
Abstract
There is no cure for diabetic nephropathy and the molecular mechanisms underlying disease aetiology remain poorly understood. While current paradigms for clinical management of diabetic nephropathy are useful in delaying disease onset and preventing its progression, they do not do so for a significant proportion of diabetic individuals, who eventually end up developing renal failure. Thus, novel therapeutic targets are needed for the treatment and prevention of the disease. MicroRNAs (miRNAs), a class of non-coding RNAs that negatively regulate gene expression, have recently been identified as attractive targets for therapeutic intervention. It is widely recognised that dysregulation of miRNA expression or action contributes to the development of a number of different human diseases, and evidence of a role for miRNAs in the aetiology of diabetic nephropathy is emerging. The discovery that modulation of miRNA expression in vivo is feasible, combined with recent results from successful clinical trials using this technology, opens the way for future novel therapeutic applications. For instance, inhibition of miRNAs that are commonly upregulated in diabetic nephropathy decreases albuminuria and mesangial matrix accumulation in animal models, suggesting that a therapeutic agent against these molecules may help to prevent the development of diabetic nephropathy. Certain challenges, including the development of safe and reliable delivery systems, remain to be overcome before miRNA-based therapeutics become a reality. However, the findings accumulated to date, in conjunction with newly emerging results, are expected to yield novel insights into the complex pathogenesis of diabetic nephropathy, and may eventually lead to the identification of improved therapeutic targets for treatment of this disease.
Similar content being viewed by others

Introduction
Diabetic nephropathy is a progressive kidney disease that develops secondary to diabetes, and is the single most common disorder leading to renal failure [1, 2]. Some degree of renal dysfunction is observed in approximately one in three patients with type 1 diabetes [3, 4], and in about half of all patients with type 2 diabetes [4, 5]. The annual incidence of diabetic nephropathy has more than doubled in the past decade, and today accounts for almost 50% of all end-stage renal disease (ESRD) [6]. Renal replacement therapy through either dialysis or kidney transplantation is necessary to control uraemia and other metabolic consequences of kidney failure in patients with ESRD. The prognosis for patients with diabetes and ESRD is grim, with less than 50% of patients surviving beyond 5 years after diagnosis [7].
Diabetic kidney disease is characterised by persistent albuminuria (>300 mg/day or >200 μg/min), progressive decline in the GFR, and elevated arterial blood pressure [8]. Findings from microscopy show tubulointerstitial fibrosis, glomerular basal membrane thickening, podocyte foot process effacement, mesangial expansion and increased extracellular matrix (ECM) [6]. Aggressive management of hypertension to reduce microalbuminuria, together with tight glycaemic control, are important therapeutic strategies for diabetic nephropathy [9]. Hypertension is most commonly managed using inhibitors of the renin–angiotensin system, which, despite being well tolerated and generally efficacious, do not cure diabetic nephropathy, nor delay disease progression in all patients [10]; thus, the need for more advanced and targeted pharmaceuticals is critical.
It is widely recognised that diabetic nephropathy can develop in response to both environmental and genetic factors, and recent studies have also shown a role for epigenetic influences, such as microRNAs (miRNAs), in the pathogenesis of the disease [11–15]. miRNAs are endogenous, single-stranded RNAs (21–25 nucleotides [nt]) that regulate gene expression either post-transcriptionally, by blocking translation or promoting cleavage of specific target mRNAs, or transcriptionally, through targeting of the promoter region [16]. Animal miRNAs silence the expression of target genes predominantly at the post-transcriptional level by imperfectly base-pairing to the 3′ untranslated region (3′UTR) of target mRNAs, leading to translation inhibition and/or mRNA deadenylation and decay [17, 18]. To date, several miRNAs have been shown to play a role in the pathogenesis of diabetes and its complications, including chronic kidney disease [19, 20]. Such findings expand our understanding of the mechanisms by which diabetic nephropathy may develop and progress and may provide a new platform upon which novel therapeutic targets and more efficient treatments for the disease may be considered. The goal of this review is to present an overview on the emerging therapeutic potential of miRNA-based treatment strategies for this disease.
miRNAs and the diabetic kidney
miRNA biogenesis
miRNAs are processed from a large (about 1,000 nt) primary miRNA (pri-miRNA) by the Drosha–DiGeorge syndrome chromosome region (DGCR) 8 microprocessor complex to an intermediate, ~70 nt stem-loop precursor (pre-miRNA) [21]. After transport of the pre-miRNA to the cytoplasm, an RNase III-type enzyme known as Dicer cleaves off the loop of the pre-miRNA generating a ~23 nt RNA duplex [22]. In animals, one strand of the final RNA product, the mature miRNA, is then loaded into the miRNA-induced silencing complex (miRISC), which guides it to either repress mRNA translation or reduce mRNA stability, following perfect binding between residues 2 and 8 at the 5′ end of the miRNA (seed region) and the 3′UTR of the target gene [17, 18]. Each miRNA can potentially downregulate the expression of a large number of different mRNAs, and each mRNA can possess multiple binding sites for one or several different miRNAs [23].
