
Abstract
Aims/hypothesis
Human complex metabolic traits are in part regulated by genetic determinants. Here we applied exome sequencing to identify novel associations of coding polymorphisms at minor allele frequencies (MAFs) >1% with common metabolic phenotypes.
Methods
The study comprised three stages. We performed medium-depth (8×) whole exome sequencing in 1,000 cases with type 2 diabetes, BMI >27.5 kg/m2 and hypertension and in 1,000 controls (stage 1). We selected 16,192 polymorphisms nominally associated (p < 0.05) with case–control status, from four selected annotation categories or from loci reported to associate with metabolic traits. These variants were genotyped in 15,989 Danes to search for association with 12 metabolic phenotypes (stage 2). In stage 3, polymorphisms showing potential associations were genotyped in a further 63,896 Europeans.
Results
Exome sequencing identified 70,182 polymorphisms with MAF >1%. In stage 2 we identified 51 potential associations with one or more of eight metabolic phenotypes covered by 45 unique polymorphisms. In meta-analyses of stage 2 and stage 3 results, we demonstrated robust associations for coding polymorphisms in CD300LG (fasting HDL-cholesterol: MAF 3.5%, p = 8.5 × 10−14), COBLL1 (type 2 diabetes: MAF 12.5%, OR 0.88, p = 1.2 × 10−11) and MACF1 (type 2 diabetes: MAF 23.4%, OR 1.10, p = 8.2 × 10−10).
Conclusions/interpretation
We applied exome sequencing as a basis for finding genetic determinants of metabolic traits and show the existence of low-frequency and common coding polymorphisms with impact on common metabolic traits. Based on our study, coding polymorphisms with MAF above 1% do not seem to have particularly high effect sizes on the measured metabolic traits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last few years, genome-wide association studies (GWAS) have led to substantial progress in mapping common genetic variation with impact on common phenotypes including those of the metabolic syndrome [1–10]. This advance has revealed hundreds of genetic determinants of human complex phenotypes [1]. Despite this progress a major part of the heritable contribution to variation in most widespread metabolic traits remains unaccounted for [11]. Thus, for type 2 diabetes and related metabolic traits it has been estimated that 10–30% of the observed heritability can be attributed to the hitherto identified variants [2, 4, 8, 10].
DNA sequencing has emerged as a powerful technology enabling detection of low-frequency and rare variation not captured by initial GWAS design and in future studies the GWAS approach may be complemented by imputation of single nucleotide polymorphisms (SNPs) from whole-genome sequencing of a subset of individuals [12]. Sequencing of all genes in the genome (exome) [13, 14] is an alternative approach relying on the hypothesis that functional disease-associated variation resides in the coding regions. Exome sequencing has proven valuable in the search for mutations responsible for Mendelian diseases [15, 16] and emerging reports suggest the benefit of applying large-scale exome sequencing to uncover variation associated with complex human traits [17, 18].
Here we present the results of a first-generation medium-pass (8×) exome sequencing approach in 2,000 Danish individuals (stage 1) with follow-up of 16,192 SNPs in 15,989 Danes (stage 2) and replication of 45 SNPs, discovered in a joint analysis of stage 1 and 2, in up to 63,896 Europeans (stage 3) (Fig. 1). To achieve sufficient statistical power a large number of the SNPs selected from stage 1 were genotyped in the much larger sample size in stage 2 making the statistical power comparable to a study where all individuals from both stage 1 and 2 are genotyped for all SNPs [19]. Our objective was to find novel associations of coding variants at minor allele frequencies (MAFs) above 1% with metabolic phenotypes.

Overview of the study. UTR, untranslated region
Methods
Study populations
Danish individuals investigated in stage 1 and 2 of the study were selected from five Danish centres (electronic supplementary material [ESM] Table 1). Exome sequencing in stage 1 (Fig. 1) was performed in 2,000 individuals. Of these, 1,000 were cases recruited based on the presence of type 2 diabetes, BMI >27.5 kg/m2 and hypertension (systolic/diastolic BP >140/90 mmHg or use of antihypertensive medication) to represent common forms of type 2 diabetes and 1,000 were control individuals who all had fasting plasma glucose <5.6 mmol/l, 2 h post-OGTT plasma glucose <7.8 mmol/l, BMI <27.5 kg/m2 and BP <140/90 mmHg (ESM Table 2). In stage 2, 16,192 SNPs were analysed in all 15,989 Danish individuals recruited from five Danish centres (ESM Table 1) in order to perform association mapping of metabolic traits. The individuals in whom exome sequencing was performed in stage 1 were, to obtain called genotypes, among the 15,989 samples genotyped in stage 2. Data from the five Danish centres were pooled in stage 2 analyses. In brief, type 2 diabetes association studies were performed in 4,854 cases defined by WHO 1999 criteria [20] and in 7,325 non-diabetic control individuals. Obesity was studied in 5,488 obese cases (BMI ≥30 kg/m2) and 4,851 lean controls (BMI <25 kg/m2) while hypertension was investigated in 7,299 cases (BP >140/90 mmHg or treated with antihypertensive medication) and 3,290 controls (BP <140/90 mmHg). In analysis of quantifiable metabolic traits, BMI and waist circumference were studied in all samples (n up to 14,819) with available phenotype data excluding individuals treated with insulin. In studies of fasting plasma glucose (n = 9,087) and fasting serum insulin (n = 8,419) all previously diagnosed and treated diabetes patients (n = 1,743) were excluded while individuals treated with lipid-lowering drugs (n = 110) were excluded in analyses of fasting lipid levels (n = 13,326). In studies of systolic and diastolic BP all individuals treated with antihypertensive medication (n = 968) were excluded leaving 12,651 individuals for analyses. Clinical samples from six different European countries were investigated in replication studies of selected SNPs (stage 3) (ESM Table 3). All participants in the study gave written informed consent. The studies were conducted in accordance with the Declaration of Helsinki II and were approved by the local Ethical Committees.
