
Abstract
Diabetes is not a single homogeneous disease but composed of many diseases with hyperglycaemia as a common feature. Four factors have, historically, been used to identify this diversity: the age at onset; the severity of the disease, i.e. degree of loss of beta cell function; the degree of insulin resistance and the presence of diabetes-associated autoantibodies. Our broad understanding of the distinction between the two major types, type 1 diabetes mellitus and type 2 diabetes mellitus, are based on these factors, but it has become apparent that they do not precisely capture the different disease forms. Indeed, both major types of diabetes have common features, encapsulated by adult-onset autoimmune diabetes and maturity-onset diabetes of the young. As a result, there has been a repositioning of our understanding of diabetes. In this review, drawing on recent literature, we discuss the evidence that autoimmune type 1 diabetes has a broad clinical phenotype with diverse therapeutic options, while the term non-autoimmune type 2 diabetes obscures the optimal management strategy because it encompasses substantial heterogeneity. Underlying these developments is a general progression towards precision medicine with the need for precise patient characterisation, currently based on clinical phenotypes but in future augmented by laboratory-based tests.
Key points | |
• The need to clarify diabetes classification, which is currently imprecise in distinguishing major disease types, using laboratory tests | |
• The importance of predictors of disease progression, including genetic, immune and metabolic features | |
• The potential for predicting therapeutic responses to provide a more personalised approach to therapy | |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The term ‘diabetes’, meaning a ‘fountain’ (of urine), is a term forged in ignorance. From the earliest years it was recognised that ‘honey urine’, that is, diabetes mellitus, was a condition distinct from other forms of polyuria. But with the discovery of insulin, and the knowledge that all patients with diabetes responded to insulin injections, a single cause, that is, insulin deficiency, seemed the likely single pathology. Subsequently, we came to the view that there were two major types of diabetes defined by two features, namely the age at diagnosis and the obligatory need for insulin therapy. However, it is clear that what you see (clinically) is neither what you get (in terms of pathogenesis), nor what you should do (therapeutically) [1–3]. This review highlights areas of uncertainty in distinguishing the two main types of diabetes, indicating how further characterisation by laboratory tests can impact disease management.
Categorising diabetes
Diseases like diabetes gain their identity from their clinical phenotype, historical precedent, genotype and specific environmental causes. This approach has led to a classification of diabetes as type 1 diabetes, type 2 diabetes, monogenic diabetes, gestational diabetes, and other specific disease forms [1, 2]. Disease classification should contribute to an understanding of (1) aetiology, (2) the natural history of that disease, (3) pathophysiology, (4) the disease consequences, and (5) treatment of the condition. Ideally, disease identity is defined by categorical features, specific (exclusive) to that type of diabetes.
Type 1 diabetes as an exclusive disease
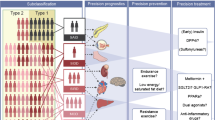
The definition of type 1 diabetes is clinically exclusive, encompassing patients with diabetes-associated autoantibodies (DAA) plus, when diagnosed outside a surveillance programme, insulin dependence. Difficulty arises when patients with clinical type 2 diabetes have immunogenetic characteristics of type 1 diabetes. These patients are diagnosed with latent autoimmune diabetes in adults (LADA), also called slowly progressive insulin-dependent diabetes (SPIDDM) or type 1.5 diabetes [3–5]. Guidelines assign all patients with DAA, including those with LADA, to type 1 diabetes. Three classes of biomarkers (immune, metabolic, genetic) characterise type 1 diabetes (Fig. 1): (1) DAA, (2) C-peptide levels as a proxy for levels of insulin secretion, and (3) HLA genotypes. Allied to childhood onset and clinical insulin dependence at diagnosis, the presence of DAA in an individual with high-disease-risk HLA genotypes and very low C-peptide levels categorically identifies type 1a diabetes [1]. However, no single factor is specific for type 1 diabetes: HLA genotypes are sensitive but not specific markers; DAA can be found in other conditions, including 1% of the normal population, as positivity is defined as a signal above the 99th percentile; age and C-peptide levels are continuous variables; and starting insulin therapy is often at the physician’s whim [6].