Role of miRNAs in diabetic nephropathy
Several recent studies have demonstrated that miRNAs play a role in normal renal development, physiology and pathophysiology. Podocyte-specific loss of Dicer activity causes foot process effacement, proteinuria and glomerulosclerosis with rapid progression to renal failure [24–26]. Similar results were observed after the inducible deletion of Drosha in podocytes [27]. Thus, it has been speculated that the global loss of miRNAs due to deletion of Dicer or Drosha contributes to the observed pathologies, and suggests a critical role of miRNAs in the maintenance of podocyte homeostasis and associated renal functions.
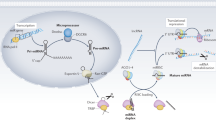
Characterisation of miRNA expression in diabetic nephropathy has emerged from both studies in animal models of diabetes and in vitro investigations of the effects of hyperglycaemia and TGF-β in different types of kidney cells in culture, including mesangial cells, podocytes and proximal tubular epithelial cells [11–13, 28, 29]. TGF-β is a central cytokine involved in cell growth and ECM production, and treatment of mouse models of diabetes with neutralising anti-TGF-β antibodies attenuates kidney hypertrophy and the increase in ECM production [30]. TGF-β can induce the expression of key miRNAs in mesangial cells and in the glomeruli of diabetic mice [11–13]. These miRNAs mediate the expression of fibrotic genes and hypertrophy associated with diabetic nephropathy (Fig. 1). Recently, Sun et al [31] reported that low doses of the cancer drug, paclitaxel, an inhibitor of TGF-β, ameliorated fibrosis in the remnant kidney model by downregulating miR-192, miR-217 and miR-377.

Role of miRNAs in diabetic nephropathy. Hyperglycaemia (HG) and TGF-β regulate the levels of miRNAs in mesangial cells and contribute to fibrosis and hypertrophy. TGF-β, transforming growth factor beta; YBX1, Y box binding protein 1; PAK1, P21/CDC42/Rac1-activated kinase 1; SOD1/2, superoxide dismutase 1 and 2; PI3K, phosphatidylinositol 3-kinase; NOX4, NADPH oxidase 4; ROS, reactive oxygen species; YWHAZ, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide; p38MAPK, p38 mitogen activated protein kinase; FN1, fibronectin 1; DN, diabetic nephropathy
Table 1 presents a list of miRNAs that are dysregulated in diabetic nephropathy and that may contribute to the development of this disease. Recently, Kim et al [32] reported that some miRNAs with low GC content, including miR-141, miR-200a and miR-29b, are selectively lost during tryzol extraction from a small number of cells. Thus, some previous findings may need to be revisited, particularly if low cell density was used, and the in vitro results have not been confirmed in tissues from animal models of diabetic nephropathy.
Kato et al [20] have also recently published a review article in which the mechanisms involving miRNA in the pathogenesis of diabetic nephropathy are discussed in great depth; therefore, we will not discuss these further here.
Therapeutic potential of miRNAs in diabetic nephropathy
Individual miRNAs often regulate the expression of multiple target genes with related functions and, thus, modulating the expression of a single miRNA can, in principle, influence an entire gene network, and consequently, exert effects on the development of certain complex diseases [33]. There are two general types of strategy for therapeutic modulation of miRNA: inhibition of activity and restoration of diminishing function. For those miRNAs whose expression is increased in diabetic nephropathy, inhibition of miRNA function may potentially restore normal regulation of target genes, while for those whose expression is lost or reduced, function could be re-established by supplementation with the miRNA of interest.
Inhibition of miRNA activity
There are presently three main strategies available for inactivating miRNA in vivo and in vitro. These are genetic knockouts, miRNA sponges and antisense oligonucleotides (ASOs) [34, 35]; each method is described in the following sections.
Genetic knockouts
To date, myriad knockout mice lacking key miRNA processing factors, such as Dicer, Drosha and argonaute 2 (Ago2), have been engineered; typically, these animals die during early gestation with severe developmental defects, suggesting a critical function for miRNA in early embryonic development [36]. With respect to renal function, mice with cell-specific inactivation of Dicer (conditional Dicer models) in podocytes, nephron progenitors, juxtaglomerular cells and proximal tubules have been developed [37], and these animals have yielded insights into the development of kidney injury. For example, mice with podocyte-specific knockout of Dicer (or Drosha) displayed significant proteinuria by 2 weeks after birth, rapid progression of glomerular and tubular injury starting at 3–4 weeks, and ESRD and death by 4–8 weeks [38]. By 3–4 weeks, electron microscopy demonstrated focal foot and wrinkling of the glomerular basal membrane (GBM), and staining of the kidneys showed decreased levels of the slit diaphragm proteins nephrin and podocin, to which the observed proteinuria was attributed. By weeks 4–8, the glomeruli became severely affected and all of the mice died [24–26, 38]. These results suggest that miRNA function is not essential for the initial development of glomeruli, but is critical for maintaining the glomerular filtration barrier [24–26]. Although similar results in mice with podocyte-specific knockout of Dicer were observed in three independent studies [24–26], rescue experiments to restore the wild-type phenotype have not yet been performed. Initial attempts to identify miRNAs responsible for the disruption of podocyte structure have suggested miR-30 family members as possible candidates [24]. In addition, miRNAs 23b, 24 and 26a may contribute to the observed phenotype in mice with specific Dicer inactivation in podocytes because they seem to be crucial for proper glomerular filtration [26]. In addition, the loss of miRNAs in nephron progenitors results in a marked decrease in nephron number [39, 40], while Dicer deletion in renin-secreting cells in the juxtaglomerular apparatus causes decreased levels of circulating renin, reduced blood pressure, interstitial fibrosis, and vascular abnormalities [41]. However, there is no developmental or functional renal defect seen in mice with disruption of Dicer in the proximal tubule. In contrast, these mice actually appear to be protected from ischaemia–reperfusion injury [42].