Exon capture, Illumina sequencing and quality control of exome sequencing outcome
Exome capture by a NimbleGen 2.1M HD array (target region 34.1 Mb, 21,810 genes) and Illumina GAII sequencing were performed on DNA from the 2,000 individuals by methods previously described [14]. Samples were not randomised in the capture and sequencing processes. The effective reads (ESM Table 4) were aligned to the human reference genome (assembly hg18, NCBI build 36.3) using SOAPaligner (http://soap.genomics.org.cn/, accessed 01/03/2009). The average sequencing depth per sample was 11× and 96% of targeted bases were covered by at least one read (ESM Fig. 1). All uniquely mapped reads were used for further analyses. The nucleotide mismatch rates were estimated from the proportion of mismatches between all bases from uniquely aligned reads. The mismatch rates ranged from 0.41% to 2.65% in the 2,000 samples with a median of 1%. To investigate the error distributions in the data, the average base quality (the Q-score) of all sequencing reads was examined. When increasing the Q-score threshold the average depth declined. The Q20 threshold (1% error rate by quality score definition) was applied in all further analyses. Further quality control was done by investigating the mismatch rates of aligned bases with different quality scores and by examining the distribution of per-base sequencing depth (ESM Fig. 2). Due to the use of the Q20 threshold and multiple hits restrictions approximately 20% of the sequencing data was discarded and the average depth per site declined from 11× to 8× (8.2× in controls, 7.7× in cases, ESM Fig. 3).
The data quality for individual samples was evaluated using called genotypes by comparing with previously genotyped SNPs and by comparing phenotypic sex with genetically determined sex estimated from the heterozygosity of the SNPs on the X-chromosome. Following bar-coding and sex comparison, 1,974 samples (986 cases and 988 controls) were available for SNP detection and association analyses.
SNP detection and allele frequency estimation in exome sequencing data
Two different approaches to obtain genotype likelihoods were applied. SOAPsnp (http://soap.genomics.org.cn/, accessed 01/03/2009) [21] was used to generate genotype likelihoods, which were used to call genotypes for quality control. For allele frequency estimation and association analysis we estimated the type-specific error rates directly from the putative polymorphic sites and used these error rates combined with the base counts at each position to obtain the genotype likelihoods [20]. As a first step in the identification of SNPs for association testing the allele frequencies of all putative polymorphic sites were estimated using the allele frequency estimator by Li et al [14]. A high error rate of 0.25% was assumed for all error types. Putative polymorphic sites were those with an allele frequency above 0.25%. Then, the allele frequencies were estimated using a maximum likelihood estimator [22], which assumes that the sites are diallelic and takes the uncertainty in the minor allele into account by summing likelihoods over all possible three minor alleles. The discovered SNPs were compared with HapMap data for overlapping SNPs and showed high concordance (ESM Fig. 4). Comparison of allele frequencies with SNPs genotyped in stage 2 (see below) showed high correlation (ESM Fig. 5).
Association analyses in stage 1
We identified 70,182 variable sites with an allele frequency higher than 1% and a total depth per site summed across all individuals above 1,000× corresponding to 0.5× per individual (Table 1, ESM Table 5). Before performing association analysis on the sequencing data we chose to include multiple stringent filters. This was done to remove SNPs that either were likely to be errors or showed bias that could be correlated with case–control status (ESM Fig. 6). Filters based on the base quality scores and based on biases observed in sequencing time were applied. The case–control association analyses were performed using a likelihood ratio test directly on the observed reads taking the uncertainty of the reads into account [22]. No covariates were included in the analysis.
Although the filtering removed the bias from the single SNP analysis, burden tests are much more sensitive to small biases because the bias accumulates when analysing multiple variants. Therefore, no burden tests were performed.
SNP selection for stage 2
SNPs were selected for genotyping in stage 2 from the exome sequencing based on three criteria: (1) SNPs nominally associated (p < 0.05) with case–control status in stage 1 were selected; (2) all SNPs annotated to one of the four annotation categories (i.e. all variants annotated as nonsense variants, non-synonymous variants, variants located in splice sites or variants in untranslated regions) were prioritised regardless of association p value; (3) synonymous variants in 192 loci previously associated with common metabolic traits at genome-wide significance. After quality control (see below), 16,192 SNPs were available for analyses. Of these, 995 SNPs were selected based on the first criterion, 13,959 based on the second criterion while 686 fulfilled both the first and the second criteria. Finally, 552 SNPs were selected based on the third criterion (Fig. 1).
Stage 2 genotyping, quality control and association analyses
SNPs selected from stage 1 were genotyped in 16,988 samples in stage 2 by a custom-designed Illumina iSelect array. Samples were randomised before genotyping. Quality control of samples included removing closely related individuals, individuals with an extreme inbreeding coefficient, individuals with a low call rate, individuals with a mislabelled sex and individuals with a high discordance rate to previously genotyped SNPs. The quality control criteria were fulfilled by 15,989 individuals. Genotypes were obtained for 18,744 SNPs. Of the SNPs discovered in stage 1, 5.1% were not polymorphic when genotyped in stage 2. The SNPs were filtered based on their MAF (>0.5%), genotype call rate (>95%), Hardy–Weinberg equilibrium (p > 10−7) or cross-hybridisation with the X-chromