
Abstract
When it comes to the epigenome, there is a fine line between clarity and confusion—walk that line and you will discover another fascinating level of transcription control. With the genetic code representing the cornerstone of rules for information that is encoded to proteins somewhere above the genome level there is a set of rules by which chemical information is also read. These epigenetic modifications show a different side of the genetic code that is diverse and regulated, hence modifying genetic transcription transiently, ranging from short- to long-term alterations. While this complexity brings exquisite control it also poses a formidable challenge to efforts to decode mechanisms underlying complex disease. Recent technological and computational advances have improved unbiased acquisition of epigenomic patterns to improve our understanding of the complex chromatin landscape. Key to resolving distinct chromatin signatures of diabetic complications is the identification of the true physiological targets of regulatory proteins, such as reader proteins that recognise, writer proteins that deposit and eraser proteins that remove specific chemical moieties. But how might a diverse group of proteins regulate the diabetic landscape from an epigenomic perspective? Drawing from an ever-expanding compendium of experimental and clinical studies, this review details the current state-of-play and provides a perspective of chromatin-dependent mechanisms implicated in diabetic complications, with a special focus on diabetic nephropathy. We hypothesise a codified signature of the diabetic epigenome and provide examples of prime candidates for chemical modification. As for the pharmacological control of epigenetic marks, we explore future strategies to expedite and refine the search for clinically relevant discoveries. We also consider the challenges associated with therapeutic strategies targeting epigenetic pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vascular disease affecting nearly all types of blood vessels is common to both type 1 and type 2 diabetes mellitus. Accelerated rates of clinically defined macrovascular complications, such as myocardial infarction and stroke, which result from large vessel atherosclerosis remain the leading causes of morbidity and premature mortality in the diabetic population. Diabetes is also associated with the occurrence of adverse microvascular complications, manifesting clinically as retinopathy, neuropathy and nephropathy.
Extensive debate surrounds the extent to which diabetic microvascular and macrovascular complications represent a continuous pathological spectrum. Closely related to this debate is the question of why not all people with diabetes complications experience more advanced forms of vascular disease. Though yet to be completely defined mechanistically, the persistent and harmful effects of antecedent hyperglycaemia may at least partly explain the variation in vascular deterioration. Nonetheless, the fundamental reasons why a proportion of diabetic individuals appear to be protected from serious complications remain poorly understood. Despite the promises of the genetic revolution, contemporary knowledge of the impact of genetic variation on diabetes does not adequately explain the disproportionate distribution and severity of diabetic vascular complications.
Realisation of novel preventative and therapeutic approaches hinges on improved characterisation of the molecular events and interactions that underlie the development and progression of diabetic vasculopathology. Interestingly, however, some in the field have shifted their research focus to understanding the post-translational and covalent chemical chromatin modifications that contribute to transcriptional regulation via structural adaptation. Insight from cultured cells and preclinical models, as well as clinical samples, has highlighted the importance of chromatin modifications in the persistent inflammatory response to glycaemic variability. Thus, epigenetics may be able to provide an explanation as to why some individuals with diabetes are predisposed to developing vascular disease and are more likely to progress to advanced stages of complications and/or develop other associated vascular pathologies.
Glycaemic memories and vascular complications of diabetes
The problem of hyperglycaemic persistence
As mentioned above, long-term inadequate glycaemic control is a major risk factor in the development of vascular complications. Despite the proclivity for patients with good metabolic control to have a significantly decreased risk for developing complications, vascular disease may still develop and progress even with intensive treatment regimens [1]. This is particularly true for individuals with a history of suboptimal glycaemic control who develop vascular disease despite good current metabolic control, a phenomenon known as ‘glycaemic memory’ or ‘legacy effect’.
The landmark Diabetes Control and Complications Trial (DCCT) was the first to demonstrate that achieving near-normal blood glucose levels ameliorates microvascular complications of type 1 diabetes [2]. Moreover, by switching both groups of study participants to the intensive insulin regimen in the wake of the successful completion of DCCT, the Epidemiology of Diabetes Interventions and Complications (EDIC) follow-up study not only confirmed the durability of the effects of glucose control on more advanced stages of complications, but also revealed that, despite stringent long-term glycaemic control, previous periods of suboptimal blood glucose continued to be a risk factor for chronic microvascular complications [3]. While provocative, these findings were not without precedent, as vascular memory of prior hyperglycaemia had been suggested by earlier studies of various experimental animal models [4,5,6]. As regards type 2 diabetes, observational studies suggest the enduring consequences of antecedent hyperglycaemia underlying vascular risk [7,8,9].
Microvascular and macrovascular complications
Although hyperglycaemia is demonstrably a principal cause of microvasculopathy—the microvasculature of the retina is particularly susceptible to excess glucose and diabetic nephropathy is not observed in the absence of hyperglycaemia [10]—the efficacy of glucose-lowering interventions to reduce cardiovascular risk is still questioned [11]. Are the pathogenic characteristics of microvascular disease also related to the development of macrovascular disease? Indeed, diabetic individuals with microvascular complications are especially prone to accelerated atherosclerosis and premature mortality [12]. Of the numerous organ systems affected by diabetes, the impact on renal function is the most pronounced. The diabetic kidney is considered a primary failing organ and its clinical features are increasingly considered to be indicative of overall vascular damage. Individuals with diabetic nephropathy endure an exceptionally high risk of cardiovascular disease, and both increased urinary albumin excretion and reduced GFR are prognostic of cardiovascular morbidity and mortality [13].
Diabetes in the GWAS era
Unlike the near inexorable progression to retinopathy, more than half of all individuals with type 1 diabetes do not develop renal complications [14]. While insufficient metabolic and haemodynamic control, as well as prolonged disease duration, may explain some cases, the fact that individuals with strict compliance can develop clinically evident nephropathy whereas many individuals with similar or worse control do not, illustrates the disproportionate distribution of the diabetic nephropathy burden [10]. Similarly, not all people with microalbuminuria progress to macroalbuminuria or end-stage renal disease (ESRD), apparently protected despite decades of chronic hyperglycaemia and haemodynamic stress. Furthermore, increased risk of renal disease aggregates in families [15], as exemplified by the finding that the incidence of nephropathy in diabetic children of individuals with diabetic nephropathy is more than three times that in children of individuals without renal disease [16, 17].
Thus, the search for genetic factors associated with diabetic nephropathy susceptibility, using initially linkage analyses, candidate gene-based approaches, and, more recently, hypothesis-free GWAS has been extensive. Yet, even with the advent of modern sequencing technologies, intensive efforts have yielded only a limited number of consistent genetic associations, and the impact on clinical management has so far been negligible [18]. Large collaborations drawing from sufficiently powered sampling, such as those recently published by the Surrogate markers for Micro-and Macro- vascular hard endpoints for Innovative diabetes Tools (SUMMIT) consortium [19], provide additional motivation for genetic studies in pursuit of the enigmatic heritability of chronic kidney disease.
