
Abstract
In mammals, the circadian timing system drives rhythms of physiology and behaviour, including the daily rhythms of feeding and activity. The timing system coordinates temporal variation in the biochemical landscape with changes in nutrient intake in order to optimise energy balance and maintain metabolic homeostasis. Circadian disruption (e.g. as a result of shift work or jet lag) can disturb this continuity and increase the risk of cardiometabolic disease. Obesity and metabolic disease can also disturb the timing and amplitude of the clock in multiple organ systems, further exacerbating disease progression. As our understanding of the synergy between the timing system and metabolism has grown, an interest has emerged in the development of novel clock-targeting pharmaceuticals or nutraceuticals for the treatment of metabolic dysfunction. Recently, the pineal hormone melatonin has received some attention as a potential chronotherapeutic drug for metabolic disease. Melatonin is well known for its sleep-promoting effects and putative activity as a chronobiotic drug, stimulating coordination of biochemical oscillations through targeting the internal timing system. Melatonin affects the insulin secretory activity of the pancreatic beta cell, hepatic glucose metabolism and insulin sensitivity. Individuals with type 2 diabetes mellitus have lower night-time serum melatonin levels and increased risk of comorbid sleep disturbances compared with healthy individuals. Further, reduced melatonin levels, and mutations and/or genetic polymorphisms of the melatonin receptors are associated with an increased risk of developing type 2 diabetes. Herein we review our understanding of molecular clock control of glucose homeostasis, detail the influence of circadian disruption on glucose metabolism in critical peripheral tissues, explore the contribution of melatonin signalling to the aetiology of type 2 diabetes, and discuss the pros and cons of melatonin chronopharmacotherapy in disease management.
Similar content being viewed by others
Introduction
The economic and health burdens of type 2 diabetes mellitus are set to reach near pandemic proportions in the next 30 years. It is predicted that by 2050, nearly 30% of the US adult population will have type 2 diabetes [1, 2]. Clinical management of type 2 diabetes includes rigorous lifestyle modifications, insulin therapy, drug treatments that promote insulin sensitisation (such as metformin) and insulin secretion, novel glucagon-like peptide-1 (GLP-1) receptor agonists, dipeptidyl peptidase (DPP)-4 inhibitors, and sodium glucose cotransporter 2 (SGLT2) inhibitors [3]. These approaches are designed to manage the symptoms of insulin resistance/beta cell dysfunction and are either used alone or in combination. Many drug therapies for diabetes are costly and in some cases have been associated with adverse events, including possible pancreatitis, hypoglycaemia and osteoporosis [4]. Thus, there remains a palpable need for new and cost-effective pharmacotherapies for diabetes that have limited additional health risks.
The internal timing system and metabolic dysfunction
In the last decade, there has been a considerable increase in our understanding of the cellular and molecular factors that contribute to diabetes development [5]. Without question, poor diet, lack of exercise, obesity and chronic insulin resistance are major contributing factors. One basic function that appears to be heavily influenced by (and also influences) obesity and metabolic disease is the internal timing system [6–10]. The timing system drives daily rhythms of physiology and behaviour, including the alternating pattern of feeding and fasting [11]. It also regulates daily rhythms of insulin secretion and glucose metabolism, either in a direct or indirect manner. In both humans and rodents, there are robust daily rhythms of glucose tolerance and insulin sensitivity [8, 12, 13]. Disturbances of the sleep/wake cycle, reduced sleep duration/quality and altered sleep architecture can impair these rhythms, likely contributing to the development of metabolic disease [14–18]. Likewise, dissociation of activity/feeding patterns from internal rhythms of hormone release and metabolic function, known as ‘environmental circadian disruption’ or ‘circadian misalignment’, can also have negative impacts on metabolism [19, 20]. Circadian disruption is commonly associated with reduced sleep quality but can occur independent of sleep loss, resulting in metabolic deficits such as hyperinsulinaemia and impaired glucose tolerance [21]. It is the prevailing view that the timing system synchronises the biochemical landscape with daily variations in the environment in order to optimise energy utilization [22]. Conversely, tipping the balance (e.g. obesity) can disturb this temporal continuity and perpetuate metabolic dysfunction [23, 24]. Though most animal studies suggest a marked influence of obesity and insulin resistance on clock function [25] , not all human clinical studies confirm this assertion [26].
What defines the crossroads between metabolic homeostasis, endocrine physiology and the timing system?
In addition to leptin and adrenal steroids, evidence reveals that pineal melatonin is a clock-driven factor that is able to modulate daily patterns of glucose metabolism [27]. Recent clinical studies have determined that melatonin can exert a marked influence on glycaemic control [28, 29]. Melatonin falls into a special class of compounds, referred to as chronobiotics, that can simultaneously affect both the timing system and metabolism, possibly via direct influence on the molecular clock [30–32]. Herein we briefly review our current understanding of molecular clock-control of glucose homeostasis, the influence of circadian disruption on glucose management and the role of melatonin as a regulator of the timing system in the aetiology of type 2 diabetes. We also detail the evidence for use of melatonin or melatonin receptor agonists in the prevention and/or management of type 2 diabetes.
The circadian timing system in mammals: from genes to hormones
Clock genes and cellular metabolism
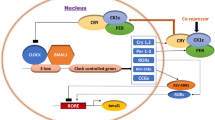
In mammals, the circadian timing system includes the coordinated activity of the central neural pacemaker, the suprachiasmatic nucleus (SCN), with target oscillators in both the brain and periphery [33]. The SCN is persistently entrained to the external environment, and the ablation of this nucleus disrupts sleep/wake, body temperature and hormonal rhythms [34, 35]. At the cellular level, rhythms depend on the activity of the molecular clock, an autoregulatory feedback loop oscillator of interacting transcription factors known as clock genes (Fig. 1) [36]. This family of transcriptional regulators includes circadian locomotor output cycles kaput (CLOCK), brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein-1 (BMAL1, also known as ARNTL1), period circadian protein (PER1, PER 2, PER 3), cryptochrome (CRY1 and CRY 2), and the nuclear receptors retinoic acid receptor-like orphan receptor alpha (RORα) and reverse thyroid receptor α (REV-ERBα). CLOCK and BMAL1 form a complex that drives Per, Cry, Rev-erbα (also known as Nr1d1) and Rora transcription. The protein products of the Per and Cry genes heterodimerise to form PER–CRY and repress BMAL1–CLOCK activity (Fig. 1). REV-ERBα and RORα synergistically control the timing and amplitude of Bmal1 (also known as Arntl) expression and provide additional stability to the molecular oscillator. Rhythmic post-translational modifications of clock proteins, including phosphorylation of the PER–CRY complex by casein kinase 1 ε/δ (CKIε/δ) (Fig. 1), introduces temporal delays that contribute to the 24 h period of the clock [37]. At the cellular level, the clock influences, and is influenced by, circadian rhythms in metabolism; daily variations in glucose uptake and metabolism influence the clock through metabolic sensors, such as adenosine monophosphate-activated protein kinase (AMPK) [38]. Further, REV-ERBα is activated by haem, levels of which vary greatly in response to nutrient availability [39]. The synergy between clock function and cellular metabolism is beyond the scope of this review and has previously been described in detail (see [22, 40]).

A schematic of the mammalian molecular clock showing clock-controlled hepatocyte- and pancreatic beta cell-specific transcripts associated with the regulation of glucose homeostasis and metabolism. A schematic of the core molecular clock is shown, including the BMAL1–CLOCK activator complex and its targets, including the PER and CRY family of repressors, REV-ERBα, RORα and clock-controlled output genes. In the hepatocyte, in addition to Glut2 (also known as Slc2a2) expression, the clock regulates genes that control: glucose metabolism and gluconeogenesis (hexokinase [Hk2], phosphoenolpyruvate carboxykinase [Pepck] and 6-phosphofructo-2-kinase/Fructose-2,6-biphosphatase 3 [Pfkfb3]); lipid metabolism (Rev-erbα, peroxisome proliferator-activated receptor gamma [Pparg], peroxisome proliferator- activated receptor gamma and coactivator 1, alpha [Pgc1α, also known as Ppargc1a]); and nuclear receptor subfamily 1, group H, member 3 (Lxr, also known as Nr1h3). In pancreatic beta cells, the clock regulates insulin secretory activity and GLUT2 (Slc2a2) expression rhythms, and also genes that control: vesicular docking and release (vesicle associated membrane protein 3 [Vamp3], syntaxin-1A (Stx1a) and syntaxin binding protein [Munc18, also known as Stxbp]); and beta cell proliferation and survival (hepatocyte nuclear factor 4 alpha [Hnf4a], insulin receptor [Insr], cyclin D1 [Ccnd1] and pancreatic and duodenal homeobox 1 [Pdx1]). The (+) and green line indicate transcriptional activation function, whereas the (−) and red lines indicate transcriptional repression. Black arrows with sine waves indicate rhythmic translation; red/green arrowhead indicates nuclear translocation of REV-ERBα and RORα; yellow circles indicate phosphorylation. B, BMAL1; C, CLOCK; CKIε/δ, casein kinase 1 ε/δ; CCG, clock-controlled genes; E-box, enhancer box; P, phosphate group; REV-ERB/ROR response element, RORE
The chronobiotic hormone, melatonin, influences metabolism
In the brain, SCN neurons receive environmental timing cues from the retina and respond by coordinating the activity of oscillators in the brain with those in the periphery [33]. It is known that this coordination is carried out in an intricate and tissue-specific manner. However, further details of the mechanisms underlying this process are poorly defined. Glucocorticoids [41], melatonin [42, 43] and direct autonomic innervation [44, 45] are endogenous signalling mechanisms by which the SCN maintains alignment of central and peripheral oscillators. Among these, melatonin is the most well studied humoral factor that affects the SCN, whilst also being affected by this region of the brain.