Individual miRNA knockouts have also been reported, including mice lacking miR-375 and miR-21. Deletion of miR-375 in mice produces hyperglycaemia, increased gluconeogenesis, and elevated pancreatic alpha cell number, indicating that this miRNA, which is highly expressed in pancreatic islets, is required for normal glucose homeostasis [43, 44]. In contrast, miR-21 knockout mice showed no overt abnormalities. In fact, these mice suffered far less interstitial fibrosis in response to kidney injury, a phenotype duplicated in wild-type mice treated with antimiR-21 oligonucleotides, showing that miR-21 contributes to fibrogenesis and epithelial injury in the kidney [45]. Although the generation of engineered animals is costly and time-consuming, the individual miRNA knockout is the only approach that guarantees a complete loss of miRNA activity, which is the primary advantage of using this method for the inhibition of miRNA activity over the other methods described in this review (see the text box).

Sponge miRNA or artificial miRNA decoys
Sponge miRNAs, also known as artificial miRNA decoys, are small RNA transcripts containing multiple copies of the target binding site for a miRNA of interest. By providing alternative binding sites, sponges effectively soak up miRNAs, and thereby prevent them from exerting effects on downstream targets. In addition, sponges can also be used to regulate miRNA activity in vitro [46] or in vivo. Typically, sponges are highly expressed transgenes that can be stably integrated into chromosomes, and are designed to be controlled by promoters whose expression is restricted to a specific cell type, tissue, or developmental stage [47]. Inhibition of miRNA expression can be long-term or transient depending on the integration of the sponge transgene in the host chromosome or the transitory location of the plasmid in the cytoplasm, respectively. The efficacy of the sponge depends on the affinity and avidity of binding sites, as well as the concentration of sponge RNAs relative to the concentration of the miRNA. The strongest available promoter for the cell type of interest should be used to maximise sponge expression [35], although when the sponge is expressed at high levels, it can inhibit the activity of the entire family of miRNAs sharing a common seed sequence [48].
The use of miRNA sponge technology in renal models is presently limited. To date, researchers have found that inhibition of miR-21 in rat and human mesangial cells using sponge technology prevented high-glucose-mediated downregulation of the tumour suppressor protein, phosphatase and tensin homologue deleted on chromosome 10 (PTEN) [49]. Interestingly, PTEN is known to contribute to some of the pathological features of diabetic nephropathy, including renal cell hypertrophy and fibronectin expression [49, 50], and the results from this study suggest that miR-21 may play a role in these processes. In addition to this study, sponge miRNA has also been used in mice to target miR-133 in the pathogenesis of cardiac hypertrophy [51], a major complication of diabetic nephropathy. In treated animals, downregulation of miR-133 increased hypertrophic markers, while in vitro overexpression of the same miRNA inhibited cardiac hypertrophy.
Although many studies have shown the effectiveness of miRNA sponges in a variety of processes, the application of this technology in the area of diabetic nephropathy has been limited and, therefore, further experiments will be necessary to prove the utility of the sponge method in vivo for the treatment of this disease.
Antisense oligonucleotides or antimiR oligonucleotides
ASOs/antimiR oligonucleotides (AMOs) are the most commonly used method to inhibit miRNA activity. Although ASO and AMO are generally considered as synonymous, the AMO can be described as a particular type of ASO against miRNA. The mechanism of inhibition is mediated by chemically-modified, single-stranded reverse complement oligonucleotides, which sequester the mature miRNA, rendering it incapable of binding to its target sequence [34]. The introduction of different chemical modifications of the sugar, the nucleobase, or the internucleotide linkages in ASOs further increases binding affinity for the target miRNA, which improves nuclease resistance, cellular uptake, and in vivo delivery [52]. Oligonucleotides with 2′ sugar modifications, including 2′-O-methyl (2′-O-Me), 2′-O-methoxyethyl (2′-O-MOE), 2′-fluoro (2′-F) and 2′,4′-methylene (locked nucleic acid or LNA) (Fig. 2) can be effective inhibitors of miRNAs in cell culture [53–57]. Among the chemical modifications tested, LNA possesses the highest affinity towards complementary RNA, which may be the reason for its unprecedented specificity relative to other modified oligonucleotides [34]. Nuclease resistance can also be strengthened by backbone modification of the phosphodiester linkages into phosphorothionate (PS) linkages in which a sulphur atom replaces one of the non-bridging oxygen atoms in the phosphate group (Fig. 2) [34].

Chemical modifications that improve the stability, biodistribution, and delivery of AMOs. The figure shows structures of the most commonly used chemical modifications in AMOs. LNA is a bicyclic RNA analogue in which the ribose is locked in a C3′-endo conformation by introduction of a 2′-O,4′-C methylene bridge. The hydroxide in the 2′ position of the ribose moiety can be replaced by a 2′-fluoro (2′-F), a 2′-O-methoxyethyl (2′-MOE) or a 2′-methyl (2′-O-Me) group to generate modified nucleotides with higher stability. In the phosphorothionate linkage, sulphur replaces one of the non-bridging oxygen atoms in the phosphate group. A morpholine ring replaces the sugar moiety in morpholino oligomers, whereas a peptide nucleic acid is a DNA analogue in which the sugar–phosphate backbone is replaced by N-(2-minoethyl)glycine units
The first miRNA inhibitors demonstrated to work in mammals were ‘antagomirs’, which are 2′-O-Me oligonucleotides with phosphorothionate linkages between nucleotides at both ends, and conjugated to cholesterol at their 3′ ends [58]. Cholesterol conjugation causes cellular uptake of the modified nucleic acid oligonucleotide by promoting its association with high- and low-density lipoproteins that can bind cell surface membrane receptors [47]. Phosphorothionate bonds promote serum protein binding, which increases in vivo distribution and bioavailability of the ASO [47]. Treatment of mice with three tail vein injections at 80 mg/kg of antagomirs against miR-16, miR-122, miR-192 and miR-194 resulted in silencing of these miRNAs in different organs, including liver and kidney [58]. It would be interesting to extend this study to test for therapeutic benefits of antagomir-192 against diabetic nephropathy, as this miRNA appears to have a central role in the development of the disease [11, 13]. The efficient inhibition of miR-122, the major miRNA found in liver, led to a 40% decrease in serum cholesterol levels in treated mice [58], demonstrating the therapeutic potential of antagomir-122 against hypercholesterolaemia, a common clinical finding in patients with diabetes. Recently, antagomirs were effectively used to knock down miR-29c in db/db mice, leading to a decrease in albuminuria and glomerular mesangial matrix expansion (Table 2) [29]. However, antagomirs are unlikely to be used clinically because they require higher doses (about 80 mg/kg) to achieve the same efficacy as other ASO strategies such as LNA, which are efficient at 2–10 mg/kg depending on the route of administration [47] (Table 2). A direct comparison of the effect of antagomir 122 vs LNA antimiR-122 was performed in C57BL/6 J mice treated with three i.p. doses of 1, 5 and 25 mg/kg [59] (Table 3). A significant decrease in the levels of miR-122 in liver, as well as a parallel increase in the expression of its target gene, Aldoa, were observed in a dose-dependent manner 48 h following the last injection with LNA antimiR-122, whereas no effect was detected in mice treated with antagomirs [59]. In addition, a 20% decrease in levels of blood total cholesterol were observed in mice treated with 1 and 5 mg/kg LNA, while raising the dose to 25 mg/kg LNA doubled the reduction in these values (i.e. 40% decrease in total cholesterol concentration). Cholesterol levels did not change in mice that received treatment with antagomirs [59]. At this time, it is not yet known whether the different effective dosages for LNA and antagomirs are the result of differences in tissue distribution or intracellular actions of the miRNA inhibitors.
Alternative ASO modifications, such as 2′-O-MOE, 2′-F, 2′-F/MOE, and LNAs (Fig. 2), have greater affinity to bind and inhibit miRNA function in vivo than 2′-O-Me RNA oligonucleotides, and all require lower doses than antagomirs to be effective [55, 56, 59–61]. Davis et al [60] compared the potency of 2′-F/MOE, 2′-O-MOE/LNA, 2′MOE, 2′-O-Me and LNAs in vivo and found that 2′-F/MOE is the most effective. The ASO chemistry dictates the fate of the targeted miRNA [34]. Thus, higher affinity oligonucleotides such as LNA/DNA, LNA/2′-O-Me and 2′-F/MOE, sequester the targeted miRNA in a heteroduplex, whereas lower affinity ASOs, such as 2′-O-Me, 2′-MOE and antagomirs promote miRNA degradation (Table 3).
In two recent reports, the inhibition of miR-192 using a low dose (2.0–2.5 mg/kg) of subcutaneously injected LNA antimiR-192 led to a significant increase in levels of the E-box repressor zinc finger E-box binding homeobox (ZEB)1/2 and decreased gene expression of collagen, TGF-β and fibronectin in normal and streptozotocin-induced diabetic mice (Table 2) [13, 14]. Furthermore, LNA antimiR-192 improved proteinuria in these mice, supporting the possibility of an anti-miRNA-base translational approach for the treatment of diabetic nephropathy.
A pioneering non-human primate study established that LNA ASOs are specific, stable, and non-toxic when administered intravenously. In this study, a decrease in plasma cholesterol was observed for more than 20 days following five daily successive i.v. injections of 10 mg/kg LNA antimiR-122 in African green monkeys [59]. The first miRNA-targeted drug to enter human clinical trials was an LNA antimiR-122 compound, miravirsen, developed by Santaris Pharma (Hørsholm, Denmark) for the treatment of hepatitis C [62]. In addition to a role in regulating cholesterol metabolism, miR-122 has been implicated as an important host factor required for effective hepatitis C virus (HCV) infection. Thus, interfering with miR-122 function may offer a new avenue for the treatment of HCV. Successful phase I and II clinical trials have assessed the efficacy, safety, and tolerability of a 4 week monotherapy treatment with LNA antimiR-122 [62]. These exciting results are likely to pave the way for the development of miRNA-based therapeutics targeting other specific diseases, including diabetic nephropathy.