Editing and interpreting chromatin modifications
Methylation is unique in the way that it is enriched at cytosine bases of the DNA template, primarily, but not exclusively, at cytosine–guanine (CpG) dinucleotides, as well as on the tails of chromatinised histones. When written by DNA methyltransferase enzymes (DNMT1, DNMT3a, and DNMT3b in humans) to the 5-carbon position of cytosine (5-methylcytosine, 5mC), the methyl modification is historically associated with transcriptional silencing by recruitment of specific factors that actively remodel the chromatin structure, as well as by the disruption of transcription factor binding sites. On the other hand, 5mC enrichment can preclude binding of transcriptional repressors such as CCCTC-binding factor (CTCF), which is associated with altered chromatin structures and thus aberrant gene activation [20]. Indeed, the precise location of the modification relative to genetically encoded regulatory elements is central to the epigenetic function of 5mC. Recent characterisation of the ten-eleven-translocation (TET) family of proteins that hydroxylate 5mC to 5-hydroxymethylcytosine (5hmC) has inspired strong interest in DNA demethylation pathways [21]. As for the role of the demethylation and chromatin remodelling by the TET proteins, the enrichment of 5hmC at the gene body is implicated in transcriptional activation. Indeed, transcriptional induction of adipocyte differentiation is dynamically regulated by the binding of CTCF to chromatin [22].
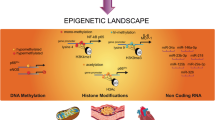
Methylation of lysine and arginine residues on histone tails is similarly associated with both transcriptional activation and repression, depending not only on the position of the substrate residue within a specified histone tail, but also the degree of modification (mono-, di-, or tri-methylation, Table 1). Trimethylated lysines at position 4 of the H3 histone tail (H3K4m3) are associated with active promoters, whereas histones methylated at H3K9 and H3K27 are predominantly enriched at repressed genes. Monomethylated H3K4 (H3K4m1) denotes distal enhancers [23], and plays a regulatory role at specific promoters [24, 25]. A dynamic network of highly specific methyl writers (methyltransferase) and erasers (demethylase) regulate these and many other sites predominantly on the tails of H3 and H4 histones [26]. Such epigenetic marks are read by multi-subunit chromatin remodelling complexes, though their precise function, including interaction with traditional transcription factors, as well as mechanisms regulating gene-specific enrichment, remain to be definitively characterised (Fig. 1).

Codified signature of the diabetic epigenome. Readers, writers and erasers in diabetes. Modification of the diabetic epigenome includes post-translational modifications to the tails of histones, carried out by histone-modifying enzymes (known a ‘writers’), such as SET7 [24, 26, 30, 102, 103, 106, 107], SETDB1 [108], SUV39H1 [109, 110], EZH2 [40, 111], KAT2A [42, 112] and GLYATL1 [113]. Experimental studies that provide mechanistic insights for specific determinants are grouped to include the enzyme and corresponding modified histone, whereas informative profiling studies using clinical cohorts are separated with examples such as SET7 [30], SUV39H1/H2 [107], H3K9 acetylation [31] and H3K9me2 [82]. The epigenetic code is dynamic and eraser enzymes are implicated in diabetes such as KDM6B [114], PHF2 [115], KDM1A [26, 108], HDAC3 [99, 116], HDAC4 [117] and HDAC7 [118]. Protein readers such as CTCF recognise post-translational histone modifications including methylation of cytosine residues in CpG dinucleotides [94, 119]. Genome readers regulate transcriptional responses and include KLF4 [38], SIRT1 [47], as well as non-CpG methylation by DNMT3B [120]. The DNA template is subject to modification and recent experimental studies have shown an association with 5mC [46, 78, 85, 121,122,123,124,125,126,127,128,129,130] and 5hmC [131]. Clinical profiling studies for DNA modification have also shown an association with 5mC [54, 79, 80, 87, 88, 132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149] and 5hmC [150]. Post-transcriptional gene regulation by RNA modifications include the writers, erasers and readers of N 6-methyladenosine (m6A). FTO [151,152,153] is an m6A eraser implicated in metabolic homeostasis [154] and is associated with type 2 diabetes [155] CARM1, coactivator associated arginine methyltransferase 1; DNMT3B, DNA methyltransferase 3B; FTO, fat mass and obesity-associated protein; GLYATL1, glycine-N-acyltransferase like 1; HNRNP, heterogeneous nuclear ribonucleoprotein; KAT2A, K (lysine) acetyltransferase 2A; KDM, lysine (K)-specific demethylase; NSD, nuclear receptor binding SET domain protein; MBD, methyl-CpG binding domain protein; PHF2, PHD finger protein 2; PRMT, protein arginine methyltransferase; SETDB, SET domain bifurcated; SETMAR, SET domain and mariner transposase fusion protein; SIRT1, sirtuin 1; SUV39H, suppressor of variegation 3-9 homologue; YTH, YTH domain protein. Blank fields in the mechanistic and profiling studies refer to either enzymes or modified determinants that were not reported in the studies listed
Further emphasising the importance of chromatinised lysine residues is histone acetylation, which promotes an open chromatin structure by electrostatic charge disruption and facilitates the assembly of transcriptional machinery [27]. The acetyl-writing activities of histone acetyltransferases (HATs) are mechanistically opposed by histone deacetylases (HDACs), a dichotomy that has been exploited for the clinical treatment of heart disease and cancer [28, 29]. Furthermore, there is obvious interplay between histone methylation and acetylation in their competition for lysine substrates. Other important though less well studied histone modifications include phosphorylation, sumoylation, ubiquitination, ADP-ribosylation, and O-GlcNAcylation.
Epigenetic changes in diabetic nephropathy
We have previously described the important role of the SET7 lysine methyltransferase in vascular endothelial cells in writing a specific high-glucose-mediated H3K4m1 signature at the promoter of the RELA gene [24, 26], which encodes the proinflammatory p65 subunit of NFκB. In accordance with the concept of glycaemic memory, this modification persisted in cultured human vascular cells and rodents beyond euglycaemic restoration. Importantly, the particular SET7-dependent H3K4m1 signature was recently identified in monocytes of diabetic individuals [30]. In addition, specific enrichment of histone acetylation was observed at several genes related to diabetes complications in DCCT/EDIC participants who received conventional treatment as compared with those who received intensive therapy [31], which may have implications for metabolic memory in vascular complications. Could similar mechanisms be responsible for the persistence and progression of diabetic nephropathy? Chromatin modifications are increasingly implicated in renal pathophysiology, and while the persistence of the majority of associations remains untested, it is evident that their influence reaches beyond roles in metabolic memory. We recently described the importance of cell-specific epigenetic changes in atherosclerosis [32] and in the section below we adopt a similar approach for diabetic nephropathy, highlighting recent key examples of chromatinised changes in podocytes and proximal epithelial cells.
Transcriptional control in podocytes
Podocytes are visceral epithelial cells that line the urinary space of the renal corpuscle. These highly specialised cells derive their name from long interdigitating foot processes that form intercellular clefts called slit pores, bridged by diaphragms consisting of podocyte-specific proteins, such as NPHS1 (also known as nephrin) and NPHS2 (also known as podocin). Structural disturbance of the slit diaphragm proteins results in insufficient filtration and proteinuria, exemplified by congenital kidney failure arising from defects in nephrin [33]. Podocyte injury and loss, through detachment, apoptosis, or epithelial to mesenchymal transition (EMT) [34], are strong predictors of diabetic nephropathy progression [35] and are closely linked to glomerulosclerosis [36].