Melatonin is considered the primary ‘chronobiotic’ hormone, regulating coordination within the timing system [46]. It is secreted from pinealocytes during the night in both nocturnal and diurnal mammals [47]. It has been proposed that this hormone may have a potential role as a therapeutic agent for metabolic disease, given its established influence on glucose homeostasis (see Fig. 2) [48]. The impact of melatonin on metabolism is almost global; amongst other processes, it influences the secretory activity of pancreatic islet cells, glucose metabolism in the liver and insulin sensitivity at target tissues [48, 49]. Strikingly, a reduced level of night-time melatonin, as assessed by 6-sulfatoxymelatonin in morning urinary void, is positively associated with an increased risk of type 2 diabetes (Fig. 2) [50]. There is an emerging link between melatonin secretion, insulin secretion and abnormal glucose metabolism, and, thus, it is reasonable to hypothesise that altered/reduced melatonin levels may contribute to the development and severity of both metabolic and sleep disturbances in patients with type 2 diabetes.

Relationship between melatonin signalling and metabolic dysfunction in type 2 diabetes. Both in individuals (a) without diabetes and (b) with diabetes, light entrains the activity of a central pacemaker in the SCN. Timing cues are relayed from the SCN to the pineal or directly to peripheral targets via β-adrenergic autonomic nervous signalling. The pineal secretion of melatonin during the night is attenuated in type 2 diabetes and is associated with hyperglycaemia and dyslipidaemia. (a) In non-diabetic individuals, night-time melatonin helps to maintain euglycaemia by sensitising the pancreas to the morning bolus of glucose. Melatonin may also regulate the release of glucagon and indirectly control the timing of GLUT2 expression in hepatocytes through its influence on the molecular clock. These melatonin-induced effects are associated with stable serum glucose, triacylglycerol and LDL-cholesterol levels. (b) In contrast, in individuals with type 2 diabetes, melatonin secretion is reduced and/or arrhythmic (indicated by oval with horizontal line) and these individuals also have reduced insulin production, excess glucagon, hyperleptinaemia and reduced GLUT2 expression in the liver. It is unclear, however, whether the depletion of melatonin directly effects the timing of the molecular clock in the SCN or peripheral tissues in these individuals. Nonetheless, these combined effects result in hyperglycaemia, insulin resistance and dyslipidaemia. Red arrows indicate the negative impacts of hypomelatonaemia on selected biomolecules and the directionality of the effect (upward pointing for enhanced and downward pointing for suppressed); question marks indicate suggested (but not proven) mechanisms. hv, light; RHT, retinohypothalamic tract; SCG, superior cervical ganglion. In both (a) and (b), black lines with square ends indicate neural signalling pathways, whereas lines ending in circles indicate hormonal signalling. Ovals containing sine waves indicate tissues/cells with autonomous clock function or rhythmic signalling processes (e.g. rhythmic output of the SCG)
The timing system and glucose homeostasis
In mammals, glucose homeostasis is one of the most fundamental physiological processes that is driven by the timing system [10, 51] since it is heavily dependent on the predictive capacity of the system to coordinate metabolic function with daily variation in nutrient uptake (feeding vs fasting) [52]. Both rodent and human studies confirm that daily rhythms of blood glucose and insulin secretion are regulated by the timing system [53–55], whilst lesions of the SCN or environmental circadian disruption can lead to insulin resistance and obesity [56, 57]. The molecular clock is essential for glucose metabolism, as evidenced by the impairment of glucose tolerance and insulin sensitivity upon disruption of core clock gene expression [51, 58–63]. Though SCN-driven neuroendocrine and autonomic outflow influence glucose homeostasis [64], peripheral clocks in the liver and pancreas also play a role in glucose management [10, 65–69]. We will avoid discussion of SCN-driven cues here, as their contribution has been reviewed elsewhere [10, 56, 64, 70, 71]. Instead, we will focus on clock function within the liver and pancreas as it relates to direct tissue level control of glucose homeostasis.
Molecular clock function in the liver and pancreas: contribution of peripheral oscillators to glucose homeostasis
Clock function in the mammalian liver, particularly in relation to glucose homeostasis, has been well described ([72, 73]). In hepatocytes, the clock contributes to gluconeogenesis, glucose transport and lipid metabolism (Fig. 1) [74]. Global and liver-targeted Bmal1 knockout mice demonstrate impaired gluconeogenesis and insulin-induced hypoglycaemia [51, 72]. Additionally, global deletion of Bmal1 results in glucose intolerance and impaired insulin secretion; however, this phenotype is not observed in mice with liver-specific Bmal1deletion. In fact, compared with mice with global Bmal1 deletion, liver-specific Bmal1 knockout mice have relative hypoglycaemia, in line with impaired gluconeogenesis [72]. Similarly, Per2 Brdm1 mutant mice (lacking a functional PER2 protein) display reduced glucose levels compared with wild-type mice during periods of fasting, indicating that PER2 can also influence glucose synthesis [10, 75]. PER2 is known to interact with PPARα and REV-ERBα, both of which regulate the expression of the gluconeogenic enzyme, glucose-6-phosphatase (G6Pase) [11]. Daily rhythms of the glucose transporter, GLUT2, in the liver are also eliminated in both Bmal1 −/− and Per2 Brdm1 mutant mice [72, 75].
Molecular clock function has been linked to the timing and amplitude of pancreatic insulin secretion [59, 76]. In the pancreas, the clock regulates target genes involved in insulin secretion, beta cell proliferation and growth, oxidative stress defences, and metabolism (Fig. 1) [77]. The insulin secretion rate (ISR) from beta cells displays a circadian rhythm and serum insulin levels vary across the 24 h day in both rodents and humans, reaching a peak during the latter half of the feeding period [53, 55, 78]. Mice with targeted disruption of the clock in beta cells present with glucose intolerance and impaired glucose-stimulated insulin secretion [59, 76]. However, beta cell mass and insulin content are normal in these mice, implicating abnormal insulin secretion over deficits in synthesis [69].
It is worth noting that single gene mutation of clock genes, particularly BMAL1 and CLOCK, are likely to have pleiotropic effects unrelated to clock function per se. Therefore, knockout studies must be considered in light of these potentially confounding off-target effects. Moreover, a majority of mouse studies are conducted using C57BL6/J mice, a congenic strain that fails to produce significant amounts of melatonin due to a genetic deficit in the rate liming enzyme for melatonin synthesis [79].
Metabolic disease and sleep
The link between sleep and diabetes/diabetic comorbidities
The most salient rhythm regulated by the internal timing system in mammals is the sleep/wake cycle [80, 81]. For some time it has been appreciated that those suffering from metabolic disease, including obesity, impaired glucose tolerance or diabetes also display comorbid sleep difficulties [82]. Most common among these conditions is sleep-disordered breathing, including obstructive sleep apnoea [83]. Insufficient sleep and/or poor sleep quality are potent risk factors for obesity and cardiometabolic disease [81, 84–86]. A large proportion of patients with insomnia (over 50%) present with comorbidities including cardiovascular disease, mental illness, obesity and diabetes [85]. Conversely, patients with type 2 diabetes often report higher rates of insomnia, with studies indicating that roughly 50% of adult diabetic individuals have insomnia compared with roughly 30% of individuals without diabetes [87]. Other sleep disorders, such as reduced sleep times/insufficient sleep, have been reported among individuals with type 2 diabetes [88]. Recently it was reported that sleep duration is a strong predictor of cardiometabolic risk score in obese adolescents [89]. A large nurses’ health study found that individuals who slept less than 5 h per night had a greater risk of being diagnosed with symptomatic diabetes [90]. If indeed short or fragmented sleep is an independent risk factor for obesity and poor glycaemic control, there is potential for improved sleep quality to ameliorate these outcomes. The reverse may also be true; treating glucose intolerance may improve sleep. In fact, data show that the insulin-sensitising drug metformin is associated with improved sleep efficiency in individuals with type 2 diabetes [91].
