
Abstract
Adverse neonatal outcomes continue to be high for mothers with type 1 and type 2 diabetes, and are far from eliminated in mothers with gestational diabetes mellitus. This is often despite seemingly satisfactory glycaemic control in the latter half of pregnancy. Here we argue that this could be a consequence of the early establishment of fetal hyperinsulinaemia, a driver that exaggerates the fetal glucose steal. Essentially, fetal hyperinsulinaemia, through its effect on lowering fetal glycaemia, will increase the glucose concentration gradient across the placenta and consequently the glucose flux to the fetus. While the steepness of this gradient and glucose flux will be greatest at times when maternal hyperglycaemia and fetal hyperinsulinaemia coexist, fetal hyperinsulinaemia will favour a persistently high glucose flux even at times when maternal blood glucose is normal. The obvious implication is that glycaemic control needs to be optimised very early in pregnancy to prevent the establishment of fetal hyperinsulinaemia, further supporting the need for pre-pregnancy planning and early establishment of maternal glycaemic control. An exaggerated glucose steal by a hyperinsulinaemic fetus could also attenuate maternal glucose levels during an OGTT, providing an explanation for why some mothers with fetuses with all the characteristics of diabetic fetopathy have ‘normal’ glucose tolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite significant advances in glycaemic control in the management of diabetes in pregnancy, adverse outcomes for the fetus are still very common, particularly for pre-existing type 1 and type 2 diabetes. For example, in a recent review of 12 studies comprising 14,099 type 1 diabetic pregnancies the RRs conferred by diabetes over the background population for perinatal mortality, congenital malformations and preterm birth were all significantly increased (3.7, 2.4 and 4.2, respectively) [1]. Of particular note, 54.2% of neonates of type 1 diabetic pregnancies were born large for gestational age (LGA) compared with 10.0% in the background population— RR of 4.5 [1]. In addition, in gestational diabetes mellitus (GDM), standard approaches to treatment, while reducing the rates of LGA, do not fully normalise neonatal outcomes, including the rates of neonatal hypoglycaemia, raised cord blood C-peptide levels and neonatal fat mass [2–4].
In this article we propose that an exaggerated fetal glucose steal phenomenon, driven by fetal hyperinsulinaemia, contributes to diabetic fetopathy, including excessive fetal fat accretion. After introducing the glucose steal phenomenon, we review what is known about the early development of insulin secretion in the fetus. We then discuss the need for tight glucose control in the first and early second trimesters to prevent fetal hyperinsulinaemia and exaggeration of the glucose steal with its implications for the fetus.
The principle of the fetal glucose steal phenomenon in normal and diabetic pregnancies
In considering determinants of glucose transfer from mother to fetus, we focus mostly on those of the mother rather than those of the fetus. Maternal glucose metabolism adapts to pregnancy so as to provide a continuous supply of glucose to the fetus. These hormone-regulated adaptations include increased hepatic glucose production in the fasting state, to ensure a glucose supply at all times, and maternal peripheral insulin resistance, which spares glucose for the fetus [5].
Mentioned rarely is the role of the fetus (and placenta) and its propensity to act as a glucose sink, stealing glucose from the mother. This process has been termed the ‘fetoplacental glucose steal phenomenon’ [6]. It was originally established in sheep [7, 8] and elaborated in detail in the rat [6]. There is also indirect evidence that it operates in humans [9, 10].
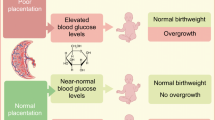
All available evidence suggests that the placenta is a passive conduit for the proportion of maternal glucose destined to reach the fetus, at least at the end of gestation [11]. Thus, the maternal-to-fetal glucose flux is mostly dictated by the maternal-to-fetal glucose concentration gradient, i.e. the glucose concentration difference between the maternal and fetal compartments. Therefore, any concentration change in either compartment will modify the concentration gradient and affect glucose flux (Fig. 1).

