
Abstract
Aims/hypothesis
Lysosomal acid lipase (LAL) hydrolyses cholesteryl esters and triacylglycerols (TG) within lysosomes to mobilise NEFA and cholesterol. Since LAL-deficient (Lal -/- ) mice suffer from progressive loss of adipose tissue and severe accumulation of lipids in hepatic lysosomes, we hypothesised that LAL deficiency triggers alternative energy pathway(s).
Methods
We studied metabolic adaptations in Lal -/- mice.
Results
Despite loss of adipose tissue, Lal -/- mice show enhanced glucose clearance during insulin and glucose tolerance tests and have increased uptake of [3H]2-deoxy-D-glucose into skeletal muscle compared with wild-type mice. In agreement, fasted Lal -/- mice exhibit reduced glucose and glycogen levels in skeletal muscle. We observed 84% decreased plasma leptin levels and significantly reduced hepatic ATP, glucose, glycogen and glutamine concentrations in fed Lal -/- mice. Markedly reduced hepatic acyl-CoA concentrations decrease the expression of peroxisome proliferator-activated receptor α (PPARα) target genes. However, treatment of Lal -/- mice with the PPARα agonist fenofibrate further decreased plasma TG (and hepatic glucose and glycogen) concentrations in Lal -/- mice. Depletion of hepatic nuclear factor 4α and forkhead box protein a2 in fasted Lal -/- mice might be responsible for reduced expression of microsomal TG transfer protein, defective VLDL synthesis and drastically reduced plasma TG levels.
Conclusions/interpretation
Our findings indicate that neither activation nor inactivation of PPARα per se but rather the availability of hepatic acyl-CoA concentrations regulates VLDL synthesis and subsequent metabolic adaptations in Lal -/- mice. We conclude that decreased plasma VLDL production enhances glucose uptake into skeletal muscle to compensate for the lack of energy supply.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lysosomal acid lipase (LAL) hydrolyses cholesteryl esters (CE) and triacylglycerols (TG), delivered to the lysosome mainly via LDL particle uptake [1], to release mono- and diacylglycerols and mobilise cholesterol and NEFA for membrane assembly, steroidogenesis and energy production. Genetic mutations of human LAL (also known as LIPA) cause an autosomal recessive lysosomal storage disorder with accumulation of CE predominantly in hepatocytes, adrenal glands, intestine and cells of the monocyte-macrophage system throughout the body [2]. Mutations in LAL cause Wolman disease (WD) or cholesteryl ester storage disease (CESD) [3–5]. WD is a rare, neonatal-onset disease with less than 1% of LAL activity, characterised by massive hepatosplenomegaly, adrenal calcifications, malabsorption, growth retardation and cachexia. Affected patients die within the first three to 12 months of life [2, 6, 7]. CESD patients have up to 5% residual LAL activity [8], which keeps the syndrome mostly unrecognised until adulthood. These patients suffer from progressive lysosomal CE and TG accumulations, which lead to the characteristic liver pathology and elevated concentrations of serum transaminases, serum LDL and TG. Premature death of individuals with CESD is due to liver failure and/or accelerated atherosclerosis rather than to chronic hyperlipidaemia [2, 9, 10].
In contrast to humans, complete loss of LAL activity in mice phenotypically resembles CESD rather than WD. LAL-deficient (Lal -/- ) mice appear normal at birth and reach the median life span of approximately 1 year [11]. Severe accumulations of CE and TG are found predominantly in the liver, spleen, small intestine and adrenals [11–13]. Lal -/- mice exhibit reduced size and body weight (BW) compared with wild-type (WT) littermates as well as progressive loss of white adipose tissue (WAT) and brown adipose tissue [12, 13]. In this study, we explored the metabolic changes caused by impaired lysosomal TG and CE hydrolysis in Lal -/- mice.
Methods
Animals
Age- and sex-matched Lal -/- and WT littermates [13] on a C57BL/6J background were maintained with unlimited access to chow and water in regular 12 h light/12 h dark cycles. For fenofibrate treatment, 5-week-old Lal -/- mice were administered 0.2% fenofibrate/chow for 4 weeks. Experimenters were blind to group assignment and outcome assessment. Animal experiments were approved by the Federal Ministry of Science, Research and Economy, Vienna, Austria. See electronic supplementary material (ESM) Methods for further details.
Lipid and hormone concentrations and fast protein liquid chromatography
Cholesterol, TG, glycerol and markers of liver injury were quantified enzymatically. Lipoprotein profiles were analysed by fast protein liquid chromatography (FPLC) [14]. Hormones were determined by ELISA. See ESM Methods for further details.
ATP analysis
Polar metabolites from mouse liver were extracted as described previously [15]. HPLC was performed on a 1100 Agilent capillary LC (Agilent Technologies, Santa Clara, CA, USA) equipped with a polyhydroxyethyl column (PolyLC Inc, Columbia, MD, USA). Solvent A (10 mmol/l ammonium acetate/water) and solvent B (10 mmol/l ammonium acetate/90% acetonitrile) were used in varying gradients. Selected ions/fragments were detected by TSQ Quantum Ultra Mass Spectrometry (Thermo Fisher Scientific, Waltham, MA, USA) in negative mode. See ESM Methods for further details.
De novo lipid synthesis
Animals were i.p. injected with [14C]acetate (5 μCi in 200 μl PBS), killed 1 h post-injection, and livers were isolated and lyophilised for 48 h. Lipid extracts were separated by thin-layer chromatography (TLC) (n-hexane:diethylether:acetic acid; 80:20:2, vol.:vol.:vol.). Radioactivity in bands corresponding to NEFA, TG, non-esterified cholesterol and CE was determined by liquid scintillation counting.
Liver acyl-CoAs
Acyl-CoAs were extracted from liver lysate homogenates using 0.5 ml of buffer (50% 0.1 mol/l KH2PO4, 50% 2-propanol; 4°C), 30 μl saturated (NH4)2SO4 and 0.5 ml acetonitrile, and centrifuged (2,500 g, 10 min, 4°C). Acyl-CoAs were determined by liquid chromatography-mass spectrometry as described previously [16]. See ESM Methods for further details.
Neutral TG hydrolase activity
Neutral TG hydrolase activity was measured as described previously [17], with minor modifications as described in ESM Methods.
Mitochondria isolation and respirometry
Liver mitochondria were isolated as described previously [18] and resuspended in medium. Mitochondria were diluted in medium plus 0.2% NEFA-free BSA, 5 mmol/l glutamate, 1 mmol/l malate, or 5 mmol/l pyruvate, 5 mmol/l succinate, 1 μmol/l rotenone. Oxygen consumption rates were measured at 37°C (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria). State 3-respiration (mitochondrial respiration due to ADP supply) and state 4o-respiration (after ATP synthase inhibition by oligomycin) were induced by 2 mmol/l ADP and 1 μg/ml oligomycin. Maximal uncoupled respiration was determined after titration of carbonyl cyanide p-trifluoromethoxyphenylhydrazone. See ESM Methods for further details.
RNA isolation and quantitative real-time PCR analysis
RNA isolation and quantitative real-time PCR were performed as described previously [19]. See ESM Methods for primer sequences and further details.
VLDL secretion
Mice (8 h-fasted) were i.p. injected with 500 mg/kg BW tyloxapol in PBS. Plasma TG were determined every hour post-injection.
Tolerance tests
Mice were i.p. injected with glucose (2 g/kg BW), insulin (0.25 U/kg BW), glucagon (140 μg/kg BW), glycerol (2 g/kg BW), pyruvate (2 g/kg BW) and glutamine (2 g/kg BW). Blood glucose levels were determined using Accu-Chek Active glucometer (Roche Diagnostics, Mannheim, Germany). See ESM Methods for further details.
[3H]2-deoxy-D-glucose uptake
Mice (6 h-fasted) were i.p. injected with 30 mg glucose and 0.5 μCi glucose per 30 g BW. Radioactivity in 40 μl plasma (15, 30 and 60 min post-injection) and tissue lysates was determined by β-counting. See ESM Methods for further details.
Quantification of metabolites by nuclear magnetic resonance spectroscopy
Liver and skeletal muscle lysates (200 μl) were mixed with methanol (400 μl), incubated at −20°C (30 min) and centrifuged. Supernatants were dried and re-dissolved in 500 μl D2O. 1H-one-dimensional-nuclear magnetic resonance experiments were performed at 310 K. Reference chemical shifts were taken from the Madison-Qingdao Metabolomics Consortium Database (http://mmcd.nmrfam.wisc.edu/) [20]. Bruker Topspin3.1 (Rheinstetten, Germany) and MestReNova10.0 software (http://mestrelab.com) were used for data acquisition, processing and analyses. See ESM Methods for further details.
In vivo MRI for body fat
MR images of anesthetised mice were acquired by 3T-MRI (Siemens Tim-Trio, Erlangen, Germany) with an eight-channel multipurpose coil (Noras MRI products, Hoechenberg, Germany).
Western blotting
Protein samples were separated by SDS-PAGE and transferred to polyvinylidene-difluoride membranes. Blots were incubated with primary antibodies followed by HRP-conjugated secondary antibodies. See ESM Methods for further details.
Subcutaneous WAT sections
Tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin. Tissue sections (4 μm) were stained with haematoxylin and eosin (H&E) and evaluated by light microscopy.
Electron microscopy
Liver sections were fixed in phosphate buffer/2.5% glutaraldehyde, washed, post-fixed in phosphate buffer/OsO4 for 1 h and 4 × 10 min in phosphate buffer. After dehydration, tissues were infiltrated (acetone and agar 100 epoxy resin, pure agar 100 epoxy resin) for 4 h, placed in agar 100 epoxy resin (8 h), transferred into embedding moulds, and allowed to polymerise (48 h, 60°C). Sections stained with lead citrate and uranyl acetate were imaged. See ESM Methods for further details.
Statistics
Since Lal -/- mice were obviously much smaller than WT mice, in vivo experiments were not blind. In vitro experiments were blind to group assignment and outcome assessment. Statistical analyses were performed using GraphPad Prism 5.04 (GraphPad Software, San Diego, CA, USA). Significant outliers were detected by Grubb’s test (http://graphpad.com/quickcalcs/Grubbs1.cfm). Significances were determined by Student’s unpaired t test and Welch correction (in case of unequal variances) for two group comparison and ANOVA followed by Bonferroni correction for multiple group comparison. Data are presented as means ± SD. Differences were considered statistically significant at p < 0.05.
Results
Decreased acyl-CoA concentrations and peroxisome proliferator-activated receptor α signalling in Lal -/- liver
We observed severe accumulation of total cholesterol (TC) and TG in livers of Lal -/- mice (Fig. 1a, b). Livers of WT mice store lipids within cytosolic lipid droplets, whereas in Lal -/- mice lipids were entrapped within lysosomes distinguished by bilayer membranes (Fig. 1a). In addition, a substantial number of cholesterol crystals were visible. TG and CE could not be hydrolysed and remained entrapped within the lysosomes (Fig. 1b). Reduced protein expression of perilipin 2 (PLIN2) and reduced neutral TG hydrolase activity indicated a reduced number of cytoplasmic lipid droplets in Lal -/- livers (Fig. 1c). In accordance, acyl-CoA concentrations were reduced in livers of Lal -/- mice, with markedly decreased acetyl-, palmitoleoyl-, oleoyl- and linoleoyl-CoA levels (Fig. 1d). Consistently, we observed decreased mRNA expression of fatty acid binding protein 1 (Fabp1) (Fig. 1e). Increased mRNA expression of cluster of differentiation 36 (Cd36) but unaltered expression of LDL receptor (Ldlr) suggested increased NEFA and unchanged LDL uptake, respectively (Fig. 1e). Moreover, de novo cholesterol synthesis was induced in Lal -/- livers as indicated by increased expression of hydroxymethylglutaryl-CoA reductase (Hmgcr) (Fig. 1e) and a moderate increase in hepatic cholesterol synthesis after i.p. injection of [14C]acetate (Fig. 1f). By contrast, de novo production of TG, CE and NEFA was significantly reduced in Lal -/- livers (Fig. 1f). mRNA levels of selected peroxisome proliferator-activated receptor α (PPARα) target genes were markedly decreased in Lal -/- compared with WT livers (Fig. 1g), which is in line with decreased acyl-CoA concentrations.

Reduced concentrations of liver acyl-CoAs in Lal -/- mice. (a) Electron micrographs of WT and Lal -/- liver. White arrows, lipid droplets; white arrowheads, fatty lysosomes; scale bar, 1 μm. (b) Hepatic TG and TC concentrations (n = 3). (c) Hepatic expression of PLIN2 with GAPDH as loading control, and hepatic neutral TG hydrolase activity. (d) Liver acyl-CoA concentrations (n = 4–5 mice aged 7–9 weeks). (e) Hepatic mRNA expression of fatty acid binding protein 1 (Fabp1), cluster of differentiation 36 (Cd36), LDL receptor (Ldlr) and 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr) (n = 5–6). (f) Hepatic incorporation of i.p. injected [14C]acetate into lipid classes after TLC separation (n = 5 mice aged 17–20 weeks); FC, non-esterified cholesterol. (g) mRNA expression of acyl-CoA thioesterase1 (Acot1), cytochrome P450, family4, subfamily a, polypeptide31 (Cyp4a31), acyl-CoA oxidase1 (Acox1), very long-chain (Vlcad) and medium chain acyl-CoA dehydrogenase (Mcad; also known as Acadm) (n = 4–5). Data represent means ± SD; *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, Student’s unpaired t test. If not stated otherwise, mice were 12–16 weeks of age. Black bars, WT mice; white bars, Lal -/- mice
Intact mitochondrial function despite decreased ATP concentrations in Lal -/- livers
Since insufficient supply of NEFA and, consequently, acyl-CoA may affect mitochondrial function and energy production, we next investigated the hepatic mitochondria of Lal -/- mice. Unaltered mitochondrial morphology (Fig. 2a) and a comparable amount of mitochondrial DNA (ESM Fig. 1a) indicated functional mitochondria. The respiratory capacity of mitochondria isolated from Lal -/- livers was decreased only for the uncoupled state when succinate was provided as substrate (Fig. 2b) but unaltered with glutamate in the presence of malate (ESM Fig. 1b) or pyruvate (ESM Fig. 1c). These data suggest that the lack of substrate (acyl-CoA) might be responsible for reduced ATP concentrations in Lal -/- livers (Fig. 2c).

Intact mitochondria and reduced ATP concentrations in Lal -/- livers. (a) Electron micrographs of mitochondria in livers of WT and Lal -/- mice aged 12 weeks; scale bar, 0.5 μm. (b) Mitochondrial respiration of mitochondria isolated from WT and Lal -/- livers (n = 4–6) using 10 mmol/l succinate/1 μmol/l rotenone as substrates. Oxygen consumption rate presented as basal mitochondrial respiration in the presence of substrates (basal), increased mitochondrial respiration due to ADP supply (state 3), after ATP synthase inhibition by oligomycin (state 4o) and after carbonyl cyanide p-trifluoromethoxyphenylhydrazone addition (uncoupled). (c) Liver ATP concentrations determined by liquid chromatography-tandem mass spectrometry (n = 5–6 mice aged 8–12 weeks). Data represent means ± SD; *p < 0.05, **p ≤ 0.01, Student’s unpaired t test. Black bars, WT mice; white bars, Lal -/- mice. OCR, oxygen consumption rate
Diminished VLDL secretion in Lal -/- mice
Since Lal -/- mice suffer from progressive loss of adipose tissue, we expected lipid metabolism during fasting to be severely affected. Interestingly, fasting failed to increase circulating TG concentrations in Lal -/- mice (Fig. 3a), whereas TC was increased in Lal -/- mice regardless of the feeding state. Plasma lipoprotein profiles of fasted Lal -/- mice revealed increased levels of LDL-cholesterol and decreased levels of HDL-cholesterol (Fig. 3b). However, in contrast to WT mice, Lal -/- mice failed to induce VLDL-TG during fasting, with redistribution of TG toward the LDL fraction (Fig. 3c). Biochemical estimation confirmed the FPLC data, showing no difference after 4 h and reduced plasma TG concentrations after 12 h of fasting (Fig. 3d).

