
Abstract
Aims/hypothesis
Generation of reduction equivalents is a prerequisite for nutrient-stimulated insulin secretion. Mitochondrial succinate dehydrogenase (SDH) fulfils a dual function with respect to mitochondrial energy supply: (1) the enzyme is part of mitochondrial respiratory chains; and (2) it catalyses oxidation of succinate to fumarate in the Krebs cycle. The aim of our study was to elucidate the significance of SDH for beta cell stimulus-secretion coupling (SSC).
Methods
Mitochondrial variables, reactive oxygen species (ROS) and cytosolic Ca2+ concentration ([Ca2+]c) were measured by fluorescence techniques and insulin release by radioimmunoassay in islets or islet cells of C57Bl/6N mice.
Results
Inhibition of SDH with 3-nitropropionic acid (3-NPA) or monoethyl fumarate (MEF) reduced glucose-stimulated insulin secretion. Inhibition of the ATP-sensitive K+ channel (KATP channel) partly prevented this effect, whereas potentiation of antioxidant defence by superoxide dismutase mimetics (TEMPOL and mito-TEMPO) or by nuclear factor erythroid 2-related factor 2 (Nrf-2)-mediated upregulation of antioxidant enzymes (oltipraz, tert-butylhydroxyquinone) did not diminish the inhibitory influence of 3-NPA. Blocking SDH decreased glucose-stimulated increase in intracellular FADH2 concentration without alterations in NAD(P)H. In addition, 3-NPA and MEF drastically reduced glucose-induced hyperpolarisation of mitochondrial membrane potential, indicative of decreased ATP production. As a consequence, the glucose-stimulated rise in [Ca2+]c was significantly delayed and reduced. Acute application of 3-NPA interrupted glucose-driven oscillations of [Ca2+]c. 3-NPA per se did not elevate intracellular ROS, but instead prevented glucose-induced ROS accumulation.
Conclusions/interpretation
SDH is an important regulator of insulin secretion and ROS production. Inhibition of SDH interrupts membrane-potential-dependent SSC, pointing to a pivotal role of mitochondrial FAD/FADH2 homeostasis for the maintenance of glycaemic control.
Similar content being viewed by others
Introduction
Nutrient-dependent activation of mitochondria is essential for physiological stimulation of insulin secretion. Succinate dehydrogenase (SDH), the catalytic subunit of complex II, adopts an exceptional position for mitochondrial regulation as this enzyme participates in both the Krebs cycle (tricarboxylic acid [TCA] cycle) and the respiratory chain. By SDH-dependent generation of reduction equivalents, metabolic flux via the TCA cycle is directly linked to oxidative phosphorylation. SDH catalyses the oxidation of succinate to fumarate, and the activity of the enzyme increases with a rise in intra-mitochondrial succinate concentration. This process is accompanied by production of FADH2. As expected, stimulation of beta cells with metabolic substrates that activate the TCA cycle (and thus complex II) is accompanied by a drop in FAD autofluorescence indicative of a shift in the balance of reduction equivalents to increased FADH2 concentration [1]. SDH activity has been measured in homogenates of rat and mouse pancreatic islets [2, 3]. As its activity in islets is ~85% lower than that in liver or exocrine pancreatic tissue [2], it may represent one of the rate limiting steps of insulin secretion.
Mutations of SDH have been linked to severe diseases, such as neuronal and neuroendocrine tumours [4–6]. For Huntington’s disease (HD), defects in complex II, III and IV of the respiratory chain have been shown in post-mortem studies [7]. Decreased expression of the FAD-binding subunit and the iron-sulphur subunit of SDH has been found in striatal biopsies of HD patients [8].
Changes in SDH have also been linked to diabetes. An influence of metabolic disorders on human SDH has been observed in muscle biopsies of patients with type 2 diabetes [9, 10]. These studies show reduced SDH activity in patients with type 2 diabetes. SDH activity is also markedly reduced in skeletal muscle cells of animals with experimentally induced or genetically encoded diabetes [11, 12]. Mitochondria of cardiomyocytes in rats with streptozotocin-induced diabetes show decreased oxygen consumption in comparison with healthy controls, resulting from reduced activity of complexes I and II. This mitochondrial dysfunction involved oxidative damage of the FAD-binding subunit of SDH, which could be reversed by insulin treatment [13].
With regard to diabetes-induced alterations of mitochondrial enzymes in pancreatic islets, only limited data are available. A case report of a patient with mitochondrial diabetes has described a marked decrease in the number of beta cells accompanied by increased beta cell SDH staining in histological samples [14]. MacDonald and colleagues observed reduced activity of mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in islets of Zucker diabetic fatty (ZDF) rats, whereas SDH activity appeared to be unchanged [15].