Sleep disturbance, circadian disruption and altered glucose metabolism
Though many studies have examined the effects of sleep disruption or deficiency on metabolism, not all sleep disorders can be considered disorders of the timing system per se. Recent efforts have been made to differentiate the effects of circadian disruption or circadian misalignment from other forms of sleep disturbance [18, 20, 21, 92]. Evidence from a clinical study of circadian disruption in controlled-laboratory conditions revealed that exposure to misalignment, as occurs during shift work or chronic jet lag, leads to hyperinsulinaemia, hyperleptinaemia and misaligned cortisol rhythms [21]. Strikingly, only 4–5 days of ‘misalignment’ was required to produce a significant increase in 2 h postprandial glucose, with almost half of the participants being categorised by the authors as prediabetic during misalignment (plasma glucose >8.06 mmol/l) relative to baseline (aligned) [21]. These effects appear to be independent of sleep loss or diminished sleep quality, suggesting that disruption of the timing system alone can lead to metabolic dysfunction [19, 20]. In support of this notion, the probability of a genetic polymorphism in the PER3 genetic locus was greater among individuals with type 2 diabetes, suggesting that altered activity of the PER3 protein, and thus the molecular clock, may be an independent risk factor for metabolic disease [93]. Finally, data show that the amount of social jet lag (a delay in mid-sleep time on ‘off days’ relative to work days) experienced is positively correlated with elevated BMI, independent of nightly sleep duration [94].
Melatonin signalling: tissue-dependent influences on glucose homeostasis and molecular clock function
As previously noted, melatonin secretion during the night (in both nocturnal and diurnal mammals) is regulated by SCN pacemaker neurons [47]. Melatonin has established and significant sleep-promoting effects [95–98]. It can also act directly on hepatocytes and pancreatic beta cells to regulate glucose homeostasis, though the precise mechanisms for these influences remain unclear.
The hepatic response to melatonin
Improvements in hepatic insulin sensitivity
Melatonin receptor type 1 (MT1) and type 2 (MT2) are expressed on hepatocytes, allowing melatonin to bind to these cells with high affinity [99, 100]. On the one hand, melatonin has been shown to induce hyperglycaemia in mice 1 h after systemic injection [99]. However, similar to metformin, melatonin can increase the activity of AMPK and potentially enhance insulin sensitivity in the liver [101]. Accordingly, in aged rats, chronic melatonin supplementation in drinking water for 8–12 weeks increased insulin sensitivity by more than twofold without changing hepatic insulin receptor expression, suggesting that these insulin-sensitising effects were mediated by target tissues outside of the liver [102].
Receptor-specific regulation of the hepatic molecular clock
Regarding the direct influence of melatonin signalling on the molecular clock, evidence demonstrates that deletion of MT1 and/or MT2 alters the timing and amplitude of clock gene expression in hepatocytes. Specifically, Muhlbauer and colleagues revealed that MT1 knockout and MT1/MT2 double knockout mice have dampened rhythms of Per1 expression, whereas single gene MT2 knockout mice have elevated Per1 mRNA in the liver [100]. This data suggests that melatonin acts differentially through MT1 and MT2 receptors to modulate liver clock gene expression. In contrast with these findings, Owino and colleagues found that deletion of melatonin receptors dampened the daily rhythm of plasma glucose in mice, though rhythms of clock gene expression in the liver were not affected [103]. Finally, Nogueira and colleagues determined that surgical removal of the pineal (i.e. pinealectomy [PINX]) from mice shifted the endogenous rhythm of RAC-alpha serine/threonine-protein kinase (Akt) phosphorylation (a marker of insulin receptor activity) and disrupted the acute liver response to insulin [104]. Although they did not examine clock gene expression, they did find that PINX altered the timing and amplitude of the known clock-regulated genes, Pepck (also known as Pck1) and Foxo1. The global response of the clock-regulated transcriptome in the liver (shown in Fig. 1) to melatonin has not been determined. Additional genome-wide studies are needed to address this gap in our knowledge. Further, a detailed examination of the influence of melatonin on transcripts directly associated with insulin signalling, glucose metabolism and beta cell function in human liver is warranted.
The pancreatic response to melatonin
Receptor-specific impact on beta cell function
MT1 and MT2 receptor expression has been detected in both rodent and human pancreatic tissue with reports suggesting that MT1 is enriched in alpha cells, whereas MT2 is limited to beta cells [105, 106]. High-throughput analysis of mRNA abundance with RNA sequencing (RNA-seq) has confirmed MT1 and MT2 expression in human islets, though equal abundance of transcript for each was reported in alpha and beta cells [107]. In the pancreatic islet, melatonin regulates a bevy of cellular activity and gene expression [108–111]. The observation that melatonin has sleep-promoting effects occurred almost in parallel with the discovery that this hormone had ‘insulin-like’ activity. Subsequent work characterised the impacts of melatonin and PINX on glucose homeostasis (for a detailed review see [109]). Data from these experiments suggest an influence of melatonin on beta cell function, although they must be interpreted with caution given the potential impact of PINX on neural substrates that regulate glucose metabolism. Nonetheless, evidence reveals that the effects of melatonin on pancreatic insulin secretion are receptor subtype dependent [109].
Insulin-stimulatory and -inhibitory effects
Using rat INS-11 insulinoma cells it was determined that activation of inhibitory G-protein coupled (Gi-coupled) MT1/MT2 receptors leads to significant reduction of cAMP synthesis, reduced activation of protein kinase A and, presumably, reduced insulin secretion [112]. Melatonin can also act through Gi-coupled MT2 receptors in INS-1 cells to lower intracellular Ca2+ levels [110, 113], which is also predicted to reduce insulin secretion. Finally, melatonin can act through Gq-coupled MT2 receptors to stimulate phospholipase C (PLC) and inositol triphosphate (IP3) activity [114], which is expected to boost insulin secretion. Together, these data suggest that melatonin can have both stimulatory and inhibitory influences on insulin secretion. Notably, in vitro melatonin administration protects beta cells from the deleterious effects of glucose toxicity, improving beta cell survival, and reducing oxidative stress responses in both INS-1 832/13 rodent beta cells and isolated human islets from individuals with type 2 diabetes [115, 116]. In vivo, chronic treatment with melatonin (10 mg kg−1 day−1) was also able to partially restore beta cell mass in a rat model of streptozotocin (STZ)-induced diabetes [117]. Moreover, insulin can boost melatonin synthesis indirectly by modulating adrenergic input to the pineal [118], suggesting that elevated insulin secretion during the day, as seen in humans, primes increases in night-time melatonin levels. Subsequently, night-time melatonin may sensitise the beta cell for the morning increase in glucose uptake [48]. Evidence for a direct influence of circulating melatonin on the molecular clock in the beta cell is exceedingly limited, but long-term treatment with the MT2 receptor agonist, ramelteon, boosted the expression of both Bmal1 and Rev-erbα mRNA in rat INS-1 cells [119].
Pre-clinical and clinical evidence for melatoninergic influences on insulin secretion and glucose metabolism: where does the clock fit in?
Preclinical evidence
Rodent models: evidence for a role for melatonin in obesity, beta cell viability and glucose homeostasis
Studies using rodent models of diabetes support a functional link between melatonin secretion, glycaemic control and pancreatic insulin secretion [109]. Reiter’s group showed that treatment with alloxan or STZ, drugs that destroy beta cells and induce diabetes, also abolishes the night-time increase in melatonin [120]. A decline in serum melatonin levels was also detected in the non-experimentally induced Goto–Kakizaki rat model of diabetes [121]. In agreement with these studies, data show that PINX in rats increased circulating glucose levels by reducing insulin concentration and increasing glucagon levels [122]. Glucose tolerance was also impaired following PINX and could be corrected by exogenous melatonin [122]. PINX reduced GLUT4 expression and responsiveness to insulin in rat adipose tissue [123] and altered the timing of gluconeogenesis, insulin sensitivity and melatonin receptor expression rhythms (especially of the core clock gene and putative melatonin receptor RORα) in the liver [124]. In a recent study, Bibak and colleagues confirmed that 6 weeks of melatonin administration (5, 10 or 20 mg/kg) reduced serum glucose and triacylglycerol levels in STZ-treated rats [125]. Additionally, melatonin treatment for 6 weeks at a concentration of 10 mg/100 ml (10% wt/vol.) in drinking water also normalised serum glucose levels in rat models of diabetes [126]. Though several rodent models have been developed, diet-induced obesity (DIO) using a high-fat diet (HFD) induces insulin resistance and gradual beta cell failure, recreating several of the phenotypic features of adult onset type 2 diabetes. Accordingly, treatment of DIO rats with melatonin attenuated weight gain and reduced glucose, leptin and triacylglycerol levels [127]. Daily injection of DIO rats with melatonin or the melatonin agonist Neu-P11 (piromelatine) also inhibited weight gain and improved insulin sensitivity, in effect countering the negative influence of DIO [128]. Finally, Sartori and colleagues reported a significant improvement in glucose tolerance in DIO mice treated with melatonin for 8 weeks [129]. These data strongly support the notion that melatonin supplementation can ameliorate obesity, attenuate beta cell depletion and improve glucose homeostasis. As described above, melatonin affects clock function in both beta cells and hepatocytes, but it is unknown if the influences of melatonin on body weight, blood glucose levels and insulin sensitivity depend on changes in clock function. Failure of exogenous melatonin to improve metabolic function in DIO mice carrying a global or conditional clock gene mutation (e.g. Bmal1 null) might suggest that the hormone’s effects are mitigated indirectly and/or independent of a direct influence on the molecular clock.