Abolished nuclear expression of HNF4α and FOXA2 and decreased VLDL synthesis in Lal -/- mice. (a) Plasma TG and TC concentrations in 12 h fasted (n = 5) and fed mice (n = 7–9). Distribution of (b) TC and (c) TG within plasma lipoproteins (n = 6); continuous lines, WT; dotted lines, Lal -/-; red lines, 4 h fasting; black lines, 12 h fasting. (d) Plasma TG concentrations of 4 h and 12 h fasted mice (n = 5). (e) VLDL secretion of 8 h fasted mice (n = 4–7) after tyloxapol injection. (f) Hepatic mRNA expression of microsomal TG transfer protein (Mttp), hepatic nuclear factor 4α (Hnf4a) and forkhead box protein a2 (Foxa2) (n = 5–6). (g) Immunoblotting against liver HNF4a and FOXA2 (both in fasted state) in cytosolic (C) and nuclear (N) fractions with GAPDH and LaminA/C as loading controls. Data represent means ± SD; *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001. (a, d, f) Student’s unpaired t test, (e) ANOVA. Mice were aged 12–16 weeks. Black bars and squares, WT mice; white bars and squares, Lal -/- mice
We next investigated VLDL secretion after inhibition of peripheral lipolysis by tyloxapol and found strongly reduced VLDL release in fasted Lal -/- mice (Fig. 3e). As expected, Lal -/- mice showed markedly increased concentrations of aminotransferases (ESM Fig. 2), an indication of liver damage. Hepatic mRNA expression of microsomal TG transfer protein (Mttp), a key player in VLDL assembly, was decreased (Fig. 3f). Drastically reduced mRNA and protein expression of the hepatic transcription factors hepatocyte nuclear factor 4a (Hnf4a) and forkhead box protein a2 (Foxa2) (Fig. 3f, g), which are involved in VLDL synthesis [21, 22], reflect a causal link between low expression levels of these transcription factors and decreased VLDL secretion in Lal -/- livers.
Increased insulin sensitivity in Lal -/- mice
Severe impairment of lipid metabolism in Lal -/- mice and reduced acyl-CoA and TG availability prompted us to investigate metabolic adaptations to defective lipid metabolism. Intraperitoneal glucose tolerance tests and insulin tolerance tests (ITT) revealed significantly improved glucose tolerance (Fig. 4a) and insulin sensitivity (Fig. 4b) in Lal -/- mice. Plasma glucose levels were reduced (Fig. 4c), whereas insulin levels were comparable between Lal -/- and WT mice (Fig. 4d). These data indicate increased usage of glucose in Lal -/- mice. Accordingly, the uptake of i.p. injected [3H]2-deoxy-D-glucose into skeletal muscles was increased by 1.9-fold in Lal -/- mice (Fig. 4e). [3H]2-deoxy-D-glucose uptake by Lal -/- livers (Fig. 4e) as well as radioactivity in plasma (Fig. 4f) were comparable between both genotypes. mRNA expression of glycolysis genes in skeletal muscles were comparable in fed (Fig. 4g) and reduced in fasted Lal -/- mice (Fig. 4h). In agreement, glucose and glycogen concentrations were decreased in fasted Lal -/- mice (Fig. 4i), whereas muscle lactate was reduced in both fed and fasted states.