Leibowitz et al [16] showed that for rat islets, SDH inhibition by 3-nitropropionic acid (3-NPA) or fumaric acid monoethyl ester (MEF) impaired insulin secretion by lowering gene transcription of preproinsulin and synthesis of proinsulin [16]. As a structural analogue of succinate, 3-NPA covalently and irreversibly binds to the active site of the enzyme thereby preventing complex-II-mediated FADH2 generation [17]. In neurological research, in which 3-NPA is widely used to damage dopaminergic neurons in animal models of HD or Parkinson’s disease [18, 19], it has been suggested that the mechanism underlying 3-NPA toxicity involves increased production of reactive oxygen species (ROS) [20].
To elucidate the role of SDH in beta cell function and to uncover the mechanisms by which the enzyme interferes with stimulus-secretion coupling (SSC), we investigated the influence of 3-NPA and MEF on mitochondrial activity, cytosolic Ca2+ concentration ([Ca2+]c), insulin release and ROS generation.
Methods
Animals and islet preparation
Islets of Langerhans or clusters of islet cells were isolated from adult C57Bl/6N mice (Janvier, Saint-Berthevin, France and Charles River, Sulzfeld, Germany). The principles of laboratory animal care were followed according to German laws. Isolation and culture were performed as described previously [21].
FAD and NAD(P)H autofluorescence
Changes in the concentration of reduction equivalents were monitored by determination of FAD and NAD(P)H autofluorescence (465 nm and 360 nm excitation wavelength, respectively). Fluorescence emission was filtered (LP515 filter) and measured by a digital camera. FAD autofluorescence was corrected for photobleaching by subtraction of a double-exponential fit curve.
Mitochondrial membrane potential, ROS and [Ca2+]c
Mitochondrial membrane potential (Δψ m) was recorded after loading the cells with rhodamine-123 (10 μg/ml, 15 min, 37°C). For quantification, data were normalised to maximal mitochondrial depolarisation induced by the uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 0.5 μmol/l). ROS accumulation was determined by oxidation of 2′,7′-dichlorodihydrofluorescein-diacetate (DCDHF-DA, 20 μmol/l, 15 min, 37°C). For determination of [Ca2+]c, cells were loaded with fura-2 acetoxymethylester (fura-2AM, 5 μmol/l, 30 min, 37°C). Fluorescence was excited at 480 nm (Δψ m and ROS) or 340 nm and 380 nm ([Ca2+]c), respectively. Fluorescence emission was filtered (LP515 filter) and measured by a digital camera.
Insulin secretion
After preparation, islets were kept overnight in RPMI medium with 11.1 mmol/l glucose. Details for steady-state incubations have been described previously [21]. Batches of five islets were incubated for 60 min at 37°C with the indicated substances. For perifusions, 50 islets were placed in a bath chamber and treated with 3 mmol/l glucose for 60 min prior to the experiment.
Solutions and chemicals
Δψ m and [Ca2+]c were measured at 37°C in a solution of pH 7.4 containing (mmol/l): 140 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES, and glucose as indicated. The medium for insulin secretion contained (mmol/l): 122 NaCl, 4.8 KCl, 2.5 CaCl2, 1.1 MgCl2 and 10 HEPES (pH 7.4). In the pre-incubation experiments, 3-NPA was added to the incubation buffer for 60 min. 3-NPA was removed during loading with fluorescence dyes. In the Ca2+ experiments (Fig. 4a–e), pre-incubation with 3-NPA was reduced to 45 min followed by 30 min loading with fura-2AM (without 3-NPA). The shortening of the pre-incubation period was intended to standardise Δψ m and [Ca2+]c experiments to a period of 75 min in buffer with 0.5 mmol/l glucose.
RPMI 1640, fetal calf serum and penicillin/streptomycin were provided by Invitrogen (Karlsruhe, Germany). Fura-2AM was from Biotrend (Köln, Germany). All other chemicals were from Sigma-Aldrich (Taufkirchen, Germany), Roth (Karlsruhe, Germany) or Merck (Darmstadt, Germany).
Presentation of results
Data were collected from islets or islet cells of at least three different mice for each series of experiments. Values are given as means ± SEM. Autofluorescence data in Fig. 1a were averaged before and after the 3-NPA-induced incremental increase in FAD autofluorescence. In Fig. 1b–g, NAD(P)H and FAD autofluorescence was analysed by comparing the averaged data directly before elevating glucose concentration to the fluorescence values reached after 15 min in 15 mmol/l glucose. Evaluation procedures for Δψ m or [Ca2+]c are described in the respective figure legends. Statistical significance was assessed by ANOVA followed by the Student–Newman–Keuls post hoc test or by the Student’s t test. A value of p ≤ 0.05 was considered to be significant.