Maternal (M) glucose is transferred to the fetus (F) across the placenta down a concentration gradient. The glucose gradient is determined by both maternal and fetal glucose levels. Maternal high glucose (MHG) and/or lower fetal glucose will steepen the gradient, leading to augmented glucose flux into the fetus. MHG is a consequence of inadequate control of maternal diabetes, which promotes increased glucose transfer to the fetus, increased fetal blood glucose, increased stimulation of insulin release from fetal islets and, consequently, fetal hyperinsulinaemia (FHI), as described by Pedersen and Osler [12]. Importantly, FHI, by increasing the rate of glucose utilisation by the fetus, will lower fetal glucose, thereby increasing the steepness of the transplacental glucose gradient and the rate of glucose transfer. In effect, MHG pushes glucose and FHI pulls glucose (the fetal glucose steal) across the placenta to the fetus. The ensuing increased glucose delivery from diabetic mothers, together with the FHI, stimulates fetal triacylglycerol formation and the deposition of excess fetal adipose tissue
Maternal glucose concentrations are determined not only by maternal factors such as the type of diabetes and how well it is managed, but also by the rate of glucose uptake in the fetus. Fetal glucose concentrations are determined by both glucose appearance, by transplacental glucose transfer from the mother, and glucose disappearance, by its uptake into fetal tissues, the latter being influenced by fetal insulin levels, fetal insulin sensitivity and fetal weight. Thus, factors on either side of the placenta determining maternal and fetal glucose concentrations are determinants of the transplacental glucose gradient (Fig. 1).
We propose that the fetal glucose steal phenomenon is exaggerated in diabetic pregnancies as a consequence of fetal hyperinsulinaemia and, in addition to maternal factors, this steal contributes to diabetic fetopathy. The hallmark of diabetic fetopathy is fetal hyperinsulinaemia, which drives fetal fat accumulation, very often resulting in an LGA birth. However, increased adiposity can also occur in infants of diabetic women who have normal birthweight [4].
Because of its dual role as a growth factor and anabolic hormone, fetal insulin has long been recognised as the major driver of excess adipose tissue deposition in fetuses of mothers with diabetes [12, 13]. Diabetic pregnancies are associated with elevated fetal insulin levels due to increased maternal glucose reaching the fetal circulation, which stimulates insulin release from the fetal pancreas (Pedersen hypothesis, Fig. 1). However, other insulin secretagogues such as amino acids and, potentially, fatty acids may also contribute [14, 15].
In newborns of diabetic mothers, glucose clearance into tissues is much faster than in newborns from normal pregnancies, often causing neonatal hypoglycaemia due to neonatal hyperinsulinaemia [16]. Hyperinsulinaemia in the fetus is likely also to accelerate glucose clearance into fetal tissues and increase the fetal glucose steal. Thus, the maternal–fetal glucose gradient can be steepened in diabetic pregnancy by both maternal (poor glycaemic control) and fetal (hyperinsulinaemia) factors, resulting in increased glucose transfer to the fetus. Furthermore, even in the presence of normal maternal glucose levels, fetal hyperinsulinaemia will still lower fetal glucose concentrations, sustaining a high glucose gradient and an exaggerated glucose steal (Fig. 1).
Early development of fetal hyperinsulinaemia
The human pancreas begins to develop 4 weeks after conception, and first insulin deposits can be found between weeks 7 and 8 [17]. In vitro fetal pancreases release insulin as early as 11 weeks of gestation [18]. Amniotic fluid insulin (AFI), which originates from loss into fetal urine, is just detectable at 12 weeks [19] and more easily measurable from 14 weeks onwards [20]. AFI can be used as a surrogate for fetal serum insulin, as AFI measured at 31 weeks has been shown to be highly correlated with cord blood insulin measured at birth (r = 0.853; p = 0.0001) [9].
While neonatal hyperinsulinaemia as a consequence of maternal diabetes is well established to occur as determined by the measurement of cord blood insulin or C-peptide from samples collected immediately after delivery [21], its onset in pregnancy is less well understood. It has long been thought that the fetal pancreas is unresponsive to glucose changes early in pregnancy [22]. While this appears to hold true for normal pregnancies, the fetal pancreas of pregnant women with diabetes is already sensitive to glucose in vitro at 12 weeks [18]. This explains why AFI levels can be elevated before the diagnosis of GDM [20, 23]. Elevated AFI concentrations, beginning at 14 weeks, are associated not only with the mother’s risk of developing GDM [20, 23] but also with the risk of the fetus having a birthweight >90th centile [20].
The cause of early fetal hyperinsulinaemia is not fully understood; however, it is highly correlated with maternal glycaemic control early in pregnancy, such that early maternal hyperglycaemia in type 1 and type 2 diabetic pregnancies, and most probably GDM, is likely to be the major driver. Roles for alterations in other maternal nutrient levels associated with by diabetes, however, may also be important. The maternal diabetic state not only leads to beta cell hyperplasia in the fetus [24, 25] but also seems to accelerate the maturation of the beta cell stimulus–secretion coupling mechanism [26]. It is of note that fat deposition in the fetus begins at around 14 weeks, which is the time of onset of early fetal hyperinsulinaemia [27].
The importance of maternal metabolism in the first trimester of pregnancy
The pathophysiological relationship between maternal glucose and fetal neonatal adiposity mediated through fetal insulin levels was proposed more than six decades ago [12] and has recently been confirmed in a large cohort of non-diabetic women of different ethnicities [28]. This raises the question about when in pregnancy the metabolic perturbations in the mother begin to influence the fetus. While this large cohort study established that it occurs at 24–28 weeks’ gestation [28], there is evidence that earlier periods may play an important role. Although body composition was not measured and, hence, body fat was not an outcome, a continuous relationship between maternal fasting glucose measured as early as 9–10 weeks’ gestation and the risk of an LGA birth has been shown [29]. Likewise, random blood glucose in the first trimester was positively correlated with proinsulin levels in the cord blood [30]. The influence of early pregnancy glycaemia is also seen in the reports of stronger associations of maternal first and second trimester glycosylated haemoglobin levels, compared with later measurements, with birthweight and occurrence of LGA [1, 31–33]. Consistent also is the finding that excessive fetal growth in pre-gestational diabetes is evident by serial ultrasound in many pregnancies from 18 to 24 weeks and that those with established LGA prior to 30 weeks are most likely to result in greater severity macrosomia at birth [31, 34, 35].
The period when the metabolic environment has an effect on fetal growth may even extend to pre-pregnancy. The influence of pre-pregnancy metabolic changes on fetal development may be mediated through modification of oocyte metabolism [36], predominantly of their mitochondria [37], through changing early embryonic growth and later growth trajectories.
Clinical consequences of the fetal glucose steal
Glucose steal and associations of early and late pregnancy glycaemic control with diabetic fetopathy
As mentioned above, stronger associations between early rather than late pregnancy glycosylated haemoglobin levels with LGA are often reported [1, 31–33]. This is not, however, a universal finding of all studies, as some show a greater effect of third trimester glycaemic control [38, 39]. Overall, these studies emphasise the importance of optimal glycaemic control through the whole of pregnancy to prevent diabetic fetopathy.
Poor glycaemic control early in pregnancy will result in the establishment of fetal hyperinsulinaemia, causing an exaggerated fetal glucose steal. As a consequence, the overactive glucose steal will increase the disposal of maternal glucose into the fetus, thus attenuating the levels of maternal hyperglycaemia. Importantly, this effect of lowering maternal glucose driven by the fetus will be greatest in pregnancies with the most hyperinsulinaemic fetuses.
Thus, the challenge for many clinicians working in the field of diabetes in pregnancy that seemingly tight late pregnancy glycaemic control often fails to prevent diabetic fetopathy, at least in type 1 and type 2 diabetic pregnancies, can be partly explained by the fetal glucose steal. This is because fetal hyperinsulinaemia will sustain an increased glucose gradient across the placenta and the glucose steal, even at times of near-normal maternal glycaemia.