Qualitative illustration of the spectrum of factors associated with different forms of diabetes, including the variable age at onset, lack of obesity, the metabolic syndrome, genetic associations, different forms of immune changes, C-peptide secretion and need for insulin therapy
MODY as an exclusive disease
While type 1 diabetes is associated with childhood-onset diabetes but most commonly diagnosed in adults, maturity onset diabetes of the young (MODY) is clinically like adult-onset diabetes but most commonly diagnosed in children, responsible for some 5% of all diabetes cases in Europe in individuals under the age of 40 [3, 7]. In a given family, index cases must be young age at onset and not insulin dependent. At least 13 genes are implicated in MODY (also known as monogenic diabetes), and genetic types of MODY have distinct causes and different clinical features [3]. Even more infrequent are other monogenic causes of diabetes, such as neonatal diabetes and maternally inherited diabetes and deafness.
Type 2 diabetes as an inclusive disease
Common chronic diseases such as type 2 diabetes tend to have a complex aetiology that cannot be encapsulated by a single feature. Indeed, type 2 diabetes broadly encompasses any form of diabetes that is not type 1 diabetes, MODY or secondary diabetes [3]. Thus, the definition of type 1 diabetes is clinically exclusive, while the definition of type 2 diabetes is inclusive, without any defining feature beyond a lack of DAA.
Seeking disease identity
Genotypes as a cause of diabetes
Genetic aetiology is the hallmark of disease classification, and identifying monogenic diabetes has transformed our ability to classify the disease. Both MODY and the ‘metabolically obese–normal weight phenotype’, associated with a variant of the IRS1 gene, are such unusual examples of genetic ‘type 2 diabetes’ [3]. The most common form of MODY in Europe is caused by mutations in the HNF1A gene (encoding hepatocyte nuclear factor 1α) gene (HNF1A), and identifying such cases is important as insulin treatment is not essential and they are hypersensitive to sulfonylureas [7]. Monogenic diabetes also encompasses autoimmune diabetes, e.g. activating germline mutations in STAT3 [8], but these monogenic forms are rare.
The major diabetes subtypes are not monogenic but polygenic. Genome-wide association studies (GWAS) have revealed that each major type of diabetes has at least 50 associated genetic factors, so it follows that the complexity of genetic susceptibility confounds efforts to genetically classify diabetes [1, 2]. Nevertheless, genetic polymorphisms associated with type 1 diabetes relate to T lymphocyte function, not beta cell function, whereas in type 2 diabetes the converse is true [1, 9]. However, a TCF7L2 polymorphism associated with insulin secretion has a strong genetic association with type 2 diabetes and with LADA [3, 10] (Fig. 1). In type 2 diabetes there is no evidence that a single gene variant influences the disease sufficiently to warrant genotyping [9]. However, further studies are under way to identify rare variants that might contribute to genetic risk.
Phenotype does not define diabetes type
The value of defining diabetes type is to guide physicians towards appropriate treatment. Diabetes used to be defined by age at onset (childhood or adult-onset), clinical phenotype (lean or obese) or treatment (insulin-dependent or not insulin-dependent). But, while these categories broadly describe groups, they are insufficient to pigeonhole individuals. The metabolic syndrome, a proxy for insulin resistance, is found in about 30% of adult type 1 diabetic patients [11]; while some childhood-onset diabetes cases have MODY or type 2 diabetes, potentially not requiring insulin [3, 7, 8]. Even diabetic ketoacidosis is neither specific nor sensitive as a marker of type 1 diabetes in that ketoacidosis occurs infrequently and can be due to type 1b diabetes or ketosis-prone diabetes, primarily a variant of type 2 diabetes, reflecting how different diseases can share common phenotypes [12].