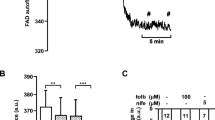
Effects of 3-NPA on reduction equivalents. Increase in FAD autofluorescence of isolated beta cells by acute application of 3-NPA (a). The trace shows a representative recording and the histogram summarises the data from all experiments. Compared with controls (b) the drop in FAD autofluorescence caused by rising glucose concentration from 0.5 (G 0.5) to 15 (G 15) mmol/l is drastically reduced after pre-incubation with 3-NPA (c) or in the presence of MEF (d). (e, f) The glucose-mediated rise in NAD(P)H fluorescence is not affected by pre-incubation with 3-NPA. Grey traces: cells were pre-incubated with 3-NPA for 1 h or perifused with MEF as indicated in the figure. Black traces: representative control experiments without SDH inhibition. (g) Summarised results of (b–f). The bars illustrate the amplitude of glucose-induced changes in autofluorescence. Negative values indicate a reduction in fluorescence intensity. The number of cells is given in the bars in each diagram. **p ≤ 0.01, ***p ≤ 0.001
Results
Inhibition of SDH reduces insulin secretion
In isolated beta cells stimulated with 15 mmol/l glucose, the specific SDH inhibitor 3-NPA [17] (1 mmol/l) irreversibly increased FAD autofluorescence, thereby indicating reduced FAD recycling via complex II (Fig. 1a). This elevation of FAD fluorescence was not prevented by dithioerythritol (DTE, 5 mmol/l), suggesting that blockade of SDH was not achieved by protein oxidation (elevation of FAD fluorescence was 18.1 ± 2.3 arbitrary fluorescence units (AU) [n = 14] with 3-NPA vs 18.1 ± 1.2 AU [n = 26] with 3-NPA and DTE, NS, data not shown). The effect of 3-NPA on FAD fluorescence was also tested with monomethyl succinate as substrate. Similar to the results obtained with glucose, the decrease in FAD fluorescence induced by methyl succinate (10 mmol/l) was countervailed by 3-NPA (n = 11; data not shown).
In cells pretreated with 3-NPA, the reduction of FAD induced by elevating glucose concentration from 0.5 to 15 mmol/l was significantly less pronounced than in control experiments without SDH inhibition (Fig. 1b, c and the right columns in Fig. 1g). As another tool to reduce SDH activity, the monoethyl ester of the Krebs cycle intermediate fumarate was used. In the presence of MEF (10 mmol/l), the glucose-induced reduction of FAD fluorescence was abolished (Fig. 1d, g). Pre-incubation with 3-NPA had no effect on the glucose-mediated rise in NAD(P)H autofluorescence (Fig. 1e, f and the left columns in Fig. 1g).
The influence of 3-NPA and MEF on the kinetics of insulin secretion was investigated by perifusion experiments. In agreement with the dose-dependent increase in FAD fluorescence with increasing concentrations of 3-NPA (Fig. 2a), insulin release decreased in response to rising concentrations of 3-NPA. Moderate inhibition of SDH by 0.1 mmol/l 3-NPA (Fig. 2b) or 5 mmol/l MEF (Fig. 2d) mainly affected the second phase of insulin release, whereas with increasing SDH inhibition (1 mmol/l 3-NPA, Fig. 2c) the first phase was also delayed and drastically reduced. Insulin release could be partly restored by the ATP-sensitive K+ channel (KATP channel) inhibitor gliclazide (10 μmol/l; Fig. 2e). Gliclazide had no effect on FAD autofluorescence (FAD fluorescence in 15 mmol/l glucose was 327 ± 10 AU, and with the addition of 10 μmol/l gliclazide it was 327 ± 10 AU [n = 9], NS, data not shown), which excludes any influence of gliclazide on FAD/FADH2 balance.

Concentration-dependent alterations of FAD and insulin release by 3-NPA and MEF. Acute application of 3-NPA dose-dependently increases FAD fluorescence (a). 3-NPA (b, c) and MEF (d) decrease insulin secretion. In (b–d) isolated islets were perifused with glucose as indicated by the horizontal bars. Grey traces: 3-NPA or MEF was added to the perifusion solution as denoted by the arrow head. Black traces: controls without SDH blocker. (e) Inhibition of insulin secretion is partly prevented by gliclazide. (f) 3-NPA has no effect on basal insulin release but reduces secretion stimulated by 8, 15 and 25 mmol/l glucose. In (e) and (f) islets were incubated for 60 min with the indicated substances. Number of cells (a) and islet preparations (e, f) is given in the bars in each diagram. The traces (b–d) summarise three to four (b, c) and five (d) individual experiments with independent islet preparations. **p ≤ 0.01, ***p ≤ 0.001; ǂ ‡significant difference to the corresponding glucose concentration without 3-NPA, p ≤ 0.05; †significant difference to 3 mmol/l glucose, p ≤ 0.001
To further elucidate the contribution of SDH to glucose-mediated insulin release, 3-NPA was tested with various glucose concentrations (Fig. 2f). 3-NPA had no effect on basal insulin release but decreased secretion stimulated by 8, 15 and 25 mmol/l glucose. With respect to FAD fluorescence, the acute effect of 1 mmol/l 3-NPA was similar in the presence of 15 and 25 mmol/l glucose (3-NPA-induced increase in FAD fluorescence: 14 ± 2 AU with 15 mmol/l glucose [n = 12] vs 9 ± 2 AU with 25 mmol/l glucose [n = 9], NS, data not shown).
Blocking SDH affects Δψ m
The effect of SDH inhibition on Δψ m was determined. Beta cells were stimulated by elevating glucose concentration from 0.5 to 15 mmol/l. This manoeuvre resulted in a hyperpolarisation of Δψ m, indicating activation of proton motive force and ATP production (Fig. 3a). In agreement with reduction of glucose-stimulated insulin release, the effect of glucose on Δψ m was significantly lower in beta cells pre-incubated with 3-NPA or treated with MEF than controls (Fig. 3a). To evaluate whether 3-NPA had an acute effect on Δψ m as observed for FAD the SDH inhibitor was applied to stimulated beta cells and changes in Δψ m were monitored. In this series of experiments, 3-NPA induced a depolarisation of Δψ m (Fig. 3b–e) that was independent of [Ca2+]c.

Influence of 3-NPA and MEF on Δψ m. (a) The hyperpolarisation of Δψ m caused by elevating glucose concentration from 0.5 (G 0.5) to 15 (G 15) mmol/l is reduced in a beta cell pretreated with 3-NPA for 1 h compared with a control cell (representative experiments). At the end of each experiment FCCP (0.5 μmol/l) was added. For evaluation, data were normalised to the maximal depolarisation achieved with FCCP. The figure summarises the normalised values for glucose-induced changes in Δψ m and the effect of 3-NPA and MEF. (b, c) Acute addition of 3-NPA leads to depolarisation of beta cell mitochondria. Nifedipine (5 μmol/l) was added to inhibit Ca2+ influx via L-type Ca2+ channels, inducing a small hyperpolarisation of Δψ m. (b) In a time-matched control condition, Δψ m remained stable in the presence of nifedipine throughout the experiment except for a slow and continuous decline in fluorescence ascribed to photobleaching. (c) 3-NPA depolarises Δψ m. Areas in grey and time scale denote the parts of the experiments that were evaluated in (d). (d) Summary of 3-NPA-induced changes in Δψ m (grey squares) vs control (black circles) after 5 and 10 min, expressed as absolute values. (e) Amplitude of changes in fluorescence 10 min after addition of 3-NPA (grey bar) vs control (Cont.; black bar). ‘0 min’ denotes Δψ m in the presence of 15 mmol/l glucose + nifedipine directly before changing bath solution. The number of cells tested is given in the bars in each diagram. *p ≤ 0.05, ***p ≤ 0.001
Effects of 3-NPA on [Ca2+]c
The results described above suggest that SDH inhibition results in less ATP generation. Consequently, 3-NPA should affect glucose-induced alterations in [Ca2+]c. To validate this hypothesis, beta cells were pre-incubated with 3-NPA. Thereafter glucose was elevated from 0.5 to 15 mmol/l and the time interval until [Ca2+]c started to rise was determined. In addition, the AUCCa was calculated for the first 10 min after increasing glucose concentration (marked as “tR” in Fig. 4a, b). These experiments showed that SDH inhibition by 3-NPA prolonged the response time of [Ca2+]c to 15 mmol/l glucose, thereby reducing the AUCCa (Fig. 4a–d). To test whether the decrease in [Ca2+]c is solely due to the lag in onset or also due to reduced Ca2+ influx during continuous stimulation, AUCCa was calculated for 5 min after the first rise in [Ca2+]c (Fig. 4e). This evaluation revealed that Ca2+ influx was significantly lower in 3-NPA treated vs control cells. The acute effect of 3-NPA on glucose-driven oscillations of [Ca2+]c was also determined. Addition of 3-NPA to glucose-stimulated beta cells terminated oscillations and reduced [Ca2+]c to basal values. The cells were still responsive to gliclazide (Fig. 4f).