Treatment of healthy 10-month-old rats with melatonin (4 μg/ml in drinking water) for 12 months attenuated age-induced hyperleptinaemia and hyperinsulinaemia [130]. Tresguerres’ group also reported that melatonin reduced age-associated increases in HOMA-IR [131]. Similar studies suggest that melatonin reduces mitochondrial dysfunction [132], inhibits oxidative enzyme activity [133], decreases apoptosis [134], improves dyslipidaemia [135] and reduces diabetes-induced oxidative stress [135]. Together, these rodent studies strengthen the assertion that reductions in melatonin promote insulin resistance and hyperglycaemia, and that these conditions can be corrected with exogenous melatonin administration.
Caveats to the preclinical evidence: is melatonin a viable option for glycaemic control?
The answer to this question may depend on when you eat and sleep relative to the timing of melatonin secretion. Results from preclinical study of melatonin’s effect on glucose homeostasis should be interpreted with caution. For one, the effective dose of melatonin is often exceedingly high (>5 mg/kg) and well above the recommended dosage used for the clinical treatment of sleep disorders (2–4 mg/day; [136]). Further, a caveat to studies in rodents is the obvious species divergence in the timing of melatonin secretion relative to food intake, peak insulin secretion and serum glucose. In both humans and rodents, melatonin levels rise during the evening, peak in the middle to latter half of the night and decline by morning [137, 138]. Of course, the dark phase is a time of energy intake in rodents, coincident with a peak in insulin sensitivity [11, 52], while in humans, the dark phase is a time of energy deficit or fasting, wherein glycogen breakdown, gluconeogenesis are elevated and insulin sensitivity is decreased [9, 139]. This temporal relationship presents a unique challenge to understanding the mechanisms whereby melatonin affects glucose metabolism. In addition to this dichotomy, we find that melatonin can have conflicting impacts on target tissues, such as the pancreatic beta cell, in both rodents and humans [49, 111, 140], and highly variable effects on systemic glucose homeostasis and insulin sensitivity [28, 29, 111, 141, 142]. In vitro, melatonin inhibits insulin secretion by lowering cAMP and cGMP production [27, 49, 143–145]. This effect is logical in humans, presuming that melatonin acts to suppress insulin during the night, effectively sensitising the beta cell in preparation for breakfast, but is more difficult to rectify in night-time eating rodents. Though useful for defining mechanisms of action at the cellular level, nocturnal rodent models may be unsuitable for the study of metabolic responses to melatonin [29]. However, study of diurnal rodent species, such as the Nile grass rat, may hold the answers to many of these questions [146]. Using these diurnal rodent models, it may be possible to more effectively examine the influence of melatonin on glucose homeostasis in a pre-clinical model with features more comparable with humans.
Clinical evidence
Melatonin levels are reduced in diabetic individuals
Though the relationship between melatonin secretion, glucose homeostasis and diabetes risk have been studied in animal models for some time, the clinical study of melatonin’s influence on these factors is less well documented. As with pre-clinical research, data from clinical studies generally support a correlation between reduced night-time melatonin and increased diabetes risk (see text box: Melatonin administration in diabetes). Studies show that normal daily rhythms of melatonin secretion are altered and/or abolished in individuals with type 2 diabetes, particularly those with autonomic neuropathy [147, 148]. Similar decrements in plasma melatonin levels during the night were also reported in diabetic patients with proliferative retinopathy relative to individuals without diabetes and diabetic individuals without retinopathy [149]. Though considerable evidence supports a positive correlation between lower night-time melatonin and increased risk of type 2 diabetes [50, 139], not all data fully support this claim. Mantele and colleagues demonstrated that night-time melatonin levels were, in fact, elevated in obese individuals (compared with lean non-diabetic individuals) and unaffected in type 2 diabetic participants [150]. However, in the same study they determined that melatonin levels were suppressed in individuals with type 2 diabetes when compared with weight-matched obese non-diabetic participants [150].

The pros and cons of melatonin therapy for glycaemic control in humans
With regard to treatment of type 2 diabetes with melatonin, the number of controlled clinical trials is somewhat limited, and the results are often mixed, with both positive and negative effects on glycaemic control being reported [27, 145, 151, 152]. Evidence from animal and human studies indicates that melatonin or synthetic melatonin receptor agonists can alter glucose homeostasis [5, 145, 153]. The study of isolated human islets from both healthy and type 2 diabetic donors has revealed that melatonin can normalise glucose-stimulated insulin secretion following exposure to hyperglycaemia [115]. Moreover, recent randomised controlled trials suggest that chronic treatment with melatonin in combination with oral blood glucose lowering agents improves fasting and postprandial glycaemic control and reduces HbA1c levels [152, 154]. Contrary to these studies, reports show that melatonin can actually impair glucose tolerance in young healthy individuals [28, 29]. These studies suggest that melatonin may be detrimental for glycaemic control and potentially advance the onset of type 2 diabetes. However, as the authors state, these studies should be interpreted with caution given that the participants were ad libitum-fed, young healthy individuals given acute melatonin treatment. Similar experiments that apply chronic melatonin treatment to obese individuals, those with impaired glucose tolerance or full type 2 diabetes are necessary to confirm and extend these observations.
Current status of melatonin/melatonin receptor agonist therapeutic use
Melatonin is available ‘over the counter’ as a non-Food and Drug Administration (FDA) regulated supplement in preparations that often contain additional components (gamma-aminobutyric acid [GABA], l-5-hydroxytryptophan [l-5-HTP], etc.), designed to enhance sleep and have a short half-life in serum [136, 155]. However, to target the melatonin signalling pathway with greater efficiency, several long-lasting high affinity MT1/MT2 receptor agonists have been developed [155–158]. These drugs, including tasimelteon (Hetlioz; [159]), ramelteon (Rozerem; [160]) and piromelatine (Neu-P11; [128]), are highly potent and selective agonists. Additionally, the compound IIK7 has been developed as a melatonin receptor agonist with 90-fold higher affinity for the MT2 receptor [161]. Like IIK7, both ramelteon and tasimelteon have a greater affinity for the MT2 receptor [162].
Clinical support for a positive (or negative) influence of these drugs on glucose homeostasis, insulin secretion and diabetes risk is lacking. While piromelatine may positively affect both insulin resistance and lipid metabolism in rodent models of type 2 diabetes [128, 162], these findings have not yet been confirmed in a controlled clinical trial. Thus, it remains to be elucidated whether melatonin or melatonin receptor agonists can be used clinically as an effective means for improving glycaemic control and normalising sleep patterns among type 2 diabetic patients, or perhaps as a preventative treatment to halt the progression of metabolic disease.
Melatonin receptor variants in humans: links with metabolic disease and risk of type 2 diabetes
The MNTR1B risk allele
Additional evidence for a clinical relationship between insulin resistance, glucose homeostasis and melatonin signalling has grown out of the search for genetic polymorphisms associated with diabetes risk. It has been reported that genetic variants of MNTR1B, which encodes the MT2 receptor, are linked to increased risk of type 2 diabetes [9] (see text box: Melatonin therapy: assess the risks!). Genome-wide association studies (GWAS) and functional cloning studies support this claim, suggesting that genetic variants of the MT2 receptor are strongly linked to impaired glucose-stimulated insulin secretion, abnormal early insulin response and increased risk of type 2 diabetes [9, 100, 106, 163, 164]. In vitro characterisation of these variants determined that many of the receptor products had diminished melatonin binding capacity [9]. In another study, the MT2 receptor variant rs10830963 (CC to CG or GG point mutation; referred to as the MNTR1B risk allele), was found in ∼30% of individuals who were at risk for type 2 diabetes. Importantly, carriers of this variant also displayed impaired glucose tolerance following an acute exposure to melatonin [28].