A proportion of patients (5–15%) have LADA and are initially not insulin-dependent but have both diabetes-associated HLA genetic risk and DAA [3–5, 13, 14]. Since DAA are characteristic of type 1 diabetes, irrespective of insulin treatment, it is an oxymoron to write about autoantibody-positive type 2 diabetes. Even patients with MODY have highly variable clinical phenotypes, some requiring insulin therapy, though most do not [3]. The burgeoning prevalence of obesity, reflecting a maladapted phenotypic plasticity, limits its value as a marker of type 2 diabetes, notably because it is increasingly found in children [2, 15].
Immunological markers and disease classification
There is extensive evidence of inflammatory changes associated with both major types of diabetes, some of which (e.g. islet antigen-specific DAA) delineate patients with more aggressive disease (Fig. 1).
There is no evidence that systemic inflammatory markers can identify each type of disease. While obesity is associated with activated macrophages and altered cytokine expression (e.g. TNF-α), monocyte gene expression is altered in both major diabetes types (including LADA as type 1 diabetes) [16]. Systemic inflammation, notably IL-6, the upstream regulator of C-reactive protein, is greater in type 2 diabetes than in type 1 diabetes (including LADA) [17]. But overlap is considerable and currently precludes any classification value.
T cells reactive to biochemically defined islet antigens and their immune mediators are associated with loss of beta cell function in type 1 diabetes [18]. Using a T cell proliferation assay, cellular immunoblotting (CI), validated to have excellent sensitivity and specificity for differentiating classical type 1 diabetes from normal controls [19], one of us (J. Palmer) initially observed that LADA patients were positive for CI, similar to type 1 diabetic patients [20]. Some patients with apparent ‘type 2 diabetes’ who were DAA negative were CI positive, and in these patients T cell reactivity to islet antigens was associated with more severe beta cell dysfunction, indeed, this association was stronger than that for autoantibody positivity [21]. While acknowledging that such immune reactivity to islet cell proteins must be validated, islet autoimmunity may contribute to a greater decline in beta cell function even in some type 2 diabetic patients (Table 1).
DAA remain the most striking markers in diabetes, even predicting autoimmune diabetes. The younger a child with the HLADR3-DQ2/DR4-DQ8 genotype, the higher the DAA risk [1]. DAA, especially to GAD65, largely define LADA, which is the most common form of type 1 diabetes [5]. Multiple autoantibodies or high titre DAA identifies patients with a more rapid loss of insulin secretory capacity, whether in prediabetes, childhood type 1 diabetes or LADA. Type 2 diabetes has no comparable disease-defining marker.
Endogenous insulin production and disease classification
Classification of diabetes in clinical practice relies on ‘salami slicing’ the continuum represented by the insulin secretion–insulin sensitivity relationship, so that those with severe loss of insulin secretion have type 1 diabetes, while those with low insulin sensitivity, but only moderate loss of insulin secretion, have type 2 diabetes. However, as with all organ-specific autoimmune diseases, damage to the target organ in type 1 diabetes is variable and highly age-dependent, being worse in childhood-onset than in adult-onset disease. Factors associated with this differential progression include age at diagnosis, HLA and non-HLA alleles, multiple DAA, high DAA titre or affinity, and T cell immune reactivity [1, 6, 13]. To distinguish patients with insulin-dependent diabetes from those who may not need insulin, e.g. MODY patients, a random serum C-peptide <200 pmol/l 3 years post diagnosis can help stratify cases for further study [22]. C-peptide is superior in children to age and obesity in discriminating between autoimmune and non-autoimmune diabetes, the areas under the receiver operating characteristic curves being 0.78, 0.68 and 0.66, respectively [15]. C-peptide levels also affect disease outcome. The Diabetes Control and Complications Trial found that 50% increased C-peptide secretion was associated with lower HbA1c, hypoglycaemia risk (down 8.2%) and risk of sustained retinopathy (down 25%) [23].
Differential metabolic progression and disease classification
Genes and rates of progression
Slower C-peptide loss in adults than children, whether with type 1 diabetes or type 2 diabetes, is probably due, in part, to a lower genetic load [1, 9]. In Europe and China, among patients with autoimmune diabetes, adults are less likely than children to have high-risk or moderate-risk HLA alleles [13, 24].
