DETECT prediction model - a DIRECT study. PLoS One 12(2):e0171816. https://doi.org/10.1371/journal.pone.0171816
Thomas EL, Fitzpatrick JA, Malik SJ, Taylor-Robinson SD, Bell JD (2013) Whole body fat: content and distribution. Prog Nucl Magn Reson Spectrosc 73:56–80. https://doi.org/10.1016/j.pnmrs.2013.04.001
OʼRegan DP, Callaghan MF, Wylezinska-Arridge M et al (2008) Liver fat content and T2*: simultaneous measurement by using breath-hold multiecho MR imaging at 3.0 T--feasibility. Radiology 247(2):550–557. https://doi.org/10.1148/radiol.2472070880
St Pierre TG, Clark PR, Chua-Anusorn W (2005) Measurement and mapping of liver iron concentrations using magnetic resonance imaging. Ann N Y Acad Sci 1054(1):379–385. https://doi.org/10.1196/annals.1345.046
Garcia-Perez I, Posma JM, Gibson R et al (2017) Objective assessment of dietary patterns by use of metabolic phenotyping: a randomised, controlled, crossover trial. Lancet Diabetes Endocrinol 5(3):184–195. https://doi.org/10.1016/S2213-8587(16)30419-3
White T, Westgate K, Hollidge S et al (2019) Estimating energy expenditure from wrist and thigh accelerometry in free-living adults: a doubly labelled water study. Int J Obes. https://doi.org/10.1038/s41366-019-0352-x
R-Core-Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Mari A, Tura A, Gastaldelli A, Ferrannini E (2002) Assessing insulin secretion by modeling in multiple-meal tests: role of potentiation. Diabetes 51(Suppl 1):S221–S226. https://doi.org/10.2337/diabetes.51.2007.S221
Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E (2002) Meal and oral glucose tests for assessment of beta -cell function: modeling analysis in normal subjects. Am J Phys Endocrinol Metab 283(6):E1159–E1166. https://doi.org/10.1152/ajpendo.00093.2002
Mari A, Ferrannini E (2008) Beta-cell function assessment from modelling of oral tests: an effective approach. Diabetes Obes Metab 10(Suppl 4):77–87. https://doi.org/10.1111/j.1463-1326.2008.00946.x
Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ (2001) A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care 24(3):539–548. https://doi.org/10.2337/diacare.24.3.539
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22(9):1462–1470. https://doi.org/10.2337/diacare.22.9.1462
Stumvoll M, Mitrakou A, Pimenta W et al (2000) Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care 23(3):295–301. https://doi.org/10.2337/diacare.23.3.295
Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA (2005) beta-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab 90(1):493–500. https://doi.org/10.1210/jc.2004-1133
Byrne MM, Sturis J, Sobel RJ, Polonsky KS (1996) Elevated plasma glucose 2 h postchallenge predicts defects in beta-cell function. Am J Phys 270:E572–E579
Novembre J, Johnson T, Bryc K et al (2008) Genes mirror geography within Europe. Nature 456(7218):98–101. https://doi.org/10.1038/nature07331
Acknowledgements
We thank all the participants and study centre staff in IMI DIRECT for their contribution to the study. Some of the data were presented as an abstract at the Precision Medicine in Type 2 Diabetes and Cardiovascular Disease meeting in 2016 (Båstad, Sweden).
Funding
This work was supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115317 (DIRECT), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution. RWK was funded by a STAR Award Novo Nordisk co-financed PhD fellowship. The work undertaken by PWF was supported in part by programme grants from the ERC-2015-CoG_NASCENT_681742 and the Swedish Research Council; strategic funding for Lund University Diabetes Centre, where some of the work described herein was performed, was provided by the Swedish Research Council, Strategic Research Area Exodiab, (Dnr 2009-1039), the Swedish Foundation for Strategic Research (IRC15-0067), the Swedish Research Council, Linnaeus grant (Dnr 349-2006-237). EP holds a Wellcome Trust Investigator award (grant reference 102820/Z/13/Z). Contributions to this work by SBru. were co-financed by the Novo Nordisk Foundation (grants NNF17OC0027594 and NNF14CC0001).
Author information
Authors and Affiliations
Consortia
Contributions
RWK and PWF wrote the manuscript. PWF, MW and EP conceptualised and designed the studies, with contributions from RWK, IMF, HJAT, JA, JDB, JK, SBru, ETD, PF, GF, RG, AHa, BJ, TJM, OP, JMS, IP, AMar, MIMcM and HR. RWK carried out the statistical analyses presented here. RWK, AK, AV, MS, KHA, SBra, CAB, AYD, FDeM, CJG, TK, AM, MHP, SPR, ELT, AT, TW, HJAT, JA, JDB, JK, JWB, SBru, ETD, PF, GF, RG, ML, TJM, OP, JMS, AMar, MIMcM, EP and PWF contributed to sample assaying, data analysis/processing and/or data quality control procedures. RWK, IMF, AHe, GNG, THH, MH, ADMK, FR, MS, TK, MR, HJAT, HV, JDB, TH, AHa, ML, OP, MW, EP and PWF contributed to data collection at study centres. PWF coordinated the overall planning and implementation of the IMI DIRECT epidemiological studies. HR and EP were the overall coordinators of the IMI DIRECT Consortium. All authors contributed to the revision of the manuscript. All authors critiqued and approved the final version of the manuscript. All authors accept responsibility for all aspects of the work insofar as ensuring that questions related to the accuracy or integrity of any part of the article are appropriately investigated and resolved. PWF and EP are the guarantors of this work and, as such, had full access to the data described here and take responsibility for the integrity of the data and its analysis.
Corresponding authors
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM Table
(PDF 124 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Koivula, R.W., Forgie, I.M., Kurbasic, A. et al. Discovery of biomarkers for glycaemic deterioration before and after the onset of type 2 diabetes: descriptive characteristics of the epidemiological studies within the IMI DIRECT Consortium. Diabetologia 62, 1601–1615 (2019). https://doi.org/10.1007/s00125-019-4906-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4906-1