The adverse effects of excess melatonin signalling: the need for a personalised medicine approach
One mechanism for these melatonin-induced negative effects in individuals with the MNTR1B risk allele was initially proposed by Lyssenko et al [106] and independently confirmed by others [165]. Tuomi and colleagues determined that overexpression of MNTR1B in carriers of the risk allele leads to elevated MT2 in pancreatic beta cells [165]. This increase in MT2 levels is proposed to heighten the insulin-inhibitory effect of melatonin. On the other hand, the same study confirmed that global deletion of the MT2 receptor in mice results in hyperinsulinaemia in response to glucose administration and reduced insulin sensitivity in the liver [165].
One can conclude from these studies that excessive or inadequate melatonin signalling via the MT2 receptor can have equally negative influences on insulin secretion and glucose homeostasis. Interestingly, Lane et al determined that, in addition to a direct influence on melatonin signalling at the beta cell, carriers of the MNTR1B risk allele have a longer period of elevated melatonin secretion during the night and that diabetes risk was greater among those carriers defined as ‘early risers’ or ‘larks’ [138]. They reasoned that a longer duration of melatonin secretion and earlier wake time may lead to melatonin-suppressed insulin secretion during times of elevated glucose intake (breakfast) and that this may dampen glucose clearance, resulting in hyperglycaemia and promoting the onset of diabetes [138]. Together, these data suggest that genetic assessment may be useful for the avoidance of potentially devastating negative impacts of melatonin or MT2 agonists on glucose metabolism, particularly among individuals with type 2 diabetes suffering from sleep disturbances.
A clinical perspective on the future of chronotherapy for metabolic disease
Multiple studies have thus far demonstrated an association between circadian disturbances or misalignment with obesity, insulin resistance and type 2 diabetes. Insulin targets, such as fat, liver and muscle, function as metabolic rheostats and depend on cell-autonomous clock function. Existing first-line drug therapies for metabolic disease and type 2 diabetes, such as the commonly used insulin sensitiser, metformin, are known to modulate molecular clock function in insulin-sensitive tissues [166–168]. Sleep disturbances predispose individuals to obesity and diabetes, and obesity predisposes individuals to sleep disturbances, instigating a vicious cycle [18, 169]. So what can we do about this? We should counsel patients destined for shift work, or already in this type of work, and those exposed to frequent trans-meridian travel, as if they were at high risk for (or already have) impaired glycaemic control, especially if they are overweight. As clinicians, we should be more mindful of sleep habits in these patients and advise them on sleep hygiene.
Central to our discourse is the following question: could melatonin, a putative central clock-dependent mediator of synchrony in the timing system, be used as a treatment for glucose dysregulation and comorbid sleep disturbances? We face a paradox here since melatonin can acutely decrease insulin secretion and enhance glucagon release [48, 49, 151] but preliminary clinical data suggest that chronic melatonin treatment improves glycaemic control and reduces HbA1c in diabetic patients [152, 154], the implication being that normalisation of circadian glucose homeostasis by chronically administered melatonin could outweigh the acute effects of melatonin. Because much of the data showing a positive effect of melatonin is pre-clinical and generated using nocturnal rodents that were administered exceedingly high concentrations of melatonin at a time of heightened food intake, it is difficult to draw solid conclusions regarding its potential efficacy in a clinical setting. Enthusiasm for melatonin in the management of diabetes is dampened by reports that melatonin impairs glucose tolerance among healthy individuals and can increase the risk of developing diabetes among those carrying the MNTR1B risk allele [28, 29, 138, 165]. Thus, care should likely be taken when providing melatonin to individuals with sleep disturbances, especially those patients who are obese, at high risk for the development of diabetes or carriers of the MNTR1B risk allele, since these individuals may be at greater risk for experiencing the negative effects of the hormone.
Conclusions
Herein, we have presented the evidence for the critical input from the timing system in the aetiology of metabolic disease, and salient evidence in support of (and against) the use of the sleep aid melatonin as a pharmacotherapy in patients with type 2 diabetes. It is clear that melatonin secretion is associated with insulin resistance in diabetic individuals and that a decline in circulating melatonin could underlie both an increased risk of type 2 diabetes and comorbid sleep disturbances. However, it is unclear whether a large clinical trial exploring the use of melatonin receptor agonists as therapeutic agents for type 2 diabetes, grossly impaired glucose tolerance and perhaps even obesity is warranted. Although the use of melatonin or melatonin agonists may eventually be ruled out as viable options, other compounds with similar antioxidant and/or clock-targeting properties may yet prove valuable for the treatment of diabetes. It is unquestionably true that the time has come for more dedicated study of chronopharmacological agents, including melatonin, in the treatment of metabolic diseases.
Change history
22 February 2018
The authors have been made aware that the following sentence is incorrect: ‘Like IIK7, both ramelteon and tasimelteon have a greater affinity for the MT2 receptor [162].’
Abbreviations
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- BMAL1:
-
Brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein-1
- CLOCK:
-
Circadian locomotor output cycles kaput
- CRY:
-
Cryptochrome
- DIO:
-
Diet-induced obesity
- Gi-coupled:
-
Inhibitory G-protein coupled
- MT1:
-
Melatonin receptor type 1
- MT2:
-
Melatonin receptor type 2
- PER:
-
Period circadian protein
- PINX:
-
Pinealectomy
- RORα:
-
Retinoic acid receptor-like orphan receptor alpha
- REV-ERBα:
-
Reverse thyroid receptor α
- STZ:
-
Streptozotocin
- SCN:
-
Suprachiasmatic nucleus
References
Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87:4–14
Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF (2010) Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metrics 8:29
Lew KN, Wick A (2015) Pharmacotherapy of type 2 diabetes mellitus: navigating current and new therapies. Medsurg Nurs 24:413–419
Nauck MA, Friedrich N (2013) Do GLP-1-based therapies increase cancer risk? Diabetes Care 36(Suppl 2):S245–S252
Srinivasan V, Ohta Y, Espino J et al (2013) Metabolic syndrome, its pathophysiology and the role of melatonin. Recent Pat Endocr Metab Immune Drug Discov 7:11–25
Huang W, Ramsey KM, Marcheva B, Bass J (2011) Circadian rhythms, sleep, and metabolism. J Clin Invest 121:2133–2141
Stenvers DJ, Jonkers CF, Fliers E, Bisschop PH, Kalsbeek A (2012) Nutrition and the circadian timing system. Prog Brain Res 199:359–376
Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH (2013) Circadian disruption leads to insulin resistance and obesity. Curr Biol 23:372–381
Karamitri A, Renault N, Clement N, Guillaume JL, Jockers R (2013) Minireview: toward the establishment of a link between melatonin and glucose homeostasis: association of melatonin MT2 receptor variants with type 2 diabetes. Mol Endocrinol 27:1217–1233
Kalsbeek A, la Fleur S, Fliers E (2014) Circadian control of glucose metabolism. Mol Metab 3:372–383
Bass J, Takahashi JS (2010) Circadian integration of metabolism and energetics. Science 330:1349–1354
Carroll KF, Nestel PJ (1973) Diurnal variation in glucose tolerance and in insulin secretion in man. Diabetes 22:333–348
Cipolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ (2014) Melatonin, energy metabolism, and obesity: a review. J Pineal Res 56:371–381
Spiegel K, Leproult R, Van Cauter E (1999) Impact of sleep debt on metabolic and endocrine function. Lancet 354:1435–1439