Restoring miRNA activity
For miRNAs whose expression is decreased during the development of diabetic nephropathy, introduction of the mature miRNA into the kidney could restore regulation of key target genes. The two primary strategies for upregulating miRNA expression are double-stranded miRNA mimics and miRNA expression vectors. These methods are discussed below.
Double-stranded miRNA mimics
Replacement of miRNA can be achieved through the introduction of miRNA mimics, short double-stranded oligonucleotides in which one strand (the guide strand) is identical to the native miRNA sequence, while the complement is referred to as the passenger strand. The double-stranded structure is required for efficient recognition and loading of the guide strand into the RISC. Of note, miRNA mimics are analogous in structure to the small interfering RNA (siRNA) molecules commonly used in gene silencing experiments [63], which can repress expression of a target mRNA with semi-complementary binding sites in its 3′UTR, much like the function of endogenous miRNAs [64]. A miR-29b mimic has been successfully used in a mouse model of cardiac fibrosis [33], and a similar approach may be employed in diabetic nephropathy to block the development of kidney fibrosis through overexpression of miR-29b (Table 1).
Although miRNA mimics are commonly used in vitro to validate miRNA targets known to be involved in the pathogenesis of diabetic nephropathy, this technology has not yet been tested in animal models of renal diseases [65]. However, similar work using siRNA approaches has shown promising results. For example, in vivo silencing of TGF-β1, a potent profibrotic cytokine well-recognised for its role in the development of diabetic nephropathy, using TGF-β1-specific synthetic siRNA injection into the renal artery (which effectively delivered siRNA into the glomeruli), decreased levels of TGF-β1 protein and delayed matrix expansion [66]. Although promising, a limitation of this approach is the short-term effect of siRNA, which is completely lost 5 to 7 days after treatment, and more experimental work in animal models of kidney disease is required to determine the potential of this technology as an miRNA-based therapeutic agent for diabetic nephropathy.
miRNA expression vectors
Therapeutic miRNA replacement can also be achieved by expressing the miRNA of interest using a vector construct. This strategy involves the expression of a short hairpin RNA (shRNA) generated from polymerase II or III promoters in a DNA or viral vector, which is then processed by Dicer before loading into RISC. The two main advantages of this approach are the potential for a more stable and long-term silencing compared with that of miRNA mimics and siRNAs, as well as the ease of expressing multiple miRNAs from the same transcript [67]. The major disadvantage is the cellular toxicity: overexpression of shRNA in rats causes hepatoxicity, organ failure and death [68]. Further, excessive expression levels of shRNA may also saturate argonaute proteins and the pre-miRNA export protein, Exportin-5, thereby limiting the amount of exogenous siRNA or miRNA that can be tolerated by a cell. In addition, the overloading of argonaute and Exportin-5 would also affect the processing of endogenous miRNAs essential for proper cellular function and survival, causing cell toxicity. However, both the toxic side-effects and the risk of over-saturating endogenous small RNA pathways can be minimised by optimising shRNA dose and sequence [69] or by using weaker or tissue-specific promoters [70–72]. This last strategy has been recently used to eliminate hepatoxicity previously observed in non-human primates treated with three shRNAs that simultaneously target conserved regions of the HCV genome [72].
Artificial miRNA expression vectors have been introduced as safer alternatives to shRNAs in vitro and in vivo [73]. These vectors contain the backbone sequence of a natural miRNA precursor, in which the stem sequence has been substituted to generate an artificial miRNA, which can efficiently and specifically silence the genes of interest [74]. The natural miRNA:miRNA* structure (where miRNA* is the passenger strand) is replaced by the artificial one, which is processed in a biogenesis pathway similar to that of natural miRNAs, and results in functional, mature artificial miRNA [75]. In contrast with shRNA, artificial miRNAs do not disrupt normal miRNA biogenesis and function because they are expressed at lower levels and are processed more efficiently, making them far less toxic. For example, in studies involving the brain, shRNA treatment caused Purkinje cell neurotoxicity, while artificial miRNA expression was well tolerated and led to effective target gene silencing in these cells [73]. These findings suggest that miRNA-based platforms are less toxic and better suited to therapeutic silencing in mammals. Although the development of artificial miRNA technology is still in its infancy, the approach nonetheless presents great potential for therapeutic intervention.
Potential challenges of miRNA-based therapeutics
The ability to manipulate miRNAs in vivo provides a potential opportunity to exploit miRNA therapeutically. However, miRNA-based therapeutics pose different challenges from those associated with classic drugs. While traditional pharmaceuticals are typically specific for a single cellular target (generally a receptor or enzyme), miRNAs, by nature, have numerous molecular targets, which, in vivo, may perturb multiple cellular functions, resulting in unintended off-target effects [63]. While the possibility of targeting multiple components of the same pathway or cellular process relevant to human disease simultaneously can be considered a strong advantage of miRNA-based therapeutics, it also raises concerns about potential toxicity in normal tissues. These toxic effects might be the result of overloading RISC with exogenous miRNA mimics, which then compete with endogenous miRNAs necessary for normal cellular function [76]. Further, depending on context, a single miRNA may yield both beneficial and pathogenic effects, and can elicit effects on unintended targets. Issues associated with biodistribution, pharmacokinetics, and cell penetration also represent potential obstacles to miRNA-based therapeutics, although, as discussed earlier, chemical modifications of synthetic nucleic acids increase the stability of the oligonucleotides in biological fluids, which may circumvent some of these concerns (Fig. 2).