Expression of the pluripotency-associated Kruppel-like factor 4 (KLF4) [37] correlates positively with NPHS1 expression and inversely with proteinuria in rodent and human podocytes [38]. Moreover, transient restoration of KLF4 in diseased glomeruli re-establishes the normal podocyte phenotype and attenuates proteinuria. KLF4 binds a specific motif on the NPHS1 promoter and upregulates gene expression by reduced methylation or demethylation of adjacent CpG sites. Methyl profiling of cultured human podocytes overexpressing KLF4 revealed widespread changes in 5mC, highlighted by reduced CpG methylation at promoters of other epithelial genes, such as NPHS2 and SYNPO (which encodes synaptopodin), in striking contrast to hypermethylation at promoters of mesenchymal genes, such as VIM (which encodes vimentin) and CTGF (which encodes connective tissue growth factor), thereby indicating KLF4-dependent determination of podocyte phenotype by gene-specific methylation. The potential implications of these findings are significant, not only for slit diaphragm maintenance but also for epithelial to mesenchymal transition observed in advanced diabetic nephropathy. The slit diaphragm proteins NPHS1 and nephrin-like 3 (NEPH3) are encoded by genes (located on chromosome 19q13.12 in a head-to-head orientation) that share a bidirectional promoter and have been shown to be dependent on 5mC for co-regulation and expression [39].
Specifically within the diabetic setting, the chromatin-dependent regulation of glucose-mediated oxidative stress in podocytes is emerging as a critical mediator of diabetic nephropathy. For example, the H3K27-methylating function of enhancer of zeste 2 repressive complex 2 subunit (EZH2) recently emerged as an important regulator of diabetic nephropathy by repressing the transcription factor PAX6 and subsequently dampening expression of the endogenous antioxidant inhibitor thioredoxin-interacting protein (TXNIP). Inhibition of EZH2 augments proteinuria, podocytopathy, glomerular Txnip expression, and renal oxidative stress in a rat model of diabetes [40]. Disease-related genes are rarely regulated by a single mechanism, but, rather, can reflect the interplay of multiple epigenetic determinants. Indeed, glucose-mediated Txnip expression is coordinated by histone acetylation and methylation in kidneys from diabetic Sur1-E1506K+/+ mice [41]. Similarly, promoter CpG hypomethylation and concomitant H3 histone hyperacetylation by the GCN5 histone acetyltransferase were found to drive protein C-dependent expression of the critical mediator of oxidative stress p66Shc in podocytes exposed to high glucose [42]. This epigenetic signature closely mirrors the specific hyperglycaemia-induced changes that activate and maintain p66Shc expression beyond restoration of euglycaemia in cultured vascular endothelial cells and diabetic mice [43], therefore representing a possible epigenetic mechanism of glycaemic memory in the podocyte. The importance of H3 acetylation for p66Shc gene regulation was further underscored by pharmacological and molecular experiments targeting class III HDACs, and specifically the NAD+-dependent SIRT1 HDAC [44].
Transcriptional control in proximal tubule epithelial and glomerular mesangial cells
Contiguous with podocytes in renal structure, proximal tubular epithelial cells (PTECs) play a crucial role in renal function, reabsorbing much of the glucose and amino acids, as well as sodium, from the glomerular filtrate. Both high glucose and abnormal protein trafficking through the glomerulus induce inflammation and tubulointerstitial lesions through PTEC activation, and the extent of interstitial fibrosis ultimately determines the rate of decline in renal function [45]. Recent in vivo investigations highlight differential 5mC patterns associated with genes such as Sglt2 (also known as Slc5a2) and G6pc, indicating that 5mC underlies selective glucose handling by PTECs in the kidney [46]. While PTECs isolated from db/db mice exhibited a comparable genome-wide methylation profile to PTECs from control animals, significant differences in 5mC were observed at genes implicated in sugar reabsorption (Slc5a2), nephropathy (Met) and hypertension (Agt) [46]. An important component of the renin-angiotensin system, Agt was shown to accumulate aberrant epigenetic changes early in the pathogenesis of diabetic nephropathy. Acetylated H3K9 was enriched at the Agt promoter as early as 5 weeks in the diabetic kidney and preceded both DNA hypomethylation and H3K9m3. The importance of H3 acetylation was further underscored by Agt transcriptional activation in response to HDAC inhibition. In contrast, these epigenetic changes were resistant to the glucose-lowering drug pioglitazone. In human diabetic nephropathy, microdissected tubuli from individuals with diabetic kidney disease exhibited differential methylation of genes implicated in fibrogenesis [47]. Central to nephron function is the network of capillaries that form the glomerulus. Activation of mesangial cells by advanced glycation end-products and high glucose increase proinflammatory and profibrotic cytokines, including angiotensin II (ANGII). Recent studies have shown that the ANGII type 1 receptor antagonist (AT1R) losartan, which is used to treat renal complications of diabetes, alters post-translational modifications on histones in glomeruli from db/db mice [48]. In mesangial cells cultured under hyperglycaemic conditions, losartan attenuates histone acetylation at RAGE (also known as AGER), PAI1 (also known as SERPINE1) and MCP-1 (also known as CCL2) promoters.
Chromatin modifications regulate inflammation in diabetic nephropathy
Kidney biopsies from experimental diabetes models or individuals with diabetes are characterised by enhanced macrophage infiltration [49, 50]. Furthermore, a localised proinflammatory response is well characterised in the vasculature and kidneys under diabetic conditions, exemplified by proinflammatory cytokine and chemokine secretion and the overproduction of reactive oxygen species [51, 52]. The local presence and contribution of activated macrophages to the aetiology of both diabetic nephropathy and cardiovascular disease [53] may point towards a general systemic role in diabetes complications. Studies have identified a potential role of chromatin modifications in monocyte-derived inflammatory gene expression in the context of diabetes, including persistent vascular complications [30, 31, 54]. Long-term memory spanning decades in vascular cells such as macrophages could potentially be explained by persistent epigenetic profiles of progenitor cells, though such an association is yet to be demonstrated. In the acute setting, chromatin modifications in monocytes have been implicated recently in functional changes associated with the exciting concept of trained innate immunity (also termed innate immune memory). This is a fascinating new field with tremendous scope and biological implications, not only with respect to inflammatory diseases, but for all diseases in which monocytes play a pathological role [55].
Trained immunity: chromatin-dependent memory
Immunological memory had classically been viewed as being characteristic of only the adaptive immune system (T and B lymphocytes). However, it has recently emerged that innate immune cells exhibit memory-like behaviour, characterised by an increased proinflammatory response to secondary infections [55,56,57]. Importantly, the trained memory of monocytes is non-specific, meaning that an encounter with a certain pathogen can also protect against infection by unrelated pathogens [56, 57]. The heightened responsiveness of monocytes is characterised by enhanced secretion of proinflammatory mediators associated with widespread changes in chromatin patterns [58]. Burgeoning interest is rapidly uncovering an intricate program of chromatin modification underlying not only training for a heightened immune response, but also tolerance to re-stimulation, which is basically the opposite of training [59]. Some aspects of this macrophage memory are at least partly dependent on the induction of latent enhancers [60], constitutively unmarked distal elements that acquire signature epigenetic features of enhancers (H3K4m1 and H3K27ac) upon stimulation with certain microbial products. Moreover, after the initial stimulation, H3K4m1 persists at decommissioned regulatory elements to mediate a faster response to re-stimulation, further emphasising the role of this specific histone modification in transcriptional memory and inflammation [24].