Age and rates of progression
Since beta cell loss is less severe in type 1 diabetes with increasing age at diagnosis, it follows that autoimmune diabetes in adults may be metabolically indistinguishable from type 2 diabetes, as recently found in LADA patients matched for BMI using a range of sophisticated tests [25]. However, the metabolic syndrome, a proxy for insulin insensitivity, is far more prevalent in type 2 diabetes (∼89%) than type 1 diabetes (∼32%), including LADA (∼42%) [11].
Immune reactivity influences clinical progression
Islet autoimmunity in early childhood, especially for multiple DAA or for insulinoma-associated antigen-2 autoantibodies, is strongly linked to rapid disease progression [1, 26]. In diabetic children, those with DAA had greater C-peptide decline (48%/year) than those without DAA-negative children (∼8%/year) [27]. Importantly, this decline was independent of demographics, HLA risk, BMI and insulin resistance; while in children with non-autoimmune diabetes, there was marked heterogeneity, suggesting a mixed group, with a mean rate of decline in C-peptide comparable to adult type 2 diabetes (∼8%/year).
We know little of the immune factors influencing disease progression in type 2 diabetes. However, a small pilot study of phenotypic ‘type 2 diabetes’ patients without either DAA or CI at baseline showed that a substantial percentage developed DAA and/or CI during 3 years’ follow-up, while positivity for CI was associated with more rapid beta cell decline [20, 21]. By implication, islet autoimmunity, distinct from LADA, may contribute to the pathophysiology of type 2 diabetes.
Altered glucose and disease progression
A recent study of type 2 diabetic patients diagnosed either by increased fasting glucose concentrations or 2 h post-load glucose concentrations, or both, had distinct cardiometabolic risk [28]. Those patients with both high fasting and 2 h glucose, compared with the other two groups, had higher BMI, higher HbA1c, greater insulin secretory decompensation and higher cardiovascular risk. By implication, diabetes and cardiovascular risk factor management should be more aggressive in the former.
Disease identity impacts disease management
For any given individual with diabetes, there is substantial variability in the nature of their disease, risk of complications and response to any given drug (Table 1, Fig. 1). More personalised disease targets, captured in recent management guidelines, matches personalised targets with personalised therapy, and, taken together, calls for more precise disease definition [29]. The management of both major types of diabetes are conventionally distinct; for one, drugs are introduced before insulin therapy and for the other the reverse, yet both involve a step-up policy in which the regimens are increasingly complex should targets not be achieved. But, with difficulty in classifying diabetes type comes uncertainty in deciding the best therapy. Patients with type 1 diabetes may not initially require insulin, especially when diagnosed as LADA. Insulin-treated patients, irrespective of diabetes type, may benefit from metformin, incretin-based agents and sodium-glucose transporter inhibitors [30]. For example, an incretin-based agent (exenatide) compared with placebo, when added to insulin glargine (A21Gly,B31Arg,B32Arg human insulin) therapy, lowered HbA1c by an additional 0.7% [31].
Autoimmunity impacts management strategy
Identifying DAA is a valuable test for disease management as they are characteristic of a form of autoimmune diabetes, especially in adults, which is often inappropriately treated and that requires more frequent review and a different perspective. Patients with autoimmune non-insulin-requiring diabetes, including LADA, should be started on insulin or an incretin-based therapy or, potentially, a combination of the two. Sulfonylurea treatment should likely be avoided, as patients on this class of drugs have a faster loss of insulin secretory capacity than when treated with insulin [32]. Autoimmunity does not equate with insulin therapy, as indicated by a Phase 2 study of adult patients with autoimmune diabetes; the combination of gliptin plus insulin was superior at sustaining C-peptide than insulin alone [33]. Although patients with typical type 1 diabetes at all ages presenting with ketoacidosis require immediate insulin therapy, not everyone with adult-onset autoimmune diabetes such as LADA, need progress to insulin therapy; in one study, 44% were still not on insulin after 12 years of disease [34]. However, disease progression to insulin treatment is more rapid in LADA than with type 2 diabetes, as reported by all major European LADA studies to date [4, 5, 35, 36]. Moreover, the former consistently have higher HbA1c levels than the latter (OR 1.8 in Sweden) [35]. Clearly, we do not manage these patients as well as those with non-insulin requiring non-autoimmune diabetes (i.e. type 2 diabetes). Finally, autoimmune diabetic patients, irrespective of age, have a substantially increased risk of thyroid and parietal cell autoimmunity [36]. The presence of DAA should encourage regular review of comorbidities and the quality of glycaemic control.
Non-autoimmune patients require variable management
As the loss of beta cell function is strongly age-dependent and young patients predominantly have autoimmune diabetes, therapeutic decisions are usually made without recourse to testing for DAA or C-peptide. This approach courts error, notably in children, in whom the prevalence of obesity and both type 1 and type 2 diabetes have increased dramatically. By measuring DAA initially and C-peptide subsequently, it is possible to identify, as appropriate, patients with MODY, type 2 diabetes in children, non-insulin-requiring autoimmune diabetes and non-autoimmune diabetes without the metabolic syndrome—potentially some 20–30% of all diabetes patients diagnosed under 40 years of age [37]. Such a definition also has utility for setting targets and therapy because cardiovascular complications differ between the major types of diabetes, e.g. guidelines offer statins to type 2 diabetes cases at an earlier age. A recent study of patients with apparent long-standing type 2 diabetes found that those with DAA or with low C-peptide did not respond as well as the remainder to a glucagon-like peptide 1 (GLP-1) agonist [38]. Patients with type 2 diabetes who do not have the metabolic syndrome (∼10%) will have a lower cardiovascular risk but are candidates for earlier insulin therapy [39]. Further studies of a range of factors implicated in disease progression should define those which merit intervention, including metabolic and oxidative stress, amyloid deposition and islet integrity associated with hyperglucagonaemia and diminished incretin effect [40] (see text box: Potential future studies in adult-onset diabetes). Even the titre of DAA can impact disease progression, e.g. in a Chinese study, low titre DAA patients had a similar rate of loss of C-peptide after diagnosis as did patients with type 2 diabetes [41, 42]. Irrespective of the type of diabetes, individuals with a high personal cardiovascular risk should be more aggressively treated for macrovascular risk factors. For example, central obesity, found in almost one-third of children with type 1 diabetes, was associated with early macrovascular risk markers [15], while a family history of diabetes in young type 2 diabetic patients is associated with an enhanced risk of decreased insulin sensitivity [43]. Research efforts could be focused on classification of diabetes subtypes in the context of non-autoimmune and non-insulin-requiring diabetes (see text box: Potential future studies in adult-onset diabetes).
Potential future studies in adult-onset diabetes | |
Genetics | Define features common to type 1 diabetes and type 2 diabetes using well-powered genome-wide association studies with well-defined phenotypes, as well as identifying rare variants through exome sequencing. Determine how genetic and epigenetic features in major types of diabetes relate to beta cell function, therapy and risk for late complications |
Natural history | Define time of onset and nature of biomarkers predictive of adult-onset autoimmune and non-autoimmune diabetes. Determine how these biomarkers relate to therapy and risk for late complications |
T cell, innate and B cell immunity | Identify novel biomarkers. Confirm and extend evidence that T and B cell autoimmunity and innate immunity may distinguish types of diabetes. Define their time of onset. Determine how these changes relate to therapy |
Metabolic function | Define metabolic changes that distinguish subtypes of diabetes, and identify appropriate therapy and potential outcome |
Pancreas | Define any distinction, including amyloid deposition, between major types of diabetes and pancreatic histology. Determine how these changes relate to therapy |
Treatment | Define optimum therapy for subtypes of diabetes |
Genetics and management strategy
The most dramatic impact of disease definition on disease management is shown by MODY (see text box: Matrix of treatment options based on laboratory tests and the potential value of each therapeutic decision). Before they are identified as having MODY, many young patients have their glucose levels managed with insulin when they could be switched to other agents including sulfonylureas [3, 7]. Moreover, their families can also carry and transmit the MODY-related gene. Hepatic nuclear factor 1α (HNF1A) MODY is one of the most common forms of MODY. A crossover trial of metformin and sulfonylureas in patients with type 2 diabetes and HNF1A MODY established that the latter have a fivefold greater response to the sulfonylurea gliclazide than to metformin (fasting plasma glucose reduction 4.7 vs 0.9 mmol/l) and a 3.9-fold greater response to gliclazide than patients with type 2 diabetes [7].
Matrix of treatment options based on laboratory tests and the potential value of each therapeutic decision | ||
Treatment | Indication | Potential value |
No treatment | • GCK (glucokinase) gene mutation | • Stop unnecessary therapy |
Sulfonylureas | • Neonatal diabetes (KCNJ11 [Kir6.2] gene mutation) | • Stop insulin, start high-dose sulfonylurea to improve glucose control and for potential neurological benefit |
Sulfonylureas | • MODY (HNF1A and HNF4A gene mutations) • CYP2C9 (Cytochrome P450 2C9 enzyme) inactivating gene mutations and TCF7L2 gene variants | • Stop insulin, start sulfonylureas to improve glucose control |
Sulfonylureas | • DAA-positive non-insulin requiring diabetes | • Contraindicated as more rapid disease progression |
Metformin | • SLC22A1, SLC22A2 and SLC47A1 gene variants | • Use metformin to improve glucose control (individuals are metformin-sensitive) |
Dipeptidyl peptidase-4 inhibitor | • DAA-positive non-insulin-requiring diabetes | • Preferable to sulfonylureas to improve glucose control |
Insulin | • Non-obese with low C-peptide and without the metabolic syndrome | • Consider early introduction of insulin therapy to improve glucose control |
Insulin | • DAA-positive non-insulin-requiring diabetes | • Use in preference to sulfonylureas to improve glucose control |
Patients vary in their tolerance and response to drugs (see text box: Matrix of treatment options based on laboratory tests and the potential value of each therapeutic decision). Medication–gene interactions for glycaemic outcomes has identified examples [44], including (1) metformin and SLC22A1, SLC22A2 and SLC47A1 loci; and (2) sulfonylureas and CYP2C9 and TCF7L2 loci. Sulfonylureas are primarily inactivated in the liver by the cytochrome P450 2C9 enzyme; as 6% of the population carry two reduced function polymorphisms, they should inactivate sulfonylureas poorly, and patients with these variants are three times more likely to have an HbA1c of below 53 mmol/mol (7%) [44]. Metformin is an organic cation actively transported by a variety of such transporters, which could account for the 34% heritability of glycaemic response to metformin [45]. In due course, the value of genetics should be realised not only in defining subtypes of diabetes, including MODY, but also by identifying differential disease outcomes and appropriate therapeutic strategies based on trials of highly selected cases.
Conclusion
We have outlined evidence that the use of laboratory tests could improve disease classification, definition of therapeutic targets and the efficacy of treatment for major types of diabetes. Clinical care is moving towards precision medicine derived from such laboratory investigations to enhance patient management
Abbreviations
- CI:
-
Cellular immunoblotting
- DAA:
-
Diabetes-associated autoantibodies
- HNF1A:
-
Hepatic nuclear factor 1α
- LADA:
-
Latent autoimmune diabetes in adults
References
Atkinson MA, Eisenbarth GS, Michels AW (2014) Type 1 diabetes. Lancet 383:69–82
Kahn SE, Cooper ME, del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383:1068–1083
Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L (2014) The many faces of diabetes: a disease with increasing heterogeneity. Lancet 383:1084–1094
Turner R, Stratton I, Horton V et al (1997) UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet 350:1288–1293
Hawa MI, Kolb H, Schloot N et al (2013) Adult-onset autoimmune diabetes in Europe is prevalent with a broad clinical phenotype: Action LADA 7. Diabetes Care 36:908–913
Brophy S, Yderstraede K, Mauricio D et al (2008) Time to insulin initiation cannot be used in defining latent autoimmune diabetes in adults. Diabetes Care 31:439–441
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT (2003) Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362:1275–1281
Flanagan SE, Haapaniemi E, Russell MA et al (2014) Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet 46:812–814
Morris AP, Voight BF, Teslovich TM et al (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44:981–990
Cervin C, Lyssenko V, Bakhtadze E et al (2008) Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes 57:1433–1437
Hawa MI, Thivolet C, Mauricio D et al (2009) Metabolic syndrome and autoimmune diabetes: Action LADA 3. Diabetes Care 32:160–164
Balasubramanyam A, Garza G, Rodriguez L et al (2006) Accuracy and predictive value of classification schemes for ketosis-prone diabetes. Diabetes Care 29:2575–2579
Zhou Z, Xiang Y, Ji L et al (2013) Frequency, immunogenetics, and clinical characteristics of latent autoimmune diabetes in China (LADA China study): a nationwide, multicenter, clinic-based cross-sectional study. Diabetes 62:543–550
Tuomi T, Carlsson A, Li H et al (1999) Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes 48:150–157
Cedillo M, Libman IM, Arena VC et al (2015) Obesity, islet cell autoimmunity, and cardiovascular risk factors in youth at onset of type 1 autoimmune diabetes. J Clin Endocrinol Metab 100:E82–E86
Padmos RC, Schloot NC, Beyan H et al (2008) Distinct monocyte gene-expression profiles in autoimmune diabetes. Diabetes 57:2768–2773
Pham MN, Hawa MI, Pfleger C et al (2011) Pro- and anti-inflammatory cytokines in latent autoimmune diabetes in adults, type 1 and type 2 diabetes patients: Action LADA 4. Diabetologia 54:1630–1638
Pfleger C, Meierhoff G, Kolb H, Schloot NC (2010) Association of T cell reactivity with beta-cell function in recent onset type 1 diabetes patients. J Autoimmun 34:127–135
Brooks-Worrell B, Warsen A, Palmer JP (2009) Improved T cell assay for identification of type 1 diabetes patients. J Immunol Methods 344:79–83
Brooks-Worrell BM, Reichow JL, Goel A, Ismail H, Palmer JP (2011) Identification of autoantibody-negative autoimmune type 2 diabetic patients. Diabetes Care 34:168–173
Goel A, Chiu H, Felton J, Palmer JP, Brooks-Worrell B (2007) T cell responses to islet antigens improves detection of autoimmune diabetes and identifies patients with more severe beta-cell lesions in phenotypic type 2 diabetes. Diabetes 56:2110–2115
Thanabalasingham G, Pal A, Selwood MP et al (2012) Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care 35:1206–1212
Lachin JM, McGee P, Palmer JP, DCCT/EDIC Research Group (2014) Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial. Diabetes 63:739–748
Howson JM, Rosinger S, Smyth DJ, Boehm BO, Todd JA (2011) Genetic analysis of adult-onset autoimmune diabetes. Diabetes 60:2645–2653
Juhl CB, Bradley U, Holst JJ, Leslie RD, Yderstraede KB, Hunter S (2014) Similar weight-adjusted insulin secretion and insulin sensitivity in short-duration late autoimmune diabetes of adulthood (LADA) and type 2 diabetes: Action LADA 8. Diabet Med 31:941–945