Currently, the main bottleneck for the development of miRNA-based treatments is the efficient delivery of a therapeutic RNA to its target tissue. Transfer of therapeutic RNA from the blood to the target tissue is a challenge because anything larger than 5 nm, including therapeutically complexed RNA, cannot cross the capillary endothelium, and will remain in circulation until filtered by the kidneys. Hepatic cells allow molecules up to 200 nm in diameter to enter and, not surprisingly, delivery of therapeutic RNAs is most successful in the liver [47]. Different delivery approaches have been developed to circumvent these challenges, and these can be broadly divided into two categories: conjugation and formulation [63]. Conjugation strategies include binding cell-penetrating peptides, antibodies, lipophilic moieties, or receptor ligands, such as cholesterol molecules (which are used in antagomirs) [58]. Formulation approaches vary substantially and include synthetic liposomes, lipid emulsions, polymers and nanoparticles. The encapsulation of nucleic acids in lipid-forming vesicles between 50 and 500 nm have been successfully used to avoid undesired filtration by the kidney and to enhance intracellular delivery [63]. Alternatively, nucleic acids or encoding plasmids can be delivered by virus, including adenovirus, lentivirus, or single-stranded cytoplasmic RNA virus [77, 78].
Despite significant advances, most nucleic acid delivery agents developed to date have only demonstrated efficacy targeting the liver, as noted above. For the kidney, an effective delivery approach is a mandatory requirement for the use of synthetic nucleic acids as therapeutics for renal diseases, including diabetic nephropathy. However, the rapid renal clearance that eliminates therapeutic RNA molecules from circulation can actually be advantageous in targeting the kidney, because tubular epithelial cells tend to reabsorb oligonucleotides from the ultrafiltrate [79]. For example, a conjugation strategy using 32P-cholesterol-siRNA bound to HDL-C or albumin, but not to LDL-C, resulted in nucleic acid accumulation in the kidney [80]. Similarly, systemic delivery of siRNA oligonucleotides conjugated with cholesterol and targeting 12/15-lipoxygenase ameliorated renal injury and prevented diabetic nephropathy in a streptozotocin-injected mouse model of type 1 diabetes [81]. More recently, antagomirs and LNA miRNA inhibitors have been effectively delivered to the kidney inducing the downregulation of different miRNAs commonly increased in diabetic nephropathy (Table 1). Together, these results provide encouraging support for the potential of miRNA-based therapeutics for the treatment of kidney diseases, including diabetic nephropathy.
Conclusions
It is now widely accepted that the dysregulation of miRNA expression or action contributes to the development of various human diseases, although evidence for the role of miRNAs in diabetic nephropathy is only now emerging. The discovery that modulation of miRNA expression in vivo is feasible, together with the recent results from successful clinical trials using this technology, opens the way for future novel therapeutic applications. Specifically, inhibition of those miRNAs that are commonly upregulated in diabetic nephropathy (i.e., miR-192 and miR-377) decreases albuminuria and mesangial matrix accumulation in animal models, and is thereby concordant with the idea that developing a therapy against these molecules may help to prevent diabetic nephropathy and/or slow its progression. One of the strengths of miRNA-based technology is the powerful ability to affect simultaneously multiple genes in the same pathway by inhibiting or mimicking a single miRNA. Various challenges remain to be overcome before miRNA-based therapeutics become a reality, including, primarily, the development of safe and reliable cell- and organ-specific delivery systems. However, the findings accumulated to date, in conjunction with newly emerging results, are expected to yield novel insights into the complex pathogenesis of diabetic nephropathy, and may eventually lead to the identification of improved therapeutic targets for treatment of this disease.