Once fetal hyperinsulinaemia is established, however, continued maternal hyperglycaemia into the third trimester will likely compound the effects of the fetal hyperinsulinaemia and glucose steal on accelerated fetal growth, as the maternal–fetal glucose gradient and glucose transfer to the fetus will be greater. This is consistent with reports that show an effect of late pregnancy glycaemia on the adverse outcome of LGA [38, 39].
Exaggerated glucose steal due to fetal hyperinsulinaemia: risk of masking GDM diagnosis by OGTT
Weiss et al assessed changes in the OGTT from 25 to 31 weeks’ gestation in women with GDM categorised according to the presence or absence of fetal hyperinsulinaemia [9]. In women with GDM without evidence of fetal hyperinsulinaemia, determined by AFI <48.6 pmol/l, glucose tolerance deteriorated from 25 to 31 weeks’ gestation (Fig. 2). By contrast, in GDM women with fetal hyperinsulinaemia (AFI >48.6 pmol/l), glucose tolerance improved during the same period (Fig. 2). This can be explained by siphoning of the glucose to the fetus in women with hyperinsulinaemic fetuses, due to the effects of an increase in glucose steal activity [9]. The placentas of hyperinsulinaemic, macrosomic fetuses might also have been larger, such that increased placental glucose consumption could also have contributed to the blunting of the OGTT glucose rise observed in the mothers. Thus, there is a risk that GDM will not be diagnosed in women with the most affected fetuses. Consistent with this, seven of 21 (33%), compared with none of 11 (0%), women with AFI >48.6 pmol/l, compared with AFI <48.6 pmol/l, delivered neonates with birthweight >4,000 g [9].

OGTTs were performed in 34 mothers with GDM at 25 and 31 weeks prior to any commencing insulin therapy. Amniocentesis was also performed at week 31 and AFI levels were measured [9]. In the mothers of the 13 normoinsulinaemic fetuses (AFI <48.6 pmol/l; solid blue line), maternal glucose tolerance deteriorated from the first to the second time point as manifested by a higher 1 h post-load glucose value (9.3 vs 10.0 mmol/l; p = 0.0006). In the mothers of the 21 hyperinsulinaemic fetuses (AFI >48.6 pmol/l; dotted red line), the glucose levels at 1 and 2 h of the repeated OGTT were lower (significant for the 1 h reading only) than at 25 weeks (1 h, 11.2 vs 9.9 mmol/l, p = 0.002; 2 h, 8.8 vs 7.7 mmol/l, p = 0.158) [9]. Figure created from data from Weiss et al [9]
The data of Weiss et al are important for two reasons (Fig. 2) [9]. First, they support the premise that an exaggerated glucose steal phenomenon operates in diabetic pregnancies complicated by fetal hyperinsulinaemia. Second, they provide a reason why some women with fetuses with all the characteristics of diabetic fetopathy are often found to have ‘normal’ glucose tolerance.
Can tight glycaemic control later in pregnancy reverse fetal hyperinsulinaemia and the fetal glucose steal?
The findings that treatment of GDM from 24 to 28 weeks can significantly reduce the rate of LGA births suggest it is possible to reverse fetal hyperinsulinaemia, at least when hyperglycaemia is mild [2, 3]. However, the rate of neonatal hypoglycaemia needing intravenous glucose therapy was not significantly reduced in the GDM treatment arms [2, 3]. Furthermore, treatment did not significantly reduce the rate of elevated cord C-peptide levels in the study where this was measured [3]. Despite normalisation of birthweight, management of GDM has not been shown to normalise excess fetal adiposity [4]. Thus, fetal hyperinsulinaemia appears to be difficult to normalise with the usual approach to managing mild maternal hyperglycaemia [2, 3].
Interestingly, in a study in which treatment of GDM with insulin was initiated according to the presence or absence of an elevated AFI concentration, the rate of elevated cord blood C-peptide levels in neonates was markedly reduced, suggesting that there is some potential to reverse fetal hyperinsulinaemia by targeting it in GDM pregnancies [40]. In pre-existing diabetes, however, when normalisation of maternal glucose levels is very difficult to achieve, fetal hyperinsulinaemia is likely to be much more difficult to reverse, as potentially even short periods of maternal hyperglycaemia will sustain it. The association between hyperglycaemic spikes in the third trimester with LGA would be consistent with this premise [38].
Future directions
Of major importance is an improved understanding of the mechanisms causing early fetal hyperinsulinaemia, as this is the driver that accelerates the fetal glucose steal in diabetic pregnancy. While it may be a consequence of maternal glycaemia only, other nutrients such as amino acids and fatty acids and/or hormones/cytokines could also contribute, particularly in the pregnancies of obese mothers in whom glucose tolerance is normal but cord blood C-peptide levels are shown to be elevated at birth [41]. It will be particularly important to determine the factors that render early glucose signalling competence to fetal islets, as this occurs earlier in diabetic pregnancies. It is already known from elegant studies of the glucokinase gene in the fetus that fetal genetic factors can alter the responsiveness of fetal islets to maternal hyperglycaemia [42]. Improved understanding of fetal (including genetic and epigenetic) factors that determine the likelihood of pathological hyperinsulinaemia in the fetus will also be useful. The role of the placenta in fetal nutrient supply early in pregnancy is entirely unknown. In particular, studies should be undertaken to address the question whether the placenta plays a passive, permissive role or whether it actively contributes to determining the amount of nutrients that will be passed on to the fetus in the early periods of gestation.
The discussion above has focused only on the fetal glucose steal. It is highly conceivable that fetal hyperinsulinaemia could also drive a fetal fatty acid and/or an amino acid steal. Furthermore, an increased supply of fatty acids and amino acids could contribute to the maintenance of fetal insulin hypersecretion and fetal hyperinsulinaemia in situations where maternal glucose levels are relatively well controlled. A fetal fatty acid steal is clearly conceivable, at least for those fatty acids which are transferred by diffusion [43]. For other fatty acids and amino acids the more complex transplacental transfer mechanisms may make a steal phenomenon more complicated.
The high likelihood that fetal hyperinsulinaemia can mask an OGTT diagnosis of GDM through the glucose steal phenomenon requires further confirmation and the clinical consequences need thorough consideration. New methodologies to reliably diagnose the presence of fetal hyperinsulinaemia should also be sought, potentially to indicate the degree of aggressiveness required to improve maternal metabolic control.
Randomised controlled clinical trials focusing on optimisation of glycaemic and metabolic control prior to and during the first trimester of pregnancy in disorders of mild hyperglycaemia, or in women who are overweight and obese even with normoglycaemia, are required to determine whether this can prevent fetal hyperinsulinaemia and excess fetal growth.
Finally, to further improve pregnancy outcomes related to fetal hyperinsulinaemia and exaggeration of the fetal glucose steal in pre-existing type 1 and type 2 diabetes, as well as in GDM, it is necessary to optimise metabolic control early in pregnancy. This will necessitate pre-pregnancy planning for women with pre-existing diabetes, as well as for those at increased risk of GDM, and better means to safely normalise glycaemia.