Knutson KL, Ryden AM, Mander BA, Van Cauter E (2006) Role of sleep duration and quality in the risk and severity of type 2 diabetes mellitus. Arch Intern Med 166:1768–1774
Van Cauter E, Knutson KL (2008) Sleep and the epidemic of obesity in children and adults. Eur J Endocrinol 159(Suppl 1):S59–S66
Van Cauter E, Spiegel K, Tasali E, Leproult R (2008) Metabolic consequences of sleep and sleep loss. Sleep Med 9(Suppl 1):S23–S28
Arble DM, Bass J, Behn CD et al (2015) Impact of sleep and circadian disruption on energy balance and diabetes: a summary of workshop discussions. Sleep 38:1849–1860
Buxton OM, Cain SW, O’Connor SP et al (2012) Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med 4:129ra143
Morris CJ, Yang JN, Garcia JI et al (2015) Endogenous circadian system and circadian misalignment impact glucose tolerance via separate mechanisms in humans. Proc Natl Acad Sci U S A 112:E2225–E2234
Scheer FA, Hilton MF, Mantzoros CS, Shea SA (2009) Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A 106:4453–4458
Marcheva B, Ramsey KM, Peek CB, Affinati A, Maury E, Bass J (2013) Circadian clocks and metabolism. Handb Exp Pharmacol: 127-155
Kohsaka A, Laposky AD, Ramsey KM et al (2007) High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab 6:414–421
Karthikeyan R, Marimuthu G, Spence DW et al (2014) Should we listen to our clock to prevent type 2 diabetes mellitus? Diabetes Res Clin Pract 106:182–190
Delezie J, Challet E (2011) Interactions between metabolism and circadian clocks: reciprocal disturbances. Ann N Y Acad Sci 1243:30–46
Otway DT, Mantele S, Bretschneider S et al (2011) Rhythmic diurnal gene expression in human adipose tissue from individuals who are lean, overweight, and type 2 diabetic. Diabetes 60:1577–1581
Peschke E, Muhlbauer E (2010) New evidence for a role of melatonin in glucose regulation. Best Pract Res Clin Endocrinol Metab 24:829–841
Rubio-Sastre P, Scheer FA, Gomez-Abellan P, Madrid JA, Garaulet M (2014) Acute melatonin administration in humans impairs glucose tolerance in both the morning and evening. Sleep 37:1715–1719
Garaulet M, Gomez-Abellan P, Rubio-Sastre P, Madrid JA, Saxena R, Scheer FA (2015) Common type 2 diabetes risk variant in MTNR1B worsens the deleterious effect of melatonin on glucose tolerance in humans. Metabolism 64:1650–1657
Solt LA, Wang Y, Banerjee S et al (2012) Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485:62–68
Burris TP, Busby SA, Griffin PR (2012) Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem Biol 19:51–59
Solt LA, Banerjee S, Campbell S, Kamenecka TM, Burris TP (2015) ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes. Endocrinology 156:869–881
Menaker M, Murphy ZC, Sellix MT (2013) Central control of peripheral circadian oscillators. Curr Opin Neurobiol 23:741–746
Stephan FK, Zucker I (1972) Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA 69:1583–1586
Moore RY, Eichler VB (1972) Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res 42:201–206
Mohawk JA, Green CB, Takahashi JS (2012) Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35:445–462
Eide EJ, Virshup DM (2001) Casein kinase I: another cog in the circadian clockworks. Chronobiol Int 18:389–398
Eckel-Mahan K, Sassone-Corsi P (2013) Metabolism and the circadian clock converge. Physiol Rev 93:107–135
Yang X, Lamia KA, Evans RM (2007) Nuclear receptors, metabolism, and the circadian clock. Cold Spring Harb Symp Quant Biol 72:387–394
Orozco-Solis R, Sassone-Corsi P (2014) Epigenetic control and the circadian clock: linking metabolism to neuronal responses. Neuroscience 264:76–87
Pezuk P, Mohawk JA, Wang LA, Menaker M (2012) Glucocorticoids as entraining signals for peripheral circadian oscillators. Endocrinology 153:4775–4783
Pevet P, Bothorel B, Slotten H, Saboureau M (2002) The chronobiotic properties of melatonin. Cell Tissue Res 309:183–191
Arendt J, Skene DJ (2005) Melatonin as a chronobiotic. Sleep Med Rev 9:25–39
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452:442–447
Sasaki H, Hattori Y, Ikeda Y et al (2016) Forced rather than voluntary exercise entrains peripheral clocks via a corticosterone/noradrenaline increase in PER2::LUC mice. Sci Rep 6:27607
Pevet P, Challet E (2011) Melatonin: both master clock output and internal time-giver in the circadian clocks network. J Physiol Paris 105:170–182
Borjigin J, Zhang LS, Calinescu AA (2012) Circadian regulation of pineal gland rhythmicity. Mol Cell Endocrinol 349:13–19
Bazwinsky-Wutschke I, Bieseke L, Muhlbauer E, Peschke E (2014) Influence of melatonin receptor signalling on parameters involved in blood glucose regulation. J Pineal Res 56:82–96
Peschke E, Bahr I, Muhlbauer E (2013) Melatonin and pancreatic islets: interrelationships between melatonin, insulin and glucagon. Int J Mol Sci 14:6981–7015
McMullan CJ, Schernhammer ES, Rimm EB, Hu FB, Forman JP (2013) Melatonin secretion and the incidence of type 2 diabetes. JAMA: J Am Med Assoc 309:1388–1396
Rudic RD, McNamara P, Curtis AM et al (2004) BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2, e377
Green CB, Takahashi JS, Bass J (2008) The meter of metabolism. Cell 134:728–742
Boden G, Ruiz J, Urbain JL, Chen X (1996) Evidence for a circadian rhythm of insulin secretion. Am J Physiol 271:E246–E252
Mari A, Camastra S, Toschi E et al (2001) A model for glucose control of insulin secretion during 24 h of free living. Diabetes 50(Suppl 1):S164–S168
Polonsky KS, Given BD, Van Cauter E (1988) Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81:442–448
Coomans CP, van den Berg SA, Lucassen EA et al (2013) The suprachiasmatic nucleus controls circadian energy metabolism and hepatic insulin sensitivity. Diabetes 62:1102–1108
Coomans CP, van den Berg SA, Houben T et al (2013) Detrimental effects of constant light exposure and high-fat diet on circadian energy metabolism and insulin sensitivity. FASEB J 27:1721–1732
Turek FW, Joshu C, Kohsaka A et al (2005) Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308:1043–1045
Marcheva B, Ramsey KM, Buhr ED et al (2010) Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466:627–631
Husse J, Hintze SC, Eichele G, Lehnert H, Oster H (2012) Circadian clock genes Per1 and Per2 regulate the response of metabolism-associated transcripts to sleep disruption. PLoS One 7, e52983
Vieira E, Marroqui L, Figueroa AL et al (2013) Involvement of the clock gene Rev-erb alpha in the regulation of glucagon secretion in pancreatic alpha-cells. PLoS One 8, e69939
Rakshit K, Qian J, Colwell CS, Matveyenko AV (2015) The islet circadian clock: entrainment mechanisms, function and role in glucose homeostasis. Diabetes Obes Metab 17(Suppl 1):115–122
Rakshit K, Hsu TW, Matveyenko AV (2016) Bmal1 is required for beta cell compensatory expansion, survival and metabolic adaptation to diet-induced obesity in mice. Diabetologia 59:734–743
La Fleur SE, Fliers E, Kalsbeek A (2014) Neuroscience of glucose homeostasis. Handb Clin Neurol 126:341–351
La Fleur SE (2003) Daily rhythms in glucose metabolism: suprachiasmatic nucleus output to peripheral tissue. J Neuroendocrinol 15:315–322
Allaman-Pillet N, Roduit R, Oberson A et al (2004) Circadian regulation of islet genes involved in insulin production and secretion. Mol Cell Endocrinol 226:59–66
Feneberg R, Lemmer B (2004) Circadian rhythm of glucose uptake in cultures of skeletal muscle cells and adipocytes in Wistar-Kyoto, Wistar, Goto-Kakizaki, and spontaneously hypertensive rats. Chronobiol Int 21:521–538
Cailotto C, La Fleur SE, Van Heijningen C et al (2005) The suprachiasmatic nucleus controls the daily variation of plasma glucose via the autonomic output to the liver: are the clock genes involved? Eur J Neurosci 22:2531–2540
Radziuk J, Pye S (2006) Diurnal rhythm in endogenous glucose production is a major contributor to fasting hyperglycaemia in type 2 diabetes. Suprachiasmatic deficit or limit cycle behaviour? Diabetologia 49:1619–1628
Bruinstroop E, Fliers E, Kalsbeek A (2014) Hypothalamic control of hepatic lipid metabolism via the autonomic nervous system. Best Pract Res Clin Endocrinol Metab 28:673–684
Kalsbeek A, Bruinstroop E, Yi CX, Klieverik L, Liu J, Fliers E (2014) Hormonal control of metabolism by the hypothalamus-autonomic nervous system-liver axis. Front Horm Res 42:1–28
Lamia KA, Storch KF, Weitz CJ (2008) Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A 105:15172–15177
Zwighaft Z, Reinke H, Asher G (2016) The liver in the eyes of a chronobiologist. J Biol Rhythm 31:115–124
Zhou D, Wang Y, Chen L et al (2016) Evolving roles of circadian rhythms in liver homeostasis and pathology. Oncotarget 7:8625–8639
Zani F, Breasson L, Becattini B et al (2013) PER2 promotes glucose storage to liver glycogen during feeding and acute fasting by inducing Gys2 PTG and G L expression. Mol Metab 2:292–305
Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ (2011) An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 54:120–124
Lee J, Liu R, de Jesus D et al (2015) Circadian control of beta-cell function and stress responses. Diabetes Obes Metab 17(Suppl 1):123–133
Delattre E, Cipolla-Neto J, Boschero AC (1999) Diurnal variations in insulin secretion and K+ permeability in isolated rat islets. Clin Exp Pharmacol Physiol 26:505–510
Ebihara S, Marks T, Hudson DJ, Menaker M (1986) Genetic control of melatonin synthesis in the pineal gland of the mouse. Science 231:491–493
Morselli LL, Guyon A, Spiegel K (2012) Sleep and metabolic function. Pflugers Arch 463:139–160
Nedeltcheva AV, Scheer FA (2014) Metabolic effects of sleep disruption, links to obesity and diabetes. Curr Opin Endocrinol Diabetes Obes 21:293–298
Barone MT, Menna-Barreto L (2011) Diabetes and sleep: a complex cause-and-effect relationship. Diabetes Res Clin Pract 91:129–137
Aurora RN, Punjabi NM (2013) Obstructive sleep apnoea and type 2 diabetes mellitus: a bidirectional association. Lancet Respir Med 1:329–338
Dikeos D, Georgantopoulos G (2011) Medical comorbidity of sleep disorders. Curr Opin Psychiatry 24:346–354
Roth T (2009) Sleep and society. Sleep Med 10(Suppl 1):S1–S2
Colwell CS, Matveyenko AV (2014) Timing is everything: implications for metabolic consequences of sleep restriction. Diabetes 63:1826–1828
Skomro RP, Ludwig S, Salamon E, Kryger MH (2001) Sleep complaints and restless legs syndrome in adult type 2 diabetics. Sleep Med 2:417–422
Gottlieb DJ, Punjabi NM, Newman AB et al (2005) Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch Intern Med 165:863–867
Iglayreger HB, Peterson MD, Liu D et al (2014) Sleep duration predicts cardiometabolic risk in obese adolescents. J Pediatr 164:1085–1090
Ayas NT, White DP, Al-Delaimy WK et al (2003) A prospective study of self-reported sleep duration and incident diabetes in women. Diabetes Care 26:380–384
Kajbaf F, Fendri S, Basille-Fantinato A et al (2014) The relationship between metformin therapy and sleep quantity and quality in patients with Type 2 diabetes referred for potential sleep disorders. Diabet Med 31:577–580
Morris CJ, Aeschbach D, Scheer FA (2011) Circadian system, sleep and endocrinology. Mol Cell Endocrinol 349:91–104
Karthikeyan R, Marimuthu G, Sooriyakumar M et al (2014) Per3 length polymorphism in patients with type 2 diabetes mellitus. Horm Mol Biol Clin Investig 18:145–149
Roenneberg T, Allebrandt KV, Merrow M, Vetter C (2012) Social jetlag and obesity. Curr Biol 22:939–943
Zhdanova IV (2005) Melatonin as a hypnotic: pro. Sleep Med Rev 9:51–65
Arendt J, Hampton S, English J, Kwasowski P, Marks V (1982) 24-hour profiles of melatonin, cortisol, insulin, C-peptide and GIP following a meal and subsequent fasting. Clin Endocrinol 16:89–95
Wright J, Aldhous M, Franey C, English J, Arendt J (1986) The effects of exogenous melatonin on endocrine function in man. Clin Endocrinol 24:375–382
Arendt J, Broadway J (1987) Light and melatonin as zeitgebers in man. Chronobiol Int 4:273–282
Poon AM, Choy EH, Pang SF (2001) Modulation of blood glucose by melatonin: a direct action on melatonin receptors in mouse hepatocytes. Biol Signals Recept 10:367–379
Muhlbauer E, Gross E, Labucay K, Wolgast S, Peschke E (2009) Loss of melatonin signalling and its impact on circadian rhythms in mouse organs regulating blood glucose. Eur J Pharmacol 606:61–71
Rui BB, Chen H, Jang L et al (2016) Melatonin upregulates the activity of AMPK and attenuates lipid accumulation in alcohol-induced rats. Alcohol Alcohol 51:11–19
Zanuto R, Siqueira-Filho MA, Caperuto LC et al (2013) Melatonin improves insulin sensitivity independently of weight loss in old obese rats. J Pineal Res 55:156–165
Owino S, Contreras-Alcantara S, Baba K, Tosini G (2016) Melatonin signaling controls the daily rhythm in blood glucose levels independent of peripheral clocks. PLoS One 11, e0148214
Nogueira TC, Lellis-Santos C, Jesus DS et al (2011) Absence of melatonin induces night-time hepatic insulin resistance and increased gluconeogenesis due to stimulation of nocturnal unfolded protein response. Endocrinology 152:1253–1263
Nagorny CL, Sathanoori R, Voss U, Mulder H, Wierup N (2011) Distribution of melatonin receptors in murine pancreatic islets. J Pineal Res 50:412–417
Lyssenko V, Nagorny CL, Erdos MR et al (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet 41:82–88
Blodgett DM, Nowosielska A, Afik S et al (2015) Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64:3172–3181
Muhlbauer E, Peschke E (2007) Evidence for the expression of both the MT1- and in addition, the MT2-melatonin receptor, in the rat pancreas, islet and beta-cell. J Pineal Res 42:105–106
Peschke E (2008) Melatonin, endocrine pancreas and diabetes. J Pineal Res 44:26–40
Stumpf I, Bazwinsky I, Peschke E (2009) Modulation of the cGMP signaling pathway by melatonin in pancreatic beta-cells. J Pineal Res 46:140–147
Muhlbauer E, Albrecht E, Hofmann K, Bazwinsky-Wutschke I, Peschke E (2011) Melatonin inhibits insulin secretion in rat insulinoma beta-cells (INS-1) heterologously expressing the human melatonin receptor isoform MT2. J Pineal Res 51:361–372
Peschke E, Fauteck JD, Musshoff U, Schmidt F, Beckmann A, Peschke D (2000) Evidence for a melatonin receptor within pancreatic islets of neonate rats: functional, autoradiographic, and molecular investigations. J Pineal Res 28:156–164
Stumpf I, Muhlbauer E, Peschke E (2008) Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic beta-cells. J Pineal Res 45:318–327
Brydon L, Roka F, Petit L et al (1999) Dual signaling of human Mel1a melatonin receptors via G(i2), G(i3), and G(q/11) proteins. Mol Endocrinol 13:2025–2038
Costes S, Boss M, Thomas AP, Matveyenko AV (2015) Activation of melatonin signaling promotes beta-cell survival and function. Mol Endocrinol 29:682–692
Park JH, Shim HM, Na AY et al (2014) Melatonin prevents pancreatic beta-cell loss due to glucotoxicity: the relationship between oxidative stress and endoplasmic reticulum stress. J Pineal Res 56:143–153
Kanter M, Uysal H, Karaca T, Sagmanligil HO (2006) Depression of glucose levels and partial restoration of pancreatic beta-cell damage by melatonin in streptozotocin-induced diabetic rats. Arch Toxicol 80:362–369
Lynch HJ, Eng JP, Wurtman RJ (1973) Control of pineal indole biosynthesis by changes in sympathetic tone caused by factors other than environmental lighting. Proc Natl Acad Sci U S A 70:1704–1707
Nishiyama K, Hirai K (2014) The melatonin agonist ramelteon induces duration-dependent clock gene expression through cAMP signaling in pancreatic INS-1 beta-cells. PLoS One 9, e102073
Champney TH, Brainard GC, Richardson BA, Reiter RJ (1983) Experimentally-induced diabetes reduces nocturnal pineal melatonin content in the Syrian hamster. Comp Biochem Physiol A Comp Physiol 76:199–201
Frese T, Bach AG, Muhlbauer E et al (2009) Pineal melatonin synthesis is decreased in type 2 diabetic Goto-Kakizaki rats. Life Sci 85:526–533
Diaz B, Blazquez E (1986) Effect of pinealectomy on plasma glucose, insulin and glucagon levels in the rat. Horm Metab Res 18:225–229
Lima FB, Machado UF, Bartol I et al (1998) Pinealectomy causes glucose intolerance and decreases adipose cell responsiveness to insulin in rats. Am J Phys 275:E934–E941
Venegas C, Garcia JA, Doerrier C et al (2013) Analysis of the daily changes of melatonin receptors in the rat liver. J Pineal Res 54:313–321
Bibak B, Khalili M, Rajaei Z, Soukhtanloo M, Hadjzadeh MA, Hayatdavoudi P (2014) Effects of melatonin on biochemical factors and food and water consumption in diabetic rats. Adv Biomed Res 3:173
Hidayat M, Maha Y, Wasim H (2015) Effect of melatonin on serum glucose and body weight in streptozotocin induced diabetes in albino rats. J Ayub Med Coll Abbottabad 27:274–276
Prunet-Marcassus B, Desbazeille M, Bros A et al (2003) Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology 144:5347–5352