Lundgren M, Lynch K, Larsson C, Elding Larsson H (2015) Cord blood insulinoma-associated protein 2 autoantibodies are associated with increased risk of type 1 diabetes in the population-based Diabetes Prediction in Skåne study. Diabetologia 58:75–78
Dabelea D, Mayer-Davis EJ, Andrews JS et al (2012) Clinical evolution of beta cell function in youth with diabetes: the SEARCH for Diabetes in Youth study. Diabetologia 55:3359–3368
Færch K, Witte DR, Tabák AG et al (2013) Trajectories of cardiometabolic risk factors before diagnosis of three subtypes of type 2 diabetes: a post-hoc analysis of the longitudinal Whitehall II cohort study. Lancet Diabetes Endocrinol 1:43–51
Raz I, Riddle MC, Rosenstock J et al (2013) Personalized management of hyperglycemia in type 2 diabetes: reflections from a Diabetes Care Editors' Expert Forum. Diabetes Care 36:1779–1788
Ludvigsson J (2014) The latest pharmacotherapy options for type 1 diabetes. Expert Opin Pharmacother 15:37–49
Buse JB, Bergenstal RM, Glass LC et al (2011) Use of twice-daily exenatide in basal insulin-treated patients with type 2 diabetes: a randomized, controlled trial. Ann Intern Med 154:103–112
Brophy S, Davies H, Mannan S, Brunt H, Williams R (2011) Interventions for latent autoimmune diabetes (LADA) in adults. Cochrane Database Syst Rev (9) Art. No.: CD006165, DOI: 10.1002/14651858.CD006165.pub3
Zhao Y, Yang L, Xiang Y et al (2014) Dipeptidyl peptidase 4 inhibitor sitagliptin maintains beta-cell function in patients with recent-onset latent autoimmune diabetes in adults: one year prospective study. J Clin Endocrinol Metab 99:E876–E880
Hawa MI, Buchan AP, Ola T et al (2014) LADA and CARDS: a prospective study of clinical outcome in established adult-onset autoimmune diabetes. Diabetes Care 37:1643–1649
Andersen CD, Bennet L, Nyström L, Lindblad U, Lindholm E, Groop L, Rolandsson O (2011) Worse glycaemic control in LADA patients than in those with type 2 diabetes, despite a longer time on insulin therapy. Diabetologia 56:252–258
Zampetti S, Capizzi M, Spoletini M et al (2012) GADA titer-related risk for organ-specific autoimmunity in LADA subjects subdivided according to gender (NIRAD study 6). J Clin Endocrinol Metab 97:3759–3765
Thunander M, Törn C, Petersson C, Ossiansson B, Fornander J, Landin-Olsson M (2012) Levels of C-peptide, body mass index and age, and their usefulness in classification of diabetes in relation to autoimmunity, in adults with newly diagnosed diabetes in Kronoberg, Sweden. Eur J Endocrinol 166:1021–1029
Jones AG, McDonald TJ, Shields BM et al (2015) Markers of β-cell failure predict poor glycemic response to GLP-1 receptor agonist therapy in type 2 diabetes. Diabetes Care. doi:10.2337/dc15-0258
Home P, Riddle M, Cefalu WT et al (2014) Insulin therapy in people with type 2 diabetes: opportunities and challenges? Diabetes Care 37:1499–1508
Halban PA, Polonsky KS, Bowden DW et al (2014) β-Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37:1751–1758
Laugesen E, Østergaard JA, Leslie RD, Danish Diabetes Academy Workshop and Workshop Speakers (2015) Latent autoimmune diabetes of the adult: current knowledge and uncertainty. Diabet Med 32:843–852
Liu L, Li X, Xiang Y et al (2015) Latent autoimmune diabetes in adults with low-titer GAD antibodies: similar disease progression with type 2 diabetes: a nationwide, multicenter prospective study (LADA China Study 3). Diabetes Care 38:16–21
Arslanian SA, Bacha F, Saad R, Gungor N (2005) Family history of type 2 diabetes is associated with decreased insulin sensitivity and an impaired balance between insulin sensitivity and insulin secretion in white youth. Diabetes Care 28:115–119
Huang C, Florez JC (2011) Pharmacogenetics in type 2 diabetes: potential implications for clinical practice. Genome Med 3:76
Zhou K, Donnelly L, Yang J et al (2014) Heritability of variation in glycaemic response to metformin: a genome-wide complex trait analysis. Lancet Diabetes Endocrinol 2:481–487
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leslie, R.D., Palmer, J., Schloot, N.C. et al. Diabetes at the crossroads: relevance of disease classification to pathophysiology and treatment. Diabetologia 59, 13–20 (2016). https://doi.org/10.1007/s00125-015-3789-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3789-z