Improved glucose tolerance and insulin sensitivity in Lal -/- mice. Plasma glucose concentrations in (a) 6 h fasted and (b) 4 h fasted mice (n = 5) after i.p. injection of (a) glucose (2 g/kg) and (b) insulin (0.25 U/kg). Concentrations of (c) plasma glucose in 6 h fasted (n = 14) and fed (n = 7) mice and (d) insulin in 4 h fasted and fed mice (n = 4). (e) Radioactivity in skeletal muscle, liver and (f) plasma of mice (n = 5) after i.p. injection of [3H]2-deoxy-d-glucose. mRNA expression of glycogen synthase1 (Gys1), glycogen phosphorylase (Pygm), hexokinase 1 (Hk1), phosphofructokinase (Pfkm), fructose-biphosphatase2 (Fbp2) and pyruvate kinase (Pkm) in skeletal muscles of (g) fed and (h) 12 h fasted mice (n = 4–5). (i) Skeletal muscle metabolites of fed and 12 h fasted mice (n = 5). Data represent means ± SD; *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001. (a, b, f) ANOVA, (c–e, g–i) Student’s unpaired t test. Mice were aged 12–16 weeks. Black bars and squares, WT mice; white bars and squares, Lal -/- mice
Decreased plasma leptin concentrations and altered expression of hepatic adiponectin and leptin receptors in Lal -/- mice
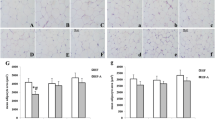
Despite comparable food intake, Lal -/- mice were lighter and gained less weight (ESM Fig. 3a, b). They lacked visceral fat and exhibited markedly reduced subcutaneous (sc) WAT at the age of 4 and 12 weeks (ESM Fig. 3c). H&E staining of scWAT sections revealed smaller adipocytes with reduced fat content (Fig. 5a) and we observed reduced inguinal scWAT mass (Fig. 5b) in Lal -/- mice. Adipose tissue fundamentally influences systemic insulin sensitivity and glucose metabolism via the adipokines leptin and adiponectin [23]. Leptin mRNA expression was significantly decreased in scWAT of Lal -/- compared with WT mice, whereas adiponectin mRNA expression was unchanged (Fig. 5c). Since fasting significantly decreases basal leptin levels [24], decreased plasma leptin concentrations (84%) in fed Lal -/- mice but comparable levels as WT mice after fasting (Fig. 5d) indicate that feeding is ineffective to increase the satiety hormone leptin in Lal -/- mice. Plasma adiponectin levels were comparable between both genotypes (Fig. 5e). mRNA expression of the leptin receptors (Leptr)1 and 2 were markedly increased in livers of fed (Fig. 5f) and fasted (Fig. 5g) Lal -/- mice. Expression of adiponectin receptor (Adipor)1 mRNA was higher and expression of Adipor2 mRNA was lower in livers of fed Lal -/- mice than WT fed mice (Fig. 5f). While Adipor1 mRNA levels were similar in fasted livers of both genotypes, transcript levels of Adipor2 were reduced in livers of Lal -/- mice compared with WT livers (Fig. 5g).

Reduced scWAT and plasma leptin concentrations in Lal -/- mice. (a) H&E staining of inguinal scWAT sections. (b) scWAT mass normalised to BW (n = 6). (c) mRNA expression of leptin (Lep) and adiponectin (Adipoq) in scWAT (n = 4–5). Plasma concentrations of (d) leptin and (e) adiponectin (n = 6). mRNA expression of the receptors of hepatic leptin (Leptr) and adiponectin (Adipor) in (f) fed and (g) 12 h fasted WT and Lal -/- mice (n = 3–5). Data represent means ± SD; *p < 0.05, **p ≤ 0.01; ***p ≤ 0.001, Student’s unpaired t test. Mice were aged 12–16 weeks. Black bars, WT mice; white bars, Lal -/- mice
Reduced liver glucose, glycogen and glutamine concentrations in Lal -/- mice
Significantly reduced plasma glycerol concentrations in fasted Lal -/- mice indicate diminished peripheral lipolysis (Fig. 6a). Reduced liver glycogen concentrations in fed Lal -/- mice (Fig. 6b) were confirmed by decreased mobilisation of glucose from glycogen after i.p. injection of glucagon (Fig. 6c). Reduced liver storage of glucose is a result of ineffective production or extensive usage. We therefore determined the ability of Lal -/- mice to produce glucose from different carbon sources. After i.p. injection of glycerol, de novo synthesised glucose reached similar maximal values after 30 min in WT and after 15 min in Lal -/- mice (Fig. 6d). Thus, glucose was cleared faster from the circulation in Lal -/- mice as shown by significantly decreased levels after 60 min, implying increased systemic glucose usage. Gluconeogenesis as measured by pyruvate tolerance test was unaltered (Fig. 6e), but drastically decreased in Lal -/- mice after i.p. injection of glutamine (Fig. 6f). In line, hepatic glucose and glutamine concentrations were markedly reduced in livers of fed Lal -/- mice (Fig. 6g), whereas lactate and pyruvate levels were comparable to those in WT mice. Decreased hepatic glucose content may be a reason why (with the exception of liver-specific phosphofructokinase [Pfkl]) the mRNA expression levels of all other liver enzymes involved in glycolysis were reduced in Lal -/- livers (Fig. 6h).

Reduced glycogen, glucose and glutamine concentrations in Lal -/- livers. (a) Plasma glycerol in fasted (n = 5) and (b) liver glycogen concentrations in fed mice (n = 8). (c–f) Glucose concentrations after i.p. injection of (c) glucagon (140 μg/kg BW), (d) glycerol (2 g/kg BW), (e) pyruvate (2 g/kg BW) and (f) glutamine (2 g/kg BW) in plasma of (c) fed and (d–f) fasted mice (n = 5–7). (g) Liver metabolites in fed mice (n = 6). (h) mRNA expression of glucokinase (Gck), phosphofructokinase (Pfkl), fructose-biphosphatase 1 (Fbp1), aldolase B (Aldob), phosphoglycerate kinase 1 (Pgk1), pyruvate kinase 1 (Pk1), glucose-6-phosphate dehydrogenase (G6pd; also known as G6pdx), glycogen phosphorylase (Pygl), glycerol-3-phosphate dehydrogenase1 (Gpd1) and pyruvate dehydrogenase (Pdhb) (n = 6–9). Data represent means ± SD; *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001. (a, b, g, h) Student’s unpaired t test, (c–f) ANOVA. Mice were aged 12–18 weeks. Black bars and squares, WT mice; white bars and squares, Lal -/- mice
PPARα activation reduces liver glucose and glycogen as well as plasma TG concentrations in Lal -/- mice
To investigate whether alterations in glucose homeostasis and VLDL synthesis in Lal -/- mice are mediated by diminished PPARα activation, Lal -/- mice were fed fenofibrate for 4 weeks. We observed decreased hepatic glucose and completely depleted glycogen content in fenofibrate-treated Lal -/- mice (Fig. 7a). Lactate concentrations in the liver were unaffected, whereas plasma lactate levels were reduced (Fig. 7b). Moreover, fenofibrate treatment reduced liver TC and plasma TG concentrations (Fig. 7c, d). We further found induced expression of the PPARα targets Cyp4a31 and Vlcad (also known as Acadvl), reduced Adipor1 transcript level and increased expression of Leptr1 (Fig. 7e). Importantly, relative mRNA expression levels of Mttp, Hnf4a and Foxa2 remained comparable between untreated and fenofibrate-treated Lal -/- mice. We conclude that defective VLDL synthesis in Lal -/- mice is a consequence of diminished hepatic HNF4α/FOXA2 signalling rather than PPARα inactivation.