Abbreviations
- 3′UTR:
-
3′ untranslated region
- AMO:
-
AntimiR oligonucleotide
- ASO:
-
Antisense oligonucleotide
- ECM:
-
Extracellular matrix
- ESRD:
-
End-stage renal disease
- LNA:
-
Locked nucleic acid
- miRISC:
-
miRNA-induced silencing complex
- miRNA:
-
MicroRNA
- nt:
-
Nucleotides
- Pre-miRNA:
-
Precursor miRNA
- PTEN:
-
Phosphatase and tensin homologue deleted on chromosome 10
- RISC:
-
RNA-induced silencing complex
- shRNA:
-
Short hairpin RNA
- siRNA:
-
Small interfering RNA
- ZEB:
-
Zinc finger E-box binding homeobox
References
Caramori ML, Mauer M (2003) Diabetes and nephropathy. Curr Opin Nephrol Hypertens 12:273–282
Gray SP, Cooper ME (2011) Alleviating the burden of diabetic nephropathy. Nat Rev Nephrol 7:71–73
Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ (2006) The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes 55:1463–1469
Thomas MC, Groop PH, Tryggvason K (2012) Towards understanding the inherited susceptibility for nephropathy in diabetes. Curr Opin Nephrol Hypertens 21:195–202
Parving HH, Lewis JB, Ravid M, Remuzzi G, Hunsicker LG (2006) Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int 69:2057–2063
Kanwar YS, Sun L, Xie P, Liu F, Chen S (2011) A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol Mech Dis 6:395–423
Blumenthal SS (2011) Evolution of treatment for diabetic nephropathy: historical progression from RAAS inhibition and onward. Postgrad Med 123:166–179
Bichu P, Nistala R, Khan A, Sowers JR, Whaley-Connell A (2009) Angiotensin receptor blockers for the reduction of proteinuria in diabetic patients with overt nephropathy: results from the AMADEO study. Vasc Health Risk Manag 5:129–140
Ayodele OE, Alebiosu CO, Salako BL (2004) Diabetic nephropathy—a review of the natural history, burden, risk factors and treatment. J Natl Med Assoc 96:1445–1454
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (1993) The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329:1456–1462
Kato M, Zhang J, Wang M, Rossi JJ et al (2007) MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A 104:3432–3437
Kato M, Putta S, Wang M et al (2009) TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol 11:881–889
Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R (2011) A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int 80:358–368
Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R (2012) Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 23:458–469
Wang Q, Wang Y, Minto AW et al (2008) MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 22:4126–4135
Kim DH, Sætrom P, Snove O, Rossi JJ (2008) MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A 42:16230–16235
Fabian MR, Cieplak MK, Frank F et al (2011) miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat Struct Mol Biol 18:1211–1217
Huntzinger E, Izaurralde E (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110
Tang X, Tang G, Ozcan S (2008) Role of microRNAs in diabetes. Biochim Biophys Acta 1179:697–701
Kato M, Park JT, Natarajan R (2012) MicroRNAs and the glomerulus. Exp Cell Res 18:993–1000
Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN (2004) The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18:3016–3027
Lee Y, Kim M, Han J et al (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Friedman RC, Farh KK, Burge CB, Bartel DP (2009) Most mammalian mRNas are conserved targets of microRNAs. Genome Res 19:92–105
Shi S, Yu L, Chiu C et al (2008) Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol 19:2159–2169
Harvey SJ, Jarad G, Cunningham J et al (2008) Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol 19:2150–2158
Ho J, Ng KH, Rosen S et al (2008) Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol 19:2069–2075
Zhdanova O, Srivastava S, Di L et al (2011) The inducible deletion of Drosha and microRNAs in mature podocytes results in a collapsing glomerulopathy. Kidney Int 80:719–730
Kato M, Wang L, Putta S, Wang M, Yuan H, Sun G (2010) Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of miR-216a, mediates TGF-{beta}-induced collagen expression in kidney cells. J Biol Chem 285:34004–34015
Long J, Wang Y, Wang W, Chang BH, Danesh FR (2011) MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J Biol Chem 286:11837–11848
Mogyorosi A, Ziyadeh FN (1999) GLUT1 and TGF-beta: the link between hyperglycaemia and diabetic nephropathy. Nephrol Dial Transplant 14:2827–2829
Sun L, Zhang D, Liu F et al (2011) Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J Pathol 225:364–377
Kim YK, Yeo J, Kim B, Ha M, Kim VN (2012) Short structured RNAs with low GC content are selectively lost during extraction from a small number of cells. Mol Cell 46:893–895
van Rooij E, Marshall WS, Olson EN (2008) Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res 103:919–928
Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S (2012) Inhibition of microRNA function by antimiR oligonucleotides. Silence 3:1–17
Ebert MS, Sharp PA (2010) MicroRNA sponges: progress and possibilities. RNA 11:2043–2050
Park CY, Choi YS, McManus MT (2010) Analysis of microRNA knockouts in mice. Hum Mol Genet 19:R169–R175
Ho J, Kreidberg JA (2012) MicroRNAs in renal development. Pediatr Nephrol. doi:10.1007/s00467-012-2204-y
Ho JJ, Marsden PA (2008) Dicer cuts the kidney. J Am Soc Nephrol 11:2043–2046
Ho J, Pandey P, Schatton T, Sims-Lucas S, Khalid M, Frank MH (2011) The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J Am Soc Nephrol 22:1053–1063
Nagalakshmi VK, Ren Q, Pugh MM, Valerius MT, McMahon AP, Yu J (2011) Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int 79:317–330
Sequeira-Lopez ML, Weatherford ET, Borges GR, Monteagudo MC, Pentz ES, Harfe BD (2010) The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol 21:460–467
Wei Q, Bhatt K, He HZ, Mi QS, Haase VH, Dong Z (2010) Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 21:756–761
Poy MN, Eliason L, Krutzfeldt J et al (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432:226–230
Poy MN, Hausser J, Trajkovski M et al (2009) miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci U S A 106:5813–5818
Chau BN, Xin C, Hartner J et al (2012) MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 4:121ra18
Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4:721–726
Broderick JA, Zamore PD (2011) MicroRNA therapeutics. Gene Ther 18:1104–1110
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798
Dey N, Das F, Mariappan MM et al (2011) MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J Biol Chem 286:25586–25603
Mahimainathan L, Das F, Venkatesan B, Choudhury GG (2006) Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 55:2115–2125
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13:613–618
Esau CC (2008) Inhibition of microRNA with antisense oligonucleotides. Methods 44:55–60
Hutvagner G, Simard MJ, Mello CC, Zamore PD (2004) Sequence-specific inhibition of small RNA function. PLoS Biol 2:E98
Meister G, Landthaler M, Dorsett Y, Tuschl T (2004) Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA 10:544–550
Davis S, Lollo B, Freier S, Esau C (2006) Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res 34:2294–2304
Orom UA, Kauppinen S, Lund AH (2006) LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 372:137–141
Vermeulen A, Robertson B, Dalby AB, Marshall WS, Karpilow J, Leake D (2007) Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA 13:723–773
Krutzfeldt J, Rajewsky N, Braich R et al (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438:685–689
Elmen J, Lindow M, Schutz S et al (2008) LNA-mediated microRNA silencing in non-human primates. Nature 452:896–899
Davis S, Propp S, Freier SM et al (2009) Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res 37:70–77
Esau C, Davis S, Murray SF et al (2006) miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98
Janssen HL, Reesink HW, Zeuzem S et al (2011) A randomized, doubleblind, placebo controlled safety and anti-viral proof of concept study of miravirsen, an oligonucleotide targeting miR-122, in treatment naive patients with genotype 1 chronic HCV infection. Hepatology 54(Suppl):1430A
van Rooij E, Purcell AL, Levin AA (2012) Developing microRNA therapeutics. Circ Res 110:496–507
Doench JG, Petersen CP, Sharp PA (2003) siRNAs can function as miRNAs. Genes Dev 17:438–442
Karolina DS, Wintour EM, Bertram J, Jeyaseelan K (2010) Riboregulators in kidney development and function. Biochimie 92:217–225
Takabatake Y, Isaka Y, Mizui M et al (2005) Exploring RNA interference as a therapeutic strategy for renal disease. Gene Ther 12:965–973
Esau CC, Monia BP (2007) Therapeutic potential for microRNAs. Adv Drug Deliv Rev 59:101–114
Grimm D, Wang L, Lee JS et al (2010) Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest 120:3106–3119
Grimm D, Streetz KL, Jopling CL et al (2006) Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441:537–541
An DS, Qin FX, Auyeung VC et al (2006) Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther 14:494–504
Giering JC, Grimm D, Storm TA, Kay MA (2008) Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol Ther 16:1630–1636
Suhy DA, Kao SC, Mao T et al (2012) Safe, long-term hepatic expression of anti-HCV shRNA in a nonhuman primate model. Mol Ther 20:1737–1749
Boudreau RL, Martins I, Davidson BL (2009) Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther 17:169–175
Zeng Y, Wagner EJ, Cullen BR (2002) Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 9:1327–1333
Hu T, Fu Q, Chen P, Ma L, Sin O, Guo D (2009) Construction of an artificial microRNA expression vector for simultaneous inhibition of multiple genes in mammalian cells. Int J Mol Sci 10:2158–2168
Bader AG, Brown D, Winkler M (2010) The promise of microRNA replacement therapy. Cancer Res 70:7027–7030
Scherr M, Venturini L, Eder M (2010) Lentiviral vector-mediated expression of pre-miRNAs and antagomiRs. Methods Mol Biol 614:175–185
Langlois RA, Shapiro JS, Pham AM, tenOever BR (2012) In vivo delivery of cytoplasmic RNA virus-derived miRNAs. Mol Ther 20:367–375
Molitoris BA, Dagher PC, Sandoval RM et al (2009) siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol 20:1754–1764
Wolfrum C, Shi S, Jayaprakash KN et al (2007) Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 25:1149–1157
Yuan H, Lanting L, Xu ZG et al (2008) Effects of cholesterol-tagged small interfering RNAs targeting 12/15-lipoxygenase on parameters of diabetic nephropathy in a mouse model of type 1 diabetes. Am J Physiol Renal Physiol 295:F605–F617
Gregory PA, Bert AG, Paterson EL et al (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Zarjou A, Yang S, Abraham E, Agarwal A, Liu G (2011) Identification of a microRNA signature in renal fibrosis: role of miR-21. Am J Physiol Renal Physiol 301:F793–F801
Du B, Ma LM, Huang MB et al (2010) High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett 584:811–816
Wang B, Komers R, Carew R et al (2012) Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol 23:252–265
Long J, Wang Y, Wang W, Chang BH, Danesh FR (2010) Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J Biol Chem 30:23457–23465
Wang B, Koh P, Winbanks C et al (2011) miR-200a prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 60:280–287
Zhang Z, Luo X, Ding S et al (2012) MicroRNA-451 regulates p38 MAPK signaling by targeting of Ywhaz and suppresses the mesangial hypertrophy in early diabetic nephropathy. FEBS Lett 586:20–26
Fu Y, Zhang Y, Wang Z et al (2010) Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol 32:581–589
Duisters RF, Tijsen AJ, Schroen B et al (2009) miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104:170–178
Krutzfeldt J, Kuwajima S, Braich R et al (2007) Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35:2885–2892
Rayner KJ, Esau CC, Hussain FN et al (2011) Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478:404–407
Obad S, dos Santos CO, Petri A et al (2011) Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 43:371–378
Lanford RE, Hildebrandt-Eriksen ES, Petri A et al (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MLA and JKD contributed to the design and drafting of this article. Both authors approved the final version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alvarez, M.L., DiStefano, J.K. Towards microRNA-based therapeutics for diabetic nephropathy. Diabetologia 56, 444–456 (2013). https://doi.org/10.1007/s00125-012-2768-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2768-x