3-NPA-induced alterations in [Ca2+]c. (a, b) Rise of [Ca2+]c of isolated beta cells in response to a change in perifusion solution from 0.5 (G 0.5) to 15 (G 15) mmol/l glucose. In (b) cells were pre-incubated with 3-NPA (1 mmol/l) for 45 min. The dotted lines in (a) and (b) indicate the response time (tR), i.e. the time interval from changing glucose concentration to the first rise in [Ca2+]c. (c–e) Evaluation of the example data shown in (a) and (b). (c) Differences in the response time tR, in (d) AUCCa is calculated for the first 10 min in 15 mmol/l glucose, and in (e) AUCCa is evaluated for 5 min after the onset of glucose-mediated increase in [Ca2+]c. (f) Acute application of 3-NPA inhibits the regular oscillations of whole islets exposed to 15 mmol/l glucose. Islets are responsive to gliclazide (10 μmol/l). One representative experiment is shown in the trace and the histogram summarises the data. The number of cells/islets tested is given in the bars in each diagram. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Pre-incub., pre-incubation; F, fluorescence
Influence of sulfonylureas and antioxidants on insulin release of 3-NPA treated islets
It has been suggested that interruption of respiratory chains at complex II leads to increased production of radicals due to incomplete electron transfer [22]. We investigated whether activation of antioxidant defence mechanisms effectively counteracts the inhibition of insulin secretion caused by 3-NPA. The SOD mimetic TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl; 1 mmol/l) and its mitochondrially targeted derivative mito-TEMPO ((2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenyl-phosphonium chloride; 10 μmol/l) [23–25] were applied during the steady-state incubation with 3-NPA. Neither drug protected against the impairment of glucose-stimulated insulin secretion (Fig. 5a, b). General upregulation of antioxidant enzymes by overnight incubation with the nuclear factor erythroid 2-related factor 2 (Nrf-2) activators oltipraz (10 μmol/l) and tert-butylhydroxyquinone (tBHQ, 30 μmol/l), or with the sulfonylurea gliclazide (10 μmol/l), which has also been reported to increase activity of antioxidant enzymes after long-term incubation [21], did not prevent the inhibitory effect of 3-NPA (Fig. 5c–e). Figure 5f clearly illustrates for oltipraz that the protocols used above efficiently protect against oxidative stress. For TEMPOL we have recently shown protection of islets against H2O2-mediated impairment of electrical activity and insulin secretion [26].

Inefficacy of mimetics and activators of antioxidant defence pathways on the secretion-inhibiting effect of 3-NPA. (a–e) 3-NPA was present during the 60 min incubation period in which insulin release was determined. (a, b) Islets were treated with TEMPOL or mito-TEMPO during the steady-state incubations. (c, d) For stimulation of Nrf-2, islets were incubated overnight in medium containing oltipraz or tBHQ. The drugs were also present during the 60 min steady-state incubation. (e) Islets were incubated with gliclazide (10 μmol/l) overnight to upregulate antioxidant enzymes. To exclude direct stimulation of insulin release by KATP channel inhibition, the drug was removed prior to the 60 min incubation. None of these experiments prevented 3-NPA-induced reduction of insulin secretion. (f) Control experiment showing that overnight incubation with oltipraz is able to prevent H2O2-induced inhibition of insulin release. The number of individual experiments is given in the bars in each diagram. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Incub., incubation
Influence of SDH inhibition on mitochondrial ROS generation
To investigate whether SDH inhibition in beta cells is accompanied by changes in ROS, intracellular ROS accumulation was analysed by monitoring DCDHF oxidation. Beta cells were incubated with 15 mmol/l glucose or with 15 mmol/l glucose and 1 mmol/l 3-NPA. Interestingly, short-term inhibition (15 min) of SDH by 3-NPA had no effect on ROS accumulation (Fig. 6a). After a 60 min incubation period with 3-NPA, intracellular ROS even decreased by ~40% (Fig. 6b). For comparison, ROS accumulation in unstimulated beta cells was also determined in this series of experiments. The data indicate that blocking SDH largely prevents endogenous, glucose-stimulated ROS production.

Determination of ROS accumulation in islet cell clusters. Cells were incubated in the presence of 15 mmol/l glucose or 15 mmol/l glucose + 1 mmol/l 3-NPA for 15 min (a) or 60 min (b) prior to determination of 2′,7′-dichlorofluorescein (DCF) fluorescence. Glucose-mediated elevation of intracellular ROS concentration was inhibited by 3-NPA. The number of cells tested is given in the bars in each diagram. (a, b) Note that the different fluorescence levels in the presence of 15 mmol/l glucose result from different incubation times. ***p ≤ 0.001
Discussion
Mitochondrial enzymes and insulin secretion
The co-action of cytosolic and mitochondrial enzymes involved in the generation of reduction equivalents and ATP is essential for pancreatic beta cells. During glycolysis, NADH is provided by substrate-level phosphorylation of glyceraldehyde-3-phosphate by glyceraldehyde-3-phosphate dehydrogenase (GAPDH). It has been shown that shuttling glycolytic NADH into the mitochondria is indispensable for glucose-mediated SSC [27]. In the TCA cycle, substrate turnover generates NAD(P)H, and SDH regulates formation of FADH2.
Consequently, impairment of mitochondrial enzymes is expected to affect the SSC of beta cells. However, as experiments inhibiting activity of the TCA cycle are not well correlated to changes in glucose-mediated insulin secretion [27, 28], the significance of the different enzymatic steps for regulation of insulin release is still unclear.
Here, we show that inhibition of SDH by 3-NPA or MEF markedly reduces glucose-stimulated insulin secretion. Kinetic measurements reveal inhibition of both phases of insulin release with the second phase of insulin secretion being affected first by reduced FADH2 supply. The effect of 3-NPA was largely prevented by the sulfonylurea gliclazide, emphasising that impairment of SSC upstream to KATP channel closure is the main mechanism for 3-NPA toxicity. To what extent modulation of SDH activity influences development of type 2 diabetes remains to be elucidated by further studies.
Mitochondrial function and [Ca2+]c
To further investigate how SDH inhibition interferes with SSC of beta cells we analysed glucose-induced alterations of [Ca2+]c. 3-NPA retarded the glucose-mediated elevation of [Ca2+]c. In addition, once started, Ca2+ influx was significantly lower in 3-NPA-pre-incubated beta cells than in untreated controls.
In astrocytes, short-term application of 3-NPA steadily increased [Ca2+]c [29]. Consequently, we tested whether acute addition of 3-NPA affects the typical oscillatory pattern of [Ca2+]c occurring during continuous stimulation with glucose. We observed a reduction of [Ca2+]c to basal values (Fig. 4f), supporting our hypothesis that inhibition of SDH reduces ATP synthesis. Subsequent opening of KATP channels leads to an interruption of the SSC. In agreement with the data obtained for insulin secretion, acute application of gliclazide effectively counteracted the influence of 3-NPA on [Ca2+]c.
To characterise the effect of SDH inhibition on mitochondrial function, we analysed FADH2, NAD(P)H and Δψ m. As expected, FAD autofluorescence was irreversibly increased by 3-NPA. Importantly, a glucose-stimulated decrease in FAD fluorescence (which is indicative of an increase in FADH2 concentration) was largely absent in beta cells pretreated with 3-NPA and in cells treated with MEF. By contrast, the lack of effect of SDH inhibition on glucose-mediated changes in NAD(P)H is astonishing as NADH generating steps of the Krebs cycle should be affected by blocking SDH. However, one must keep in mind that fluorescence-optical determination of NAD(P)H covers several processes (Krebs cycle, glycolysis and rate of respiration). Obviously, these pathways balance each other without any consequence on the net effect. The question arising from these results is whether altered FAD/FADH2 balance without any changes in NAD(P)H is sufficient to influence mitochondrial ATP synthesis. In neurons it was reported that inhibition of SDH by 3-NPA reduced ATP by ~25% within the following 2 h. This effect was not accompanied by apoptosis, indicating that the drop in ATP concentration resulted from impaired cell function and not from loss of cell mass [30]. In our experiments, changes in Δψ m were determined and taken as a measure of altered activity of mitochondrial respiratory chains. Our data show that SDH inhibition drastically reduces the ability of glucose to hyperpolarise Δψ m (Fig. 3a). Compared with the effects observed by Leibowitz et al [16], who tested 3-NPA in rat islets and observed complete inhibition of glucose-induced hyperpolarisation of Δψ m, the effect of 3-NPA was less pronounced in our experiments. This discrepancy might be due to the experimental design: we switched between 15 and 0.5 mmol/l glucose whereas Leibowitz et al [16] compared the effect of 3-NPA on Δψ m at 16.7 and 3 mmol/l glucose (levels at which respiratory chains are already partly activated).
To further analyse how 3-NPA influences Δψ m, we tested whether 3-NPA-induced inhibition of mitochondrial respiratory chains requires Ca2+ influx. In experiments performed in the presence of an L-type Ca2+ channel blocker, we observed a gradual depolarisation of Δψ m, indicating Ca2+-independent action of 3-NPA on mitochondria.