Reduced plasma TG and liver glucose and glycogen in fenofibrate-treated Lal -/- mice. Five-week-old Lal -/- mice were administered fenofibrate (0.2%) for 4 weeks. (a) Hepatic lactate, glucose and glycogen, and (b) plasma lactate and glucose concentrations (n = 5). TG and TC concentrations in (c) liver and (d) plasma (n = 4). (e) mRNA expression of cytochrome P450, family4, subfamily a, polypeptide 31 (Cyp4a31), very long-chain chain acyl-CoA dehydrogenase (Vlcad), adiponectin receptors (Adipor1 and 2), leptin receptor 1 (Leptr1), hepatic nuclear factor 4α (Hnf4a), forkhead box protein a2 (Foxa2) and microsomal TG transfer protein (Mttp) (n = 3–4). Data represent means ± SD; *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001. Student’s unpaired t test. Black bars, Lal -/- mice; white bars, fenofibrate-treated Lal -/- mice
Discussion
The appearance of hepatic ‘fatty lysosomes’ in insulin-sensitive Lal -/- mice contrasts with hepatosteatosis, in which accumulation of cytoplasmic lipid droplets is accompanied by insulin resistance [25–27]. Liver is the main organ for VLDL assembly and secretion and thus influences whole body TG and cholesterol homeostasis [28]. Skop et al have already suggested the involvement of LAL in VLDL synthesis [29]. The authors linked the process to autophagy, since inhibition of lysosomal activity by chloroquine decreased VLDL secretion in vitro. Our in vivo data, however, indicate that defective VLDL synthesis in Lal -/- mice is a consequence of decreased hepatic availability of acyl-CoAs, which then leads to downregulation of PPARα signalling, nuclear exclusion of HNFα and FOXA2, decreased Mttp expression, reduced TG synthesis, and eventually futile lipidation of ApoB [21, 22, 30, 31]. In general, PPARα activation mediates lipid oxidation and reduces ectopic lipid storage, thereby counteracting insulin resistance [32]. Interestingly, Lal -/- mice show reduced expression of PPARα target genes, yet produce less VLDL, which contrasts with the increased secretion of VLDL observed in Ppara -/- mice [33]. However, fenofibrate treatment did not normalise the phenotype but further decreased plasma TG (and hepatic glucose and glycogen) concentrations in Lal -/- mice. These findings indicate that neither activation nor inactivation of PPARα per se but rather the availability of hepatic acyl-CoAs regulates VLDL synthesis and subsequent metabolic adaptations.
HNF4α, a key regulator of various metabolic pathways, is classified as an orphan receptor despite NEFA being suggested as its endogenous ligand [34]. In fact, Yuan et al demonstrated that HNF4α is selectively associated with linoleic acid in mammalian cells and in the liver of fed mice [35], indicating that linoleic acid is at least one possible endogenous ligand for HNF4α. Nuclear exclusion of HNF4α together with a 60% reduction in 18:2-CoA in livers of Lal -/- mice suggest that lysosomal mobilisation of linoleic acid is involved in VLDL synthesis via the HNF4α pathway. Accordingly, protein expression of FOXA2, which is controlled by insulin signalling [36] and promotes VLDL synthesis [21], was significantly reduced in Lal -/- liver. Besides reduced VLDL secretion, Lal -/- mice had increased insulin sensitivity compared with their WT littermates. This finding is in accordance with studies in animal models [37, 38] and type 2 diabetes patients [39], indicating a connection between insulin resistance and VLDL overproduction. We thus propose that reduced plasma VLDL is one of the features to induce a shift from NEFA to glucose utilisation, resulting in improved insulin sensitivity as observed by enhanced uptake of glucose in skeletal muscles of these mice. Although non-significant, a 35% increase of skeletal muscle glycogen in fed Lal -/- mice indicates enhanced storage of glucose for energy supply during fasting.