The non-specific trained memory of monocytes is thought to have beneficial effects in numerous immunological settings, including vaccination programmes [61]. In contrast, trained immunity may play a maladaptive role in chronic inflammatory (metabolic) diseases such as atherosclerosis [62, 63]. Especially since microbial training of monocytes not only enhances responsiveness to subsequent pathogens, but also primes transcription of chemokines and scavenger receptors to promote foam cell formation [62, 64]. This suggests an important link between trained immunity and metabolic diseases. Even more relevant are experiments demonstrating that non-pathogen-related pro-atherosclerotic metabolites such as oxidised LDL (oxLDL) and lipoprotein(a) [Lp(a)] can also induce trained immunity [64, 65]. Monocytes trained by oxLDL or Lp(a) exhibit a long-term pro-atherogenic monocyte phenotype, which is associated with specific and persistent H3K4m3 enrichment at activated promoters, and is accordingly attenuated by pan-methyltransferase inhibition [64, 65].
Chronic hyperglycaemia associates with monocyte activation, induced directly by glucose or by other endogenous compounds associated with hyperglycaemia, such as AGEs [66, 67]. Whether glucose or AGEs promote epigenetic reprogramming of monocytes, macrophages or progenitor cells remains to be determined; however, this could play an important role in the phenomenon of hyperglycaemic memory in individuals with diabetes [68].
Immunometabolism and diabetic vascular complications
The epigenetic reprogramming of monocytes clearly involves marked changes in cellular metabolism, which is determined by their activation status [58, 69]. Specifically, oxidative phosphorylation is used as a primary metabolic process by resting cells, which contrasts with a profound switch to aerobic glycolysis (Warburg effect) upon activation [69]. The glycolytic switch is under the control of the Akt–mammalian target of rapamycin (mTOR)– hypoxia-inducible factor-1α (HIF1α) pathway and aimed at optimising immune cell function, including macromolecular synthesis and enhanced cytokine production. Recent multilevel -omics analysis revealed that glycolysis, glutaminolysis and cholesterol synthesis are non-redundant pathways for the induction of trained immunity by the microbial cell wall component β-glucan [70].
Interestingly, the metabolic state of the cell is linked to a particular epigenetic program [71]. Indeed, the intracellular changes in the metabolic milieu may in fact drive the epigenetic reprogramming of monocytes during trained immunity [72]. For example, accumulation of fumarate in β-glucan-trained cells, due to glutamine replenishment of the tricarboxylic acid cycle, integrates immune and metabolic circuits with epigenetic regulation by inhibiting the lysine demethylase 5 (KDM5) histone demethylase. Moreover, fumarate induces epigenetic reprogramming similar to β-glucan-mediated trained immunity [70].
The relationship between metabolic processes and epigenetic changes has so far been studied predominantly in the field of cancer research [73], but could potentially have major implications for cellular behaviour in disturbed metabolic environments such as diabetes [74]. Assuming that chronic hyperglycaemia increases glucose availability as a substrate for innate immune cells, this may affect intracellular metabolism (e.g. stimulate glycolysis), including changes in intermediate metabolites that promote epigenetic changes [68]. Chromatin-dependent immunological training by fumarate [70] is particularly relevant for inflammation in diabetic nephropathy because this metabolite was recently shown to accumulate in the kidneys of diabetic rats [75].
Predisposition, progression and prognosis
The heritability of epigenomic signatures continues to be intensely debated, though several plausible mechanisms of transgenerational transmission of acquired phenotypes have been described (reviewed elsewhere [76]), including metabolic traits [77]. Recent studies demonstrate that epigenetic regulation underlying phenotypic determinants of adult metabolic health is influenced in utero and by the early postnatal environment [74, 78]. The potential for shared environmental exposures to impart similar epigenetic patterns among related individuals cannot discount the influence of genetic variation. Perhaps the phenotypic consequences of some susceptible genes are only revealed under specific patterns of epi-regulation induced by environmental variation or diabetes-specific processes, thereby confounding their discovery by traditional GWAS. Previously unmarked genomic regulatory elements can be commissioned by stimulus-dependent persistent chromatin modifications [60], and similar undiscovered mechanisms could be responsible for a latent susceptibility to diabetic nephropathy or exacerbate the effects of disease-associated genetic variants.
Expansion of high-throughput sequencing technologies to include the profiling of chromatin modifications on a genome-wide scale represents a new approach towards understanding predisposition to diabetic nephropathy. Studies comparing diabetic individuals with and without diabetic nephropathy reported differential DNA methylation at numerous genes, including several previously identified by GWAS [54, 79, 80]. One example is the gene encoding unc-13 homologue B (UNC13B), which is associated with glucose-mediated apoptosis in glomerular cells [81], and is hypermethylated near its transcription start site in peripheral blood cells of type 2 diabetes patients with diabetic nephropathy [79]. Furthermore blood derived from individuals with type 1 diabetes revealed an association between diabetic nephropathy and differential methylation at genes involved in mitochondrial function [80]. This study compared methylation patterns in African-American and Hispanic diabetic individuals with ESRD and diabetic people without nephropathy. Interestingly, participants with ESRD being treated with haemodialysis show significantly reduced methylation.
Immune cell subtypes are distinguished by epigenetic profiles [82], and differences between monocytes of different individuals are relatively stable [83], consistent with monocytes being an appropriate and practical source of material. Such a profiling strategy provides opportunities to gain greater insight into gene-regulating events specific to the pathological properties of monocytes in diabetes complications, as well as other vascular cell types. Epigenomes derived from peripheral blood [79] and saliva [80] exhibit marked differences between individuals with and without diabetic nephropathy, indicating, at least in principle, the utility of such a proxy for disease risk. However, the importance of cell-specific epigenomes cannot be overlooked [32].
While certainly an attractive approach to understanding complex disease phenotypes, several challenges limit the interpretability of epigenomic profiles. Notably, the aforementioned studies have used methylation array hybridisation technology that predominantly assays promoters, and to date there are no unbiased profiles of DNA methylation in the context of diabetic nephropathy. Genome coverage is critical to understand differential methylation outside promoter regions. For example, the application of next-generation sequencing generates comprehensive maps of DNA methylation data, thereby reducing the limitations often attributed to array composition [54]. Contrary to popular belief as it specifically pertains to the field of oncology, not all methylation sites are born equal [84]. While it is appreciated that genes are repressed by methylation, repression is not strictly restricted to promoters of genes and highlights the importance of methodological detection [85].