3-NPA and oxidative stress
Elevated ROS levels in response to 3-NPA have been reported for different cells [20, 31] and isolated mitochondria [22]. Bacsi and colleagues [22] demonstrated that the influence of 3-NPA on ROS critically depends on the cell type: while 3-NPA augmented intracellular ROS in cell lines of alveolar epithelial cells and parenchymal liver cells it was ineffective in macrophages. For pheochromocytoma cells, it has been shown that short-term application of 3-NPA induces a concentration-dependent elevation of ROS [32], whereas others did not detect any effect in the same cell line [33].
As oxidative stress plays an important role in beta cell failure during development and progression of type 2 diabetes [34], we monitored ROS accumulation. Our data demonstrate that 3-NPA had no acute effect on intracellular ROS and even reduced ROS concentration after an exposure time of 1 h (Fig. 6). In line with this result, the rise in FAD (which is indicative of SDH inhibition by 3-NPA) was not prevented by the thiol-group reducing agent DTE. The fact that different antioxidant strategies could not prevent the inhibitory effect of 3-NPA on insulin release is also in agreement with a mechanism that does not involve elevated ROS levels.
There is increasing evidence that a certain amount of ROS is necessary to maintain the physiologic function of pancreatic islets. Low concentrations of H2O2 have been shown to augment insulin release of unstimulated islets. Moreover, the increase in ROS coinciding with glucose stimulation seems to be a prerequisite for adequate secretion [35]. Interestingly, a 1 h application of 3-NPA completely prevented glucose-mediated ROS elevation. At present, we do not know the extent to which the elimination of glucose-triggered ROS production contributes to the inhibitory effect of 3-NPA on insulin release; however, it is tempting to speculate that in addition to decreased ATP production, the omission of other signalling factors (such as mitochondrially derived H2O2) takes part in this scenario.
In summary, our data demonstrate that inhibition of SDH is sufficient to drastically weaken the efficiency of glucose to hyperpolarise beta cell mitochondria. The resulting decrease in ATP synthesis interrupts SSC, and consequently reduces insulin secretion.
Abbreviations
- Δψ m :
-
Mitochondrial membrane potential
- AU:
-
Arbitrary fluorescence units
- [Ca2+]c :
-
Cytosolic Ca2+ concentration
- DCDHF:
-
2′,7′-Dichlorodihydrofluorescein
- DTE:
-
Dithioerythritol
- FCCP:
-
Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- Fura-2AM:
-
Fura-2 acetoxymethylester
- HD:
-
Huntington’s disease
- KATP channel:
-
ATP-sensitive K+ channel
- mito-TEMPO:
-
(2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenyl-phosphonium chloride
- MEF:
-
Monoethyl fumarate
- 3-NPA:
-
3-Nitropropionic acid
- Nrf-2:
-
Nuclear factor erythroid 2-related factor 2
- ROS:
-
Reactive oxygen species
- SDH:
-
Succinate dehydrogenase
- SSC:
-
Stimulus-secretion coupling
- tBHQ:
-
Tert-butylhydroxyquinone
- TEMPOL:
-
4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
- TCA:
-
Tricarboxylic acid
References
Van de Winkel M, Pipeleers D (1983) Autofluorescence-activated cell sorting of pancreatic islet cells: purification of insulin-containing B cells according to glucose-induced changes in cellular redox state. Biochem Biophys Res Commun 114:835–842
Lenzen S, Panten U (1983) Characterization of succinate dehydrogenase and alpha-glycerophosphate dehydrogenase in pancreatic islets. Biochem Med 30:349–356
Rasschaert J, Malaisse WJ (1993) Hexose metabolism in pancreatic islets: succinate dehydrogenase activity in islet homogenates. Cell Biochem Funct 11:155–158
Lendvai N, Pawlosky R, Bullova P et al (2014) Succinate-to-fumarate ratio as a new metabolic marker to detect the presence of SDHB/D-related paraganglioma: initial experimental and ex vivo findings. Endocrinology 155:27–32
Astuti D, Douglas F, Lennard TW et al (2001) Germline SDHD mutation in familial phaeochromocytoma. Lancet 357:1181–1182
Doyle LA, Hornick JL (2014) Gastrointestinal stromal tumours: from KIT to succinate dehydrogenase. Histopathology 64:53–67
Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH (1996) Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39:385–389
Benchoua A, Trioulier Y, Zala D et al (2006) Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell 17:1652–1663
Oberbach A, Bossenz Y, Lehmann S et al (2006) Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care 29:895–900
He J, Watkins S, de Kelley (2001) Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 50:817–823