WAT is a primary contributor to metabolic regulation during feeding and fasting. Although lipodystrophy is generally associated with insulin resistance [40], Lal -/- mice show reduced plasma glucose and enhanced glucose usage. Despite substantial loss of WAT mass in Lal -/- mice, plasma concentrations of adiponectin were comparable, whereas leptin levels were profoundly decreased after feeding. These findings indicate that Lal -/- mice have a constant energy demand due to the unavailability of NEFA from WAT cytosolic lipid droplets. Diminished VLDL synthesis induces depletion of liver energy storage pools as reflected by reduced liver glycogen, glucose and glutamine concentrations. Decreased liver glucose levels may be causative for lower hepatic mRNA expression of genes involved in glycolysis, particularly of Gck and Pk1 (also known as Pklr). The liver provides glucose through gluconeogenesis from non-carbohydrate precursors during prolonged fasting [41]. Lal -/- mice, however, are unable to produce glucose from external glutamine, suggesting that the degradation of glutamine to pyruvate is defective. However, gluconeogenesis per se is functional, as shown by unaltered pyruvate tolerance tests. Glycerol tolerance test revealed earlier maximal glucose levels and glucose was cleared faster from the circulation, confirming enhanced glucose utilisation in Lal -/- mice.
We conclude that defective lysosomal hydrolysis of CE and TG decreases hepatic acyl-CoA and ATP concentrations. Consequently, abolished nuclear expression of HNF4α and FOXA2 lead to reduced Mttp expression and attenuated VLDL secretion, which in turn induces insulin sensitivity (Fig. 8). During the past 30–40 years, our understanding of proteins and lipids that influence VLDL assembly and secretion has changed tremendously [42]. We argue that LAL should be included as another critical player, which regulates VLDL and glucose metabolism.

Decreased hepatic acyl-CoA availability reduces VLDL synthesis and triggers insulin sensitivity in Lal -/- mice. Inhibited hepatic lysosomal hydrolysis of CE and TG due to LAL deficiency leads to reduced abundance of cellular acyl-CoA and ATP. Consequently, decreased expression of the PPARα targets, HNF4α, FOXA2 and Mttp results in defective hepatic VLDL secretion. Decreased plasma glucose and leptin, and reduced liver glucose, glycogen and glutamine concentrations in fed Lal -/- mice indicate increased energy demand and extensive systemic usage of glucose. Treatment with the PPARα agonist fenofibrate further reduces plasma TG, liver glucose and glycogen concentrations in Lal -/- mice, indicating that VLDL synthesis and insulin sensitivity is PPARα-independent. Decreased VLDL production might be a consequence of reduced hepatic acyl-CoA concentrations, particularly the HNF4a ligand linoleic acid (18:2). Glucose tolerance and insulin sensitivity are increased to compensate for decreased energy availability
Abbreviations
- BW:
-
Body weight
- CE:
-
Cholesteryl ester
- CESD:
-
Cholesteryl ester storage disease
- FPLC:
-
Fast protein liquid chromatography
- H&E:
-
Haematoxylin and eosin
- ITT:
-
Insulin tolerance test
- LAL:
-
Lysosomal acid lipase
- PLIN2:
-
Perilipin 2
- PPARα:
-
Peroxisome proliferator-activated receptor α
- scWAT:
-
subcutaneous white adipose tissue
- TC:
-
Total cholesterol
- TG:
-
Triacylglycerol
- TLC:
-
Thin-layer chromatography
- WAT:
-
White adipose tissue
- WD:
-
Wolman disease
- WT:
-
Wild-type
References
Brown MS, Goldstein JL (1976) Receptor-mediated control of cholesterol metabolism. Science 191:150–154
Assmann G, Seedorf U (1995) Acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited disease, 7th edn. McGraw and Hill, New York, pp 2563–2587
Burke JA, Schubert WK (1972) Deficient activity of hepatic acid lipase in cholesterol ester storage disease. Science 176:309–310
Patrick AD, Lake BD (1969) Deficiency of an acid lipase in Wolman’s disease. Nature 222:1067–1068
Sloan HR, Fredrickson DS (1972) Enzyme deficiency in cholesteryl ester storage disease. J Clin Invest 51:1923–1926
Wolman M, Sterk VV, Gatt S, Frenkel M (1961) Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics 28:742–757
Marshall WC, Ockenden BG, Fosbrooke AS, Cumings JN (1969) Wolman’s disease. A rare lipidosis with adrenal calcification. Arch Dis Child 44:331–341
Brown MS, Sobhani MK, Brunschede GY, Goldstein JL (1976) Restoration of a regulatory response to low density lipoprotein in acid lipase-deficient human fibroblasts. J Biol Chem 251:3277–3286
Seedorf U, Wiebusch H, Muntoni S et al (1995) A novel variant of lysosomal acid lipase (Leu336-->Pro) associated with acid lipase deficiency and cholesterol ester storage disease. Arterioscler Thromb Vasc Biol 15:773–778
Pisciotta L, Fresa R, Bellocchio A et al (2009) Cholesteryl Ester Storage Disease (CESD) due to novel mutations in the LIPA gene. Mol Genet Metab 97:143–148
Aqul A, Lopez AM, Posey KS et al (2014) Hepatic entrapment of esterified cholesterol drives continual expansion of whole body sterol pool in lysosomal acid lipase-deficient mice. Am J Physiol Gastrointest Liver Physiol 307:G836–847
Du H, Duanmu M, Witte D, Grabowski GA (1998) Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 7:1347–1354
Du H, Heur M, Duanmu M et al (2001) Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res 42:489–500
Chandak PG, Obrowsky S, Radovic B et al (2011) Lack of acyl-CoA:diacylglycerol acyltransferase 1 reduces intestinal cholesterol absorption and attenuates atherosclerosis in apolipoprotein E knockout mice. Biochim Biophys Acta 1811:1011–1020