Abbreviations
- AFI:
-
Amniotic fluid insulin
- GDM:
-
Gestational diabetes mellitus
- LGA:
-
Large for gestational age
References
Colstrup M, Mathiesen ER, Damm P, Jensen DM, Ringholm L (2013) Pregnancy in women with type 1 diabetes: have the goals of st. Vincent declaration been met concerning foetal and neonatal complications? J Matern Fetal Neonatal Med 26:1682–1686
Crowther CA, Hiller JE, Moss JR et al (2005) Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med 352:2477–2486
Landon MB, Spong CY, Thom E et al (2009) A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med 361:1339–1348
Catalano PM, Thomas A, Huston-Presley L, Amini SB (2003) Increased fetal adiposity: a very sensitive marker of abnormal in utero development. Am J Obstet Gynecol 189:1698–1704
Catalano PM (2014) Trying to understand gestational diabetes. Diabet Med 31:273–281
Nolan CJ, Proietto J (1994) The feto-placental glucose steal phenomenon is a major cause of maternal metabolic adaptation during late pregnancy in the rat. Diabetologia 37:976–984
Jodarski GD, Shanahan MF, Rankin JH (1985) Fetal insulin and placental 3-o-methyl glucose clearance in near-term sheep. J Dev Physiol 7:251–258
Molina RD, Carver TD, Hay WW Jr (1993) Ontogeny of insulin effect in fetal sheep. Pediatr Res 34:654–660
Weiss PA, Scholz HS, Haas J, Tamussino KF (2001) Effect of fetal hyperinsulinism on oral glucose tolerance test results in patients with gestational diabetes mellitus. Am J Obstet Gynecol 184:470–475
Marconi AM, Davoli E, Cetin I et al (1993) Impact of conceptus mass on glucose disposal rate in pregnant women. Am J Physiol 264:E514–E518
Desoye G, van Poppel M (2015) The feto-placental dialogue and diabesity. Best Pract Res Clin Obstet Gynaecol 29:15–23
Pedersen J, Osler M (1961) Hyperglycemia as the cause of characteristic features of the foetus and newborn of diabetic mothers. Dan Med Bull 8:78–83
Enzi G, Inelmen EM, Caretta F, Villani F, Zanardo V, DeBiasi F (1980) Development of adipose tissue in newborns of gestational-diabetic and insulin-dependent diabetic mothers. Diabetes 29:100–104
Milner RD, Ashworth MA, Barson AJ (1972) Insulin release from human foetal pancreas in response to glucose, leucine and arginine. J Endocrinol 52:497–505
Prentki M, Matschinsky FM, Madiraju SR (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18:162–185
Molsted-Pedersen L (1972) Aspects of carbohydrate metabolism in newborn infants of diabetic mothers. I. Intravenous glucose tolerance tests (a) distribution and means of K values, and (b) correlation between K value and birth weight. Acta Endocrinol (Copenh) 69:174–188
Piper K, Brickwood S, Turnpenny LW et al (2004) Beta cell differentiation during early human pancreas development. J Endocrinol 181:11–23
Reiher H, Fuhrmann K, Noack S et al (1983) Age-dependent insulin secretion of the endocrine pancreas in vitro from fetuses of diabetic and nondiabetic patients. Diabetes Care 6:446–451
Weiss PA, Purstner P, Winter R, Lichtenegger W (1984) Insulin levels in amniotic fluid of normal and abnormal pregnancies. Obstet Gynecol 63:371–375
Carpenter MW, Canick JA, Hogan JW, Shellum C, Somers M, Star JA (2001) Amniotic fluid insulin at 14-20 weeks’ gestation: association with later maternal glucose intolerance and birth macrosomia. Diabetes Care 24:1259–1263
Desoye G, Hofmann HH, Weiss PA (1992) Insulin binding to trophoblast plasma membranes and placental glycogen content in well-controlled gestational diabetic women treated with diet or insulin, in well-controlled overt diabetic patients and in healthy control subjects. Diabetologia 35:45–55
Adam PA, Teramo K, Raiha N, Gitlin D, Schwartz R (1969) Human fetal insulin metabolism early in gestation. Response to acute elevation of the fetal glucose concentration and placental transfer of human insulin-I-131. Diabetes 18:409–416
Tisi DK, Burns DH, Luskey GW, Koski KG (2011) Fetal exposure to altered amniotic fluid glucose, insulin, and insulin-like growth factor-binding protein 1 occurs before screening for gestational diabetes mellitus. Diabetes Care 34:139–144
Van Assche FA (1997) Symmetric and asymmetric fetal macrosomia in relation to long-term consequences. Am J Obstet Gynecol 177:1563–1564
Van Assche FA, Holemans K, Aerts L (2001) Long-term consequences for offspring of diabetes during pregnancy. Br Med Bull 60:173–182