Shaping the diabetes epigenome
Altered DNA methylation patterns at specific loci can distinguish phenotypic cases from controls to reveal possible causal mechanisms. However, the cross-sectional approach can also be confounded by reverse causation, where the interrogated epigenomes are shaped by (rather than cause) the disease [86], as described recently in studies of BMI [87] and type 1 diabetes [88]. Exerting an even greater influence is the DNA sequence itself, which is estimated to account for up to 80% of inter-individual DNA methylation [86]. To circumvent both issues of causality, epigenome profiling strategies should be reconsidered to include concurrent genotyping and transcriptome profiling of the same cells from a single individual [86].
Inter-individual epigenetic differences may prove to be valuable predictive biomarkers of diabetic nephropathy susceptibility and development. Progression of atherosclerosis can indeed be associated with the degree of DNA methylation within plaques [89], and prospective approaches could provide similar insight into the role of chromatin modifications in diabetic nephropathy, with potential prognostic applications. Sampling of individuals prior and subsequent to disease onset permits the discovery of epigenetic changes that precede and possibly even predict the overt phenotype, while reducing the effects of genetic variation that confound cross-sectional studies [86]. Further strengthening the relationship between epigenetics and metabolic memory, recent longitudinal profiling of monocyte DNA methylation from the same individuals at 7–6 year intervals identified loci-specific differential DNA methylation established during the DCCT that persists for several years during the EDIC Study. A noteworthy discovery was the persistent hypomethylation of nephropathy-associated TXNIP, an effect that was replicated in a cell culture model of hyperglycaemic variability [54].
As discussed previously, 5mC is a powerful regulatory determinant, and given that 60–90% of the cytosines in the adult vertebrate cell contain CpG methylation, the prospect that many other functional sites within the genome are potentially altered would seem highly likely, although there is limited experimental evidence of this (Fig. 2). The most direct mechanism by which CpG methylation could alter gene expression would be the prevention or enhancement of the binding of transcriptional machinery at ubiquitous consensus binding sites. For example, CTCF is a chromatin insulator that serves to regulate access of distant enhancers to promoters [90]. Indeed, CTCF binding is methylation sensitive [91], binding imprinted control regions only at the unmethylated parental allele to regulate specific gene expression patterns [92]. CTCF binding also protects regions from DNA methylation [93]. This diversity in methylation distribution provides a direct mechanism to regulate gene expression, offering a simple and elegant means of controlling key target genes in signalling complexes and core pathways implicated in type 2 diabetes [94]. The alternative possibility is that transcriptional repressors that assemble on chromatin recognise the methylation moiety together with other transcriptional components [95]. Indeed, methylation-specific binding proteins potentially serve to control gene expression of many functional sites within the genome in a chromatin context, thereby offering an attractive model to explain the capacity of gene function more effectively [96].

Epigenetic and metabolic memory nexus. Persistent epigenetic changes in the context of transient, medium- and long-term metabolic memory. In certain cell types, hyperglycaemic memory exists. For example, in response to hyperglycaemia, SET7 methyltransferase enzyme writes mono-methylation of histone H3 lysine 4 (H3K4me1) in vascular endothelial cells, and this methylation is retained over the long term. The effectiveness of transient hyperglycaemic stimuli to tightly control the expression of target genes implicated in vascular dysfunction and inflammation relies on ROS-mediated pathways. Indeed, a hyperglycaemic sensor (SET7) and the transfer of a chemical group (mono-methylation on H3K4) to a gene are two common principles implicated in metabolic memory. These regulatory events serve to remodel chromatin to precisely decorate target genes such as the p65 subunit of NFkB, with H3K4me1 corresponding with persistent transcriptional activation [24, 26, 30, 102]. The long-term contribution of hyperglycaemic signalling cues derived from the monocytes of type 1 diabetic individuals from the DCCT and EDIC trials show HbA1c levels and H3K9ac are also tightly linked [31]. The control of genes related to the NFkB inflammatory pathway by modifications exhibits common principles of epigenetic control that serves as a paradigm for metabolic memory. The mechanism conferring H3K9ac and metabolic memory remains poorly characterised. Clearly, this explains only part of the epigenetic complexity, in the metabolic memory example and using monocytes derived from the DCCT and EDIC trials, genomic modification, specifically, 5mC is also an important determinant in the control of gene expression [54]. The mechanism underlying 5mC modification in metabolic memory remains poorly characterised. Recent advances in our understanding of the mechanisms of genomic modification show 5hmC, a DNA base derived from 5mC by oxidation by TET enzymes, is implicated in transposon activity associated with exposure to adverse in utero programming and gestational diabetes [150]. These findings emphasise that cytosine residue modification plays an important role in the regulation of genes implicated in diabetes
Targeting chromatin modifications in the clinic
With increased exploration of chromatin modifications in various medical contexts, it may be possible in the future not only to track causal mechanisms of vascular disease, but also to capture an individual patient’s position within a complex spectrum of pathophysiological processes, thereby supporting tailored approaches to the anticipation and prevention of diabetic complications [97]. The epigenome is responsive to internal and external stimuli as diverse as disease-specific processes, nutrition [74] and exercise [98]. Exploitation of this plasticity has fast become a novel avenue of investigation to improve dysregulated gene function. Whereas metabolic manipulation of the chromatin landscape is a relatively recent suggestion, pharmacological compounds that modulate epigenetic regulators have a longer history in the clinic. Common to both strategies is the imperative challenge of specificity.
The emerging picture of epigenetic regulation is one of remarkable complexity. Defining the tissue-specific relative contributions of epigenetic writers and erasers remains an unresolved but critical issue for developing novel therapeutic epigenetic modulators. Whereas HDAC3 deletion from the macrophage is vasculo-protective [99], deletion of the same enzyme from endothelial cells exacerbates macrovascular disease [100]. Many enzymes that modify histones also target specific amino acids on other proteins, including transcription factors, to post-translationally regulate their stability and activity, with major implications for the interpretation of gene expression profiles. In addition to writing H3K4m1, SET7 methylates a variety of transcription factors in different cellular contexts [101], and is therefore implicated in chromatin-dependent and chromatin-independent gene regulation [102, 103]. Furthermore, evidence is emerging to suggest that histone-modifying enzymes can post-translationally modify and regulate each other [74], meaning that a single enzyme could potentially influence many distinct modifications. Finally, the mechanisms driving loci-specific enrichment of chromatin modifications remain largely uncharacterized, though several examples of non-coding RNA and transcription factor co-recruitment have recently emerged [104, 105]. The respective repertoire of gene-localising mechanisms is likely to reflect cell type specificity of distinct gene programs.
Despite these formidable challenges, the epigenome is rich in opportunity. Compounds that target epigenetic pathways are increasingly investigated pre-clinically, and drugs that are already used in clinical management of diabetes may impact the epigenetic landscape. For example, metformin prevents trained immunity by the Bacillus Calmette–Guérin (BCG) vaccine via mTOR inhibition and suppression of glycolysis [69]. Whether this holds true for immunological training in the context of diabetes remains to be elucidated.