She M, Deng X, Guo Z et al (2009) NEU-P11, a novel melatonin agonist, inhibits weight gain and improves insulin sensitivity in high-fat/high-sucrose-fed rats. Pharmacol Res 59:248–253
Sartori C, Dessen P, Mathieu C et al (2009) Melatonin improves glucose homeostasis and endothelial vascular function in high-fat diet-fed insulin-resistant mice. Endocrinology 150:5311–5317
Wolden-Hanson T, Mitton DR, McCants RL et al (2000) Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology 141:487–497
Cuesta S, Kireev R, Garcia C, Rancan L, Vara E, Tresguerres JA (2013) Melatonin can improve insulin resistance and aging-induced pancreas alterations in senescence-accelerated prone male mice (SAMP8). Age 35:659–671
Agil A, El-Hammadi M, Jimenez-Aranda A et al (2015) Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J Pineal Res 59:70–79
Nayki U, Onk D, Balci G et al (2016) The effect of melatonin on oxidative stress and apoptosis in experimental diabetes mellitus-related ovarian injury. Gynecol Endocrinol 32:421–426
Winiarska K, Dzik JM, Labudda M et al (2016) Melatonin nephroprotective action in Zucker diabetic fatty rats involves its inhibitory effect on NADPH oxidase. J Pineal Res 60:109–117
Nishida S, Segawa T, Murai I, Nakagawa S (2002) Long-term melatonin administration reduces hyperinsulinemia and improves the altered fatty-acid compositions in type 2 diabetic rats via the restoration of Delta-5 desaturase activity. J Pineal Res 32:26–33
Gooneratne NS, Edwards AY, Zhou C, Cuellar N, Grandner MA, Barrett JS (2012) Melatonin pharmacokinetics following two different oral surge-sustained release doses in older adults. J Pineal Res 52:437–445
Lewy AJ, Sack RL, Blood ML, Bauer VK, Cutler NL, Thomas KH (1995) Melatonin marks circadian phase position and resets the endogenous circadian pacemaker in humans. Ciba Found Symp 183:303–317
Lane JM, Chang AM, Bjonnes AC et al (2016) Impact of common diabetes risk variant in MTNR1B on sleep, circadian, and melatonin physiology. Diabetes 65:1741–1751
McMullan CJ, Curhan GC, Schernhammer ES, Forman JP (2013) Association of nocturnal melatonin secretion with insulin resistance in nondiabetic young women. Am J Epidemiol 178:231–238
de Lima LM, dos Reis LC, de Lima MA (2001) Influence of the pineal gland on the physiology, morphometry and morphology of pancreatic islets in rats. Braz J Biol 61:333–340
Peschke E, Schucht H, Muhlbauer E (2010) Long-term enteral administration of melatonin reduces plasma insulin and increases expression of pineal insulin receptors in both Wistar and type 2-diabetic Goto-Kakizaki rats. J Pineal Res 49:373–381
Pai SA, Majumdar AS (2014) Protective effects of melatonin against metabolic and reproductive disturbances in polycystic ovary syndrome in rats. J Pharm Pharmacol 66:1710–1721
Bazwinsky-Wutschke I, Wolgast S, Muhlbauer E, Albrecht E, Peschke E (2012) Phosphorylation of cyclic AMP-response element-binding protein (CREB) is influenced by melatonin treatment in pancreatic rat insulinoma beta-cells (INS-1). J Pineal Res 53:344–357
Muhlbauer E, Albrecht E, Bazwinsky-Wutschke I, Peschke E (2012) Melatonin influences insulin secretion primarily via MT(1) receptors in rat insulinoma cells (INS-1) and mouse pancreatic islets. J Pineal Res 52:446–459
Sharma S, Singh H, Ahmad N, Mishra P, Tiwari A (2015) The role of melatonin in diabetes: therapeutic implications. Arch Endocrinol Metab 59:391–399
Challet E (2007) Minireview: entrainment of the suprachiasmatic clockwork in diurnal and nocturnal mammals. Endocrinology 148:5648–5655
Tutuncu NB, Batur MK, Yildirir A et al (2005) Melatonin levels decrease in type 2 diabetic patients with cardiac autonomic neuropathy. J Pineal Res 39:43–49
O’Brien IA, Lewin IG, O’Hare JP, Arendt J, Corrall RJ (1986) Abnormal circadian rhythm of melatonin in diabetic autonomic neuropathy. Clin Endocrinol 24:359–364
Hikichi T, Tateda N, Miura T (2011) Alteration of melatonin secretion in patients with type 2 diabetes and proliferative diabetic retinopathy. Clin Ophthalmol 5:655–660
Mantele S, Otway DT, Middleton B et al (2012) Daily rhythms of plasma melatonin, but not plasma leptin or leptin mRNA, vary between lean, obese and type 2 diabetic men. PLoS One 7, e37123
Peschke E, Bahr I, Muhlbauer E (2015) Experimental and clinical aspects of melatonin and clock genes in diabetes. J Pineal Res 59:1–23
Garfinkel D, Zorin M, Wainstein J, Matas Z, Laudon M, Zisapel N (2011) Efficacy and safety of prolonged-release melatonin in insomnia patients with diabetes: a randomized, double-blind, crossover study. Diabetes Metab Syndr Obes 4:307–313
Cardinali DP, Cano P, Jimenez-Ortega V, Esquifino AI (2011) Melatonin and the metabolic syndrome: physiopathologic and therapeutical implications. Neuroendocrinology 93:133–142
Hussain SA, Khadim HM, Khalaf BH, Ismail SH, Hussein KI, Sahib AS (2006) Effects of melatonin and zinc on glycemic control in type 2 diabetic patients poorly controlled with metformin. Saudi Med J 27:1483–1488
Hardeland R, Poeggeler B, Srinivasan V, Trakht I, Pandi-Perumal SR, Cardinali DP (2008) Melatonergic drugs in clinical practice. Arzneimittelforschung 58:1–10
Hardeland R (2009) New approaches in the management of insomnia: weighing the advantages of prolonged-release melatonin and synthetic melatoninergic agonists. Neuropsychiatr Dis Treat 5:341–354
Srinivasan V, Pandi-Perumal SR, Trahkt I et al (2009) Melatonin and melatonergic drugs on sleep: possible mechanisms of action. Int J Neurosci 119:821–846
Liu J, Clough SJ, Hutchinson AJ, Adamah-Biassi EB, Popovska-Gorevski M, Dubocovich ML (2016) MT1 and MT2 melatonin receptors: a therapeutic perspective. Annu Rev Pharmacol Toxicol 56:361–383
Dhillon S, Clarke M (2014) Tasimelteon: first global approval. Drugs 74:505–511
Miyamoto M (2009) Pharmacology of ramelteon, a selective MT1/MT2 receptor agonist: a novel therapeutic drug for sleep disorders. CNS Neurosci Ther 15:32–51
Fisher SP, Sugden D (2009) Sleep-promoting action of IIK7, a selective MT2 melatonin receptor agonist in the rat. Neurosci Lett 457:93–96
Laudon M, Frydman-Marom A (2014) Therapeutic effects of melatonin receptor agonists on sleep and comorbid disorders. Int J Mol Sci 15:15924–15950
Bonnefond A, Clement N, Fawcett K et al (2012) Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet 44:297–301
Lyssenko V, Groop L (2009) Genome-wide association study for type 2 diabetes: clinical applications. Curr Opin Lipidol 20:87–91
Tuomi T, Nagorny CL, Singh P et al (2016) Increased melatonin signaling is a risk factor for type 2 diabetes. Cell Metab 23:1067–1077
Um JH, Yang S, Yamazaki S et al (2007) Activation of 5′-AMP-activated kinase with diabetes drug metformin induces casein kinase Iepsilon (CKIepsilon)-dependent degradation of clock protein mPer2. J Biol Chem 282:20794–20798
Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC (2011) Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes Metab 13:1097–1104
Barnea M, Haviv L, Gutman R, Chapnik N, Madar Z, Froy O (2012) Metformin affects the circadian clock and metabolic rhythms in a tissue-specific manner. Biochim Biophys Acta 1822:1796–1806
Dashti HS, Scheer FA, Jacques PF, Lamon-Fava S, Ordovas JM (2015) Short sleep duration and dietary intake: epidemiologic evidence, mechanisms, and health implications. Adv Nutr 6:648–659
Acknowledgements
The authors gratefully acknowledge the editorial assistance of A. L. Mereness and S. Grewal (Division of Endocrinology, Diabetes and Metabolism, School of Medicine and Dentistry, University of Rochester School of Medicine, Rochester, NY, USA).
Author contributions
MTS and SDW conceived the idea for the manuscript. ACF, SUM, MY, SDW and MTS wrote the manuscript. ACF and MTS prepared the figures. All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Duality of interests
SDW receives lecture and other fees from Sanofi-Aventis. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Additional information
A correction to this article is available online at https://doi.org/10.1007/s00125-018-4577-3.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 566 kb)
Rights and permissions
About this article

Cite this article
Forrestel, A.C., Miedlich, S.U., Yurcheshen, M. et al. Chronomedicine and type 2 diabetes: shining some light on melatonin. Diabetologia 60, 808–822 (2017). https://doi.org/10.1007/s00125-016-4175-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4175-1