Mintz DH, Chez RA, Hutchinson DL (1972) Subhuman primate pregnancy complicated by streptozotocin-induced diabetes mellitus. J Clin Invest 51:837–847
Poissonnet CM, Burdi AR, Bookstein FL (1983) Growth and development of human adipose tissue during early gestation. Early Hum Dev 8:1–11
The HAPO Study Cooperative Research Group (2009) Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study: associations with neonatal anthropometrics. Diabetes 58:453–459
Riskin-Mashiah S, Younes G, Damti A, Auslender R (2009) First-trimester fasting hyperglycemia and adverse pregnancy outcomes. Diabetes Care 32:1639–1643
Luo ZC, Delvin E, Fraser WD et al (2010) Maternal glucose tolerance in pregnancy affects fetal insulin sensitivity. Diabetes Care 33:2055–2061
Gold AE, Reilly R, Little J, Walker JD (1998) The effect of glycemic control in the pre-conception period and early pregnancy on birth weight in women with IDDM. Diabetes Care 21:535–538
Raychaudhuri K, Maresh MJ (2000) Glycemic control throughout pregnancy and fetal growth in insulin-dependent diabetes. Obstet Gynecol 95:190–194
Rey E, Attie C, Bonin A (1999) The effects of first-trimester diabetes control on the incidence of macrosomia. Am J Obstet Gynecol 181:202–206
Wong SF, Lee-Tannock A, Amaraddio D, Chan FY, McIntyre HD (2006) Fetal growth patterns in fetuses of women with pregestational diabetes mellitus. Ultrasound Obstet Gynecol 28:934–938
Kerssen A, de Valk HW, Visser GH (2007) Increased second trimester maternal glucose levels are related to extremely large-for-gestational-age infants in women with type 1 diabetes. Diabetes Care 30:1069–1074
Gu L, Liu H, Gu X, Boots C, Moley KH, Wang Q (2015) Metabolic control of oocyte development: linking maternal nutrition and reproductive outcomes. Cell Mol Life Sci 72:251–271
Turner N, Robker RL (2015) Developmental programming of obesity and insulin resistance: does mitochondrial dysfunction in oocytes play a role? Mol Hum Reprod 21:23–30
Damm P, Mersebach H, Rastam J et al (2014) Poor pregnancy outcome in women with type 1 diabetes is predicted by elevated HbA1c and spikes of high glucose values in the third trimester. J Matern Fetal Neonatal Med 27:149–154
Glinianaia SV, Tennant PW, Bilous RW, Rankin J, Bell R (2012) HbA1c and birthweight in women with pre-conception type 1 and type 2 diabetes: a population-based cohort study. Diabetologia 55:3193–3203
Weiss PA, Hofmann HM, Kainer F, Haas JG (1988) Fetal outcome in gestational diabetes with elevated amniotic fluid insulin levels. Dietary versus insulin treatment. Diabetes Res Clin Pract 5:1–7
Catalano PM, McIntyre HD, Cruickshank JK et al (2012) The hyperglycemia and adverse pregnancy outcome study: associations of GDM and obesity with pregnancy outcomes. Diabetes Care 35:780–786
Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S (1998) Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 19:268–270
Dancis J, Jansen V, Kayden HJ, Bjornson L, Levitz M (1974) Transfer across perfused human placenta. 3. Effect of chain length on transfer of free fatty acids. Pediatr Res 8:796–799
Acknowledgements
Open access funding provided by Medical University of Graz. The authors wish to thank P. Catalano, P. Damm, D. Sacks and M. van Poppel for critically reading the manuscript and making valuable suggestions and comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The research of GD has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013), EarlyNutrition project, grant agreement no. 289346, and Vitamin D and Lifestyle Intervention for Gestational Diabetes Mellitus Prevention (DALI) project, grant agreement no. 289364. The research of CJN has received funding from the National Health and Medical Research Council (project grant 418077) and the Canberra Hospital Private Practice Fund.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
Both authors were responsible for drafting the article and revising it critically for important intellectual content. Both authors approved the version to be submitted.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Desoye, G., Nolan, C.J. The fetal glucose steal: an underappreciated phenomenon in diabetic pregnancy. Diabetologia 59, 1089–1094 (2016). https://doi.org/10.1007/s00125-016-3931-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3931-6