Conclusion
Increasingly accessible technologies that permit unbiased acquisition of genome-wide patterns boast the capacity to transform our understanding of the chromatin landscape in the occurrence and progression of complex diseases. The immense potential for epigenetics to explain many aspects of diabetic vascular complications is evident in recent scientific literature. By sensitising the genome to environmental variation, these molecular signatures shape diverse phenotypes and functional programs. Chromatin modifications influence deleterious changes in gene expression that, under some circumstances, can endure improvements in metabolic management. Similarly, persistent epigenetic modifications drive the non-specific memory of proinflammatory macrophages, a process that is increasingly implicated in vascular disease and could prove to be instrumental to resolving the important debate as to whether diabetic microvascular and macrovascular pathology share a common pathology. Epigenomic profiling of circulating cells may further shed light on the phenotypic variation and disproportionate burden of diabetic vascular complications. Combined with classical genetic approaches, epigenomic profiling has potential to identify molecular trajectories underlying diabetic vascular disease development. While the extent that pathological chromatin changes can be manipulated in human diabetic complications remains to be established, the clinical applicability of epigenetic interventions will be greatly advanced by a deeper understanding of the cell type-specific functions and interactions of chromatin-modifying machinery in the diabetic vasculature.
Abbreviations
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- BCG:
-
Bacillus Calmette–Guérin vaccine
- CpG:
-
Cytosine–guanine dinucleotide
- CTCF:
-
CCCTC-binding factor
- DCCT:
-
Diabetes Control and Complications Trial
- DNMT:
-
DNA methyltransferase
- EDIC:
-
Epidemiology of Diabetes Interventions and Complications
- ESRD:
-
End-stage renal disease
- EZH2:
-
Enhancer of zeste 2 repressive complex 2 subunit
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- KLF4:
-
Kruppel-like factor 4
- mTOR:
-
Mammalian target of rapamycin
- oxLDL:
-
Oxidised low-density lipoprotein
- Lp(a):
-
Lipoprotein(a)
- NPHS1:
-
Nephrin
- PTEC:
-
Proximal tubular epithelial cell
- TET:
-
Ten-eleven translocation
- TXNIP:
-
Thioredoxin-interacting protein
References
Thomas MC, Groop PH, Tryggvason K (2012) Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr Opin Nephrol Hypertens 21:195–202
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
DCCT/EDIC The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group (2000) Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med 342:381–389
Hammes HP, Klinzing I, Wiegand S, Bretzel RG, Cohen AM, Federlin K (1993) Islet transplantation inhibits diabetic retinopathy in the sucrose-fed diabetic Cohen rat. Invest Ophthalmol Vis Sci 34:2092–2096
Roy S, Sala R, Cagliero E, Lorenzi M (1990) Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci U S A 87:404–408
Engerman RL, Kern TS (1987) Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36:808–812
Gaede P, Oellgaard J, Carstensen B et al (2016) Years of life gained by multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: 21 years follow-up on the Steno-2 randomised trial. Diabetologia 59:2298–2307
Hayward RA, Reaven PD, Wiitala WL et al (2015) Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 372:2197–2206
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Thomas MC (2016) Epigenetic mechanisms in diabetic kidney disease. Curr Diab Rep 16:31
Mannucci E, Dicembrini I, Lauria A, Pozzilli P (2013) Is glucose control important for prevention of cardiovascular disease in diabetes? Diabetes Care 36(Suppl 2):S259–S263
Orasanu G, Plutzky J (2009) The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol 53:S35–S42
Sasso FC, Chiodini P, Carbonara O et al (2012) High cardiovascular risk in patients with type 2 diabetic nephropathy: the predictive role of albuminuria and glomerular filtration rate. The NID-2 Prospective Cohort Study. Nephrol Dial Transplant 27:2269–2274
Chawla A, Chawla R, Jaggi S (2016) Microvasular and macrovascular complications in diabetes mellitus: distinct or continuum? Indian J Endocrinol Metab 20:546–551
The Diabetes Control and Complications Trial Research Group (1997) Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. Diabetes 46:1829–1839
Quinn M, Angelico MC, Warram JH, Krolewski AS (1996) Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 39:940–945
Seaquist ER, Goetz FC, Rich S, Barbosa J (1989) Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med 320:1161–1165
Ma RC, Cooper ME (2017) Genetics of diabetic kidney disease—from the worst of nightmares to the light of dawn? J Am Soc Nephrol 28:389–393
Sandholm N, Van Zuydam N, Ahlqvist E et al (2017) The genetic landscape of renal complications in type 1 diabetes. J Am Soc Nephrol 28:557–574
Flavahan WA, Drier Y, Liau BB et al (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529:110–114
Breiling A, Lyko F (2015) Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin 8:24
Dubois-Chevalier J, Oger F, Dehondt H et al (2014) A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res 42:10943–10959
Heintzman ND, Stuart RK, Hon G et al (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39:311–318
El-Osta A, Brasacchio D, Yao D et al (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205:2409–2417
Cheng J, Blum R, Bowman C et al (2014) A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell 53:979–992
Brasacchio D, Okabe J, Tikellis C et al (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58:1229–1236
Fu LL, Tian M, Li X et al (2015) Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 6:5501–5516
Yoon S, Eom GH (2016) HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 52:1–11
McKinsey TA (2012) Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 52:303–319
Paneni F, Costantino S, Battista R et al (2015) Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet 8:150–158
Miao F, Chen Z, Genuth S et al (2014) Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 63:1748–1762
Keating ST, Plutzky J, El-Osta A (2016) Epigenetic changes in diabetes and cardiovascular risk. Circ Res 118:1706–1722
Yoshizawa C, Kobayashi Y, Ikeuchi Y et al (2016) Congenital nephrotic syndrome with a novel NPHS1 mutation. Pediatr Int 58:1211–1215
Yamaguchi Y, Iwano M, Suzuki D et al (2009) Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis 54:653–664
Pagtalunan ME, Miller PL, Jumping-Eagle S et al (1997) Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99:342–348
Petermann AT, Pippin J, Krofft R et al (2004) Viable podocytes detach in experimental diabetic nephropathy: potential mechanism underlying glomerulosclerosis. Nephron Exp Nephrol 98:e114–e123
Takahashi K, Tanabe K, Ohnuki M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Hayashi K, Sasamura H, Nakamura M et al (2014) KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J Clin Invest 124:2523–2537
Ristola M, Arpiainen S, Saleem MA, Holthofer H, Lehtonen S (2012) Transcription of nephrin-Neph3 gene pair is synergistically activated by WT1 and NF-kappaB and silenced by DNA methylation. Nephrol Dial Transplant 27:1737–1745
Siddiqi FS, Majumder S, Thai K et al (2016) The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J Am Soc Nephrol 27:2021–2034
De Marinis Y, Cai M, Bompada P et al (2016) Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 89:342–353
Bock F, Shahzad K, Wang H et al (2013) Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 110:648–653
Paneni F, Mocharla P, Akhmedov A et al (2012) Gene silencing of the mitochondrial adaptor p66Shc suppresses vascular hyperglycemic memory in diabetes. Circ Res 111:278–289
Zhou S, Chen HZ, Wan YZ et al (2011) Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 109:639–648