Yuan M, Breitkopf SB, Yang X, Asara JM (2012) A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7:872–881
Magnes C, Suppan M, Pieber TR et al (2008) Validated comprehensive analytical method for quantification of coenzyme A activated compounds in biological tissues by online solid-phase extraction LC/MS/MS. Anal Chem 80:5736–5742
Schweiger M, Eichmann TO, Taschler U, Zimmermann R, Zechner R, Lass A (2014) Measurement of lipolysis. Methods Enzymol 538:171–193
Frezza C, Cipolat S, Scorrano L (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2:287–295
Schlager S, Goeritzer M, Jandl K et al (2015) Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J Leukoc Biol 98:837–850
Cui Q, Lewis IA, Hegeman AD et al (2008) Metabolite identification via the Madison Metabolomics Consortium Database. Nat Biotechnol 26:162–164
Wolfrum C, Stoffel M (2006) Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab 3:99–110
Sheena V, Hertz R, Nousbeck J, Berman I, Magenheim J, Bar-Tana J (2005) Transcriptional regulation of human microsomal triglyceride transfer protein by hepatocyte nuclear factor-4alpha. J Lipid Res 46:328–341
Knights AJ, Funnell AP, Pearson RC, Crossley M, Bell-Anderson KS (2014) Adipokines and insulin action: a sensitive issue. Adipocyte 3:88–96
Sinha MK, Opentanova I, Ohannesian JP et al (1996) Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short-term fasting. J Clin Invest 98:1277–1282
Comert B, Mas MR, Erdem H et al (2001) Insulin resistance in non-alcoholic steatohepatitis. Dig Liver Dis 33:353–358
Luyckx FH, Lefebvre PJ, Scheen AJ (2000) Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab 26:98–106
Tankurt E, Biberoglu S, Ellidokuz E et al (1999) Hyperinsulinemia and insulin resistance in non-alcoholic steatohepatitis. J Hepatol 31:963
Tiwari S, Siddiqi SA (2012) Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol 32:1079–1086
Skop V, Cahova M, Papackova Z et al (2012) Autophagy-lysosomal pathway is involved in lipid degradation in rat liver. Physiol Res 61:287–297
Koo SH, Montminy M (2006) Fatty acids and insulin resistance: a perfect storm. Mol Cell 21:449–450
Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ (2001) Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 21:1393–1403
Haluzik MM, Haluzik M (2006) PPAR-alpha and insulin sensitivity. Physiol Res 55:115–122
Linden D, Alsterholm M, Wennbo H, Oscarsson J (2001) PPARalpha deficiency increases secretion and serum levels of apolipoprotein B-containing lipoproteins. J Lipid Res 42:1831–1840
Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE (2002) Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem 277:37973–37976
Yuan X, Ta TC, Lin M et al (2009) Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One 4:e5609
Puigserver P, Rodgers JT (2006) Foxa2, a novel transcriptional regulator of insulin sensitivity. Nat Med 12:38–39
Adiels M, Olofsson SO, Taskinen MR, Boren J (2008) Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol 28:1225–1236
Meshkani R, Adeli K (2009) Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin Biochem 42:1331–1346
Krauss RM (2004) Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 27:1496–1504
Asterholm IW, Halberg N, Scherer PE (2007) Mouse Models of Lipodystrophy Key reagents for the understanding of the metabolic syndrome. Drug Discov Today Dis Models 4:17–24
Pilkis SJ, Granner DK (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54:885–909
Sundaram M, Yao Z (2010) Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) 7:35
Acknowledgements
Open access funding provided by Medical University of Graz. The authors thank S. Rainer and A. Ibovnik (Medical University of Graz, Austria) for excellent technical assistance, R. Schreiber (University of Graz, Austria) for providing the PLIN2 antibody, E. Bernhart for providing the Lamin A/C antibody (Medical University of Graz, Austria) and I. Hindler (Medical University of Graz, Austria) for the care of the mice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Austrian Science Fund FWF (DK-MCD W1226, P27070, P22832, SFB-LIPOTOX F3004) (DKr), the PhD programme ‘Molecular Medicine’ of the Medical University of Graz and BioTechMed-Graz. TM was supported by the Bavarian Ministry of Sciences, Research and the Arts (BioSysNet), the German Research Foundation (MA 5703/1-1), the Centre for Integrated Protein Science, Munich, the President’s International Fellowship Initiative of CAS (No: 2015VBB045) and the National Natural Science Foundation of China (No. 31450110423).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
BR and DK conceived and designed the study and wrote the paper. BR, NV, CL, SS, MG, JVP, MK, DKo, JR, MW, TT, MS, LG, CM, CD and TM contributed to data collection and analyses. RBG, SF, ES, HD, WFG and TM contributed to study design and data interpretation. All authors critically revised the manuscript and have read and approved the final version. DK is responsible for the integrity of the work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 189 kb)
ESM Fig. 1
(PDF 53 kb)
ESM Fig. 2
(PDF 58 kb)
ESM Fig. 3
(PDF 187 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Radović, B., Vujić, N., Leopold, C. et al. Lysosomal acid lipase regulates VLDL synthesis and insulin sensitivity in mice. Diabetologia 59, 1743–1752 (2016). https://doi.org/10.1007/s00125-016-3968-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3968-6