Eshima H, Tanaka Y, Sonobe T et al (2013) In vivo imaging of intracellular Ca2+ after muscle contractions and direct Ca2+ injection in rat skeletal muscle in diabetes. Am J Physiol Regul Integr Comp Physiol 305:R610–R618
Adachi T, Kikuchi N, Yasuda K et al (2007) Fibre type distribution and gene expression levels of both succinate dehydrogenase and peroxisome proliferator-activated receptor-gamma coactivator-1alpha of fibres in the soleus muscle of Zucker diabetic fatty rats. Exp Physiol 92:449–455
Lashin OM, Szweda PA, Szweda LI, Romani AM (2006) Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med 40:886–896
Kobayashi T, Nakanishi K, Nakase H et al (1997) In situ characterization of islets in diabetes with a mitochondrial DNA mutation at nucleotide position 3243. Diabetes 46:1567–1571
MacDonald MJ, Tang J, Polonsky KS (1996) Low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of Zucker diabetic fatty rats. Diabetes 45:1626–1630
Leibowitz G, Khaldi MZ, Shauer A et al (2005) Mitochondrial regulation of insulin production in rat pancreatic islets. Diabetologia 48:1549–1559
Coles CJ, Edmondson DE, Singer TP (1979) Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem 254:5161–5167
Herrera-Mundo N, Sitges M (2010) Mechanisms underlying striatal vulnerability to 3-nitropropionic acid. J Neurochem 114:597–605
Ayala-Pena S (2013) Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic Biol Med 62:102–110
Choi YJ, Om JY, Kim NH et al (2013) Heat shock transcription factor-1 suppresses apoptotic cell death and ROS generation in 3-nitropropionic acid-stimulated striatal cells. Mol Cell Biochem 375:59–67
Gier B, Krippeit-Drews P, Sheiko T et al (2009) Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J Clin Invest 119:3246–3256
Bacsi A, Woodberry M, Widger W et al (2006) Localization of superoxide anion production to mitochondrial electron transport chain in 3-NPA-treated cells. Mitochondrion 6:235–244
Oprescu AI, Bikopoulos G, Naassan A et al (2007) Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes 56:2927–2937
Tang C, Han P, Oprescu AI et al (2007) Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes 56:2722–2731
Lim S, Rashid MA, Jang M et al (2011) Mitochondria-targeted antioxidants protect pancreatic beta-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell Physiol Biochem 28:873–886
Schönecker S, Kraushaar U, Düfer M et al (2014) Long-term culture and functionality of pancreatic islets monitored using microelectrode arrays. Integr Biol (Camb) 6:540–544
Eto K, Tsubamoto Y, Terauchi Y et al (1999) Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283:981–985
MacDonald MJ, Brown LJ, Longacre MJ, Stoker SW, Kendrick MA (2013) Knockdown of both mitochondrial isocitrate dehydrogenase enzymes in pancreatic beta cells inhibits insulin secretion. Biochim Biophys Acta 1830:5104–5111
Deshpande SB, Fukuda A, Nishino H (1997) 3-Nitropropionic acid increases the intracellular Ca2+ in cultured astrocytes by reverse operation of the Na+-Ca2+ exchanger. Exp Neurol 145:38–45
Pang Z, Geddes JW (1997) Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci 17:3064–3073
Wang J, Green PS, Simpkins JW (2001) Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem 77:804–811
Mandavilli BS, Boldogh I, van Houten B (2005) 3-nitropropionic acid-induced hydrogen peroxide, mitochondrial DNA damage, and cell death are attenuated by Bcl-2 overexpression in PC12 cells. Brain Res Mol Brain Res 133:215–223
Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP (1998) Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci 18:4439–4450
Drews G, Krippeit-Drews P, Düfer M (2010) Oxidative stress and beta-cell dysfunction. Pflugers Arch 460:703–718
Pi J, Zhang Q, Fu J et al (2010) ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol 244:77–83
Acknowledgements
We thank M. Arning (University of Münster, Münster, Germany) and I. Breuning (University of Tübingen, Tübingen, Germany) for skilful technical assistance.
Funding
The work was supported by grants from the Deutsche Forschungsgemeinschaft (GD and MD).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AE, PSM, CB and SU acquired and analysed data and revised the manuscript. PKD analysed data, revised the manuscript and contributed to discussion. GD designed the concept, revised the manuscript and contributed to discussion. MD designed the concept, acquired and analysed data and wrote the manuscript. All authors approved the manuscript. MD is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Edalat, A., Schulte-Mecklenbeck, P., Bauer, C. et al. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia 58, 1532–1541 (2015). https://doi.org/10.1007/s00125-015-3577-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3577-9