Tang SC, Lai KN (2012) The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol Dial Transplant 27:3049–3056
Marumo T, Yagi S, Kawarazaki W et al (2015) Diabetes induces aberrant DNA methylation in the proximal tubules of the kidney. J Am Soc Nephrol 26:2388–2397
Hasegawa K, Wakino S, Simic P et al (2013) Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med 19:1496–1504
Reddy MA, Sumanth P, Lanting L et al (2014) Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int 85:362–373
Furuta T, Saito T, Ootaka T et al (1993) The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis 21:480–485
Sassy-Prigent C, Heudes D, Mandet C et al (2000) Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes 49:466–475
Wada J, Makino H (2016) Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol 12:13–26
Tanaka M, Masuda S, Matsuo Y et al (2016) Hyperglycemia and inflammatory property of circulating monocytes are associated with inflammatory property of carotid plaques in patients undergoing carotid endarterectomy. J Atheroscler Thromb 23:1212–1221
Kon V, Linton MF, Fazio S (2011) Atherosclerosis in chronic kidney disease: the role of macrophages. Nat Rev Nephrol 7:45–54
Chen Z, Miao F, Paterson AD et al (2016) Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A 113:E3002–E3011
Netea MG, Joosten LA, Latz E et al (2016) Trained immunity: a program of innate immune memory in health and disease. Science 352:aaf1098
Kleinnijenhuis J, Quintin J, Preijers F et al (2012) Bacille Calmette-Guérin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A 109:17537–17542
Quintin J, Saeed S, Martens JH et al (2012) Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12:223–232
Saeed S, Quintin J, Kerstens HH et al (2014) Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345:1251086
Novakovic B, Habibi E, Wang SY et al (2016) Beta-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167:1354–1368.e1314
Ostuni R, Piccolo V, Barozzi I et al (2013) Latent enhancers activated by stimulation in differentiated cells. Cell 152:157–171
Blok BA, Arts RJ, van Crevel R, Benn CS, Netea MG (2015) Trained innate immunity as underlying mechanism for the long-term, nonspecific effects of vaccines. J Leukoc Biol 98:347–356
Bekkering S, Joosten LA, van der Meer JW, Netea MG, Riksen NP (2013) Trained innate immunity and atherosclerosis. Curr Opin Lipidol 24:487–492
Christ A, Bekkering S, Latz E, Riksen NP (2016) Long-term activation of the innate immune system in atherosclerosis. Semin Immunol 28:384–393
Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP (2014) Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol 34:1731–1738
van der Valk FM, Bekkering S, Kroon J et al (2016) Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 134:611–624
Yan SF, Ramasamy R, Schmidt AM (2008) Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab 4:285–293
Shanmugam N, Reddy MA, Guha M, Natarajan R (2003) High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 52:1256–1264
van Diepen JA, Thiem K, Stienstra R, Riksen NP, Tack CJ, Netea MG (2016) Diabetes propels the risk for cardiovascular disease: sweet monocytes becoming aggressive? Cell Mol Life Sci 73:4675–4684
Cheng SC, Quintin J, Cramer RA et al (2014) mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345:1250684
Arts RJ, Novakovic B, Ter Horst R et al (2016) Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab 24:807–819
Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell 48:612–626
Cheng SC, Joosten LA, Netea MG (2014) The interplay between central metabolism and innate immune responses. Cytokine Growth Factor Rev 25:707–713
Kaelin WG Jr, McKnight SL (2013) Influence of metabolism on epigenetics and disease. Cell 153:56–69
Keating ST, El-Osta A (2015) Epigenetics and metabolism. Circ Res 116:715–736
de Oliveira AA, de Oliveira TF, Bobadilla LL et al (2017) Sustained kidney biochemical derangement in treated experimental diabetes: a clue to metabolic memory. Sci Rep 7:40544
Sharma A (2017) Transgenerational epigenetics: integrating soma to germline communication with gametic inheritance. Mech Ageing Dev 163:15–22
Cropley JE, Eaton SA, Aiken A et al (2016) Male-lineage transmission of an acquired metabolic phenotype induced by grand-paternal obesity. Mol Metab 5:699–708
Khurana I, Kaspi A, Ziemann M et al (2016) DNA methylation regulates hypothalamic gene expression linking parental diet during pregnancy to the offspring’s risk of obesity in Psammomys obesus. Int J Obes 40:1079–1088
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA (2010) Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genet 3:33
Sapienza C, Lee J, Powell J et al (2011) DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 6:20–28
Tregouet DA, Groop PH, McGinn S et al (2008) G/T substitution in intron 1 of the UNC13B gene is associated with increased risk of nephropathy in patients with type 1 diabetes. Diabetes 57:2843–2850
Miao F, Wu X, Zhang L, Riggs AD, Natarajan R (2008) Histone methylation patterns are cell-type specific in human monocytes and lymphocytes and well maintained at core genes. J Immunol 180:2264–2269
Reinius LE, Acevedo N, Joerink M et al (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7:e41361
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome–biological and translational implications. Nat Rev Cancer 11:726–734
Pirola L, Balcerczyk A, Tothill RW et al (2011) Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 21:1601–1615
Birney E, Smith GD, Greally JM (2016) Epigenome-wide association studies and the interpretation of disease-omics. PLoS Genet 12:e1006105
Richmond RC, Sharp GC, Ward ME et al (2016) DNA methylation and BMI: investigating identified methylation sites at HIF3A in a causal framework. Diabetes 65:1231–1244
Rakyan VK, Beyan H, Down TA et al (2011) Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 7:e1002300
Valencia-Morales Mdel P, Zaina S, Heyn H et al (2015) The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genet 8:7
Lewis A, Murrell A (2004) Genomic imprinting: CTCF protects the boundaries. Curr Biol 14:R284–R286
Kanduri C, Pant V, Loukinov D et al (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10:853–856
Demars J, Shmela ME, Khan AW et al (2014) Genetic variants within the second intron of the KCNQ1 gene affect CTCF binding and confer a risk of Beckwith-Wiedemann syndrome upon maternal transmission. J Med Genet 51:502–511
Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS (2004) Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science 303:238–240
Stitzel ML, Sethupathy P, Pearson DS et al (2010) Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab 12:443–455
El-Osta A, Kantharidis P, Zalcberg JR, Wolffe AP (2002) Precipitous release of methyl-CpG binding protein 2 and histone deacetylase 1 from the methylated human multidrug resistance gene (MDR1) on activation. Mol Cell Biol 22:1844–1857
Harikrishnan KN, Chow MZ, Baker EK et al (2005) Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet 37:254–264
McCarthy MI (2015) Genomic medicine at the heart of diabetes management. Diabetologia 58:1725–1729
King-Himmelreich TS, Schramm S, Wolters MC et al (2016) The impact of endurance exercise on global and AMPK gene-specific DNA methylation. Biochem Biophys Res Commun 474:284–290
Hoeksema MA, Gijbels MJ, Van den Bossche J et al (2014) Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med 6:1124–1132
Zampetaki A, Zeng L, Margariti A et al (2010) Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 121:132–142
Keating ST, El-Osta A (2013) Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics 8:361–372
Okabe J, Orlowski C, Balcerczyk A et al (2012) Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res 110:1067–1076
Keating ST, Ziemann M, Okabe J, Khan AW, Balcerczyk A, El-Osta A (2014) Deep sequencing reveals novel Set7 networks. Cell Mol Life Sci 71:4471–4486
Lian Y, Wang J, Feng J et al (2016) Long non-coding RNA IRAIN suppresses apoptosis and promotes proliferation by binding to LSD1 and EZH2 in pancreatic cancer. Tumour Biol 37:14929–14937
Battistelli C, Cicchini C, Santangelo L et al (2017) The snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 36:942–955
Yuan H, Reddy MA, Deshpande S et al (2016) Epigenetic histone modifications involved in profibrotic gene regulation by 12/15-lipoxygenase and its oxidized lipid products in diabetic nephropathy. Antioxid Redox Signal 24:361–375
Syreeni A, El-Osta A, Forsblom C et al (2011) Genetic examination of SETD7 and SUV39H1/H2 methyltransferases and the risk of diabetes complications in patients with type 1 diabetes. Diabetes 60:3073–3080
Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R, Parrizas M (2010) Histone demethylase LSD1 regulates adipogenesis. J Biol Chem 285:30034–30041
Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R (2008) Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A 105:9047–9052
Hardikar AA, Satoor SN, Karandikar MS et al (2015) Multigenerational undernutrition increases susceptibility to obesity and diabetes that is not reversed after dietary recuperation. Cell Metab 22:312–319
Chen H, Gu X, Su IH et al (2009) Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23:975–985
Sakai M, Tujimura-Hayakawa T, Yagi T et al (2016) The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat Commun 7:13147
Vecellio M, Spallotta F, Nanni S et al (2014) The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in the human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes 63:2132–2147
Gallagher KA, Joshi A, Carson WF et al (2015) Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 64:1420–1430
Okuno Y, Ohtake F, Igarashi K et al (2013) Epigenetic regulation of adipogenesis by PHF2 histone demethylase. Diabetes 62:1426–1434
Sathishkumar C, Prabu P, Balakumar M et al (2016) Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenetics 8:125
Wang X, Liu J, Zhen J et al (2014) Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int 86:712–725
Daneshpajooh M, Bacos K, Bysani M et al (2017) HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia 60:116–125
Bell AC, Felsenfeld G (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405:482–485
Barres R, Osler ME, Yan J et al (2009) Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab 10:189–198
Pennarossa G, Maffei S, Campagnol M, Tarantini L, Gandolfi F, Brevini TA (2013) Brief demethylation step allows the conversion of adult human skin fibroblasts into insulin-secreting cells. Proc Natl Acad Sci U S A 110:8948–8953
Volkmar M, Dedeurwaerder S, Cunha DA et al (2012) DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 31:1405–1426
Ling C, Del Guerra S, Lupi R et al (2008) Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51:615–622
Kirchner H, Sinha I, Gao H et al (2016) Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol Metab 5:171–183
Babu M, Durga Devi T, Makinen P et al (2015) Differential promoter methylation of macrophage genes is associated with impaired vascular growth in ischemic muscles of hyperlipidemic and type 2 diabetic mice: genome-wide promoter methylation study. Circ Res 117:289–299
Radford EJ, Ito M, Shi H et al (2014) In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 345:1255903
Dayeh T, Volkov P, Salo S et al (2014) Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 10:e1004160
Yang BT, Dayeh TA, Volkov PA et al (2012) Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol 26:1203–1212
Park JH, Stoffers DA, Nicholls RD, Simmons RA (2008) Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 118:2316–2324
Multhaup ML, Seldin MM, Jaffe AE et al (2015) Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab 21:138–149
Kalani A, Kamat PK, Tyagi N (2015) Diabetic stroke severity: epigenetic remodeling and neuronal, glial, and vascular dysfunction. Diabetes 64:4260–4271
Volkov P, Bacos K, Ofori JK et al (2017) Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 66:1074–1085
Soriano-Tarraga C, Jimenez-Conde J, Giralt-Steinhauer E et al (2016) Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet 25:609–619
Kulkarni H, Kos MZ, Neary J et al (2015) Novel epigenetic determinants of type 2 diabetes in Mexican-American families. Hum Mol Genet 24:5330–5344
Orozco LD, Morselli M, Rubbi L et al (2015) Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab 21:905–917
Gu T, Falhammar H, Gu HF, Brismar K (2014) Epigenetic analyses of the insulin-like growth factor binding protein 1 gene in type 1 diabetes and diabetic nephropathy. Clin Epigenetics 6:10
Ruchat SM, Houde AA, Voisin G et al (2013) Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 8:935–943
Mudry JM, Lassiter DG, Nylen C et al (2016) Insulin and glucose alter death-associated protein kinase 3 (DAPK3) DNA methylation in human skeletal muscle. Diabetes 66:651–662
Volkov P, Olsson AH, Gillberg L et al (2016) A genome-wide mQTL analysis in human adipose tissue identifies genetic variants associated with DNA methylation, gene expression and metabolic traits. PLoS One 11:e0157776
Clarke-Harris R, Wilkin TJ, Hosking J et al (2014) PGC1alpha promoter methylation in blood at 5-7 years predicts adiposity from 9 to 14 years (EarlyBird 50). Diabetes 63:2528–2537
Zeng L, Kanwar YS, Amro N et al (2003) Epigenetic and genetic analysis of p16 in dermal fibroblasts from type 1 diabetic patients with nephropathy. Kidney Int 63:2094–2102
Paul DS, Teschendorff AE, Dang MA et al (2016) Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun 7:13555
Reichetzeder C, Dwi Putra SE, Pfab T et al (2016) Increased global placental DNA methylation levels are associated with gestational diabetes. Clin Epigenetics 8:82
Bacos K, Gillberg L, Volkov P et al (2016) Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun 7:11089
Ronn T, Volkov P, Gillberg L et al (2015) Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 24:3792–3813
Yuan W, Xia Y, Bell CG et al (2014) An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat Commun 5:5719
Nilsson E, Jansson PA, Perfilyev A et al (2014) Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63:2962–2976
El Hajj N, Pliushch G, Schneider E et al (2013) Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 62:1320–1328
Ling C, Poulsen P, Simonsson S et al (2007) Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest 117:3427–3435
Sun M, Song MM, Wei B et al (2016) 5-Hydroxymethylcytosine-mediated alteration of transposon activity associated with the exposure to adverse in utero environments in human. Hum Mol Genet 25:2208–2219
Frayling TM, Timpson NJ, Weedon MN et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Fischer J, Koch L, Emmerling C et al (2009) Inactivation of the Fto gene protects from obesity. Nature 458:894–898
Church C, Moir L, McMurray F et al (2010) Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet 42:1086–1092
Wu W, Feng J, Jiang D et al (2017) AMPK regulates lipid accumulation in skeletal muscle cells through FTO-dependent demethylation of N6-methyladenosine. Sci Rep 7:41606
Shen F, Huang W, Huang JT et al (2015) Decreased N 6-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J Clin Endocrinol Metab 100:E148–E154
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors acknowledge support from the National Health and Medical Research Council (NHMRC) (APP1113188, APP1082572). JAvD is supported by a Veni Grant from The Netherlands Organization for Scientific Research (91616083) and received funding from the Dutch Diabetes Foundation (2013.81.1674). NPR is supported by a dr. E. Dekker grant from the Netherlands Heart Foundation (2012T051), and received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 667837.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Electronic supplementary material
ESM Downloadable slideset
(PPTX 771 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keating, S.T., van Diepen, J.A., Riksen, N.P. et al. Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia 61, 6–20 (2018). https://doi.org/10.1007/s00125-017-4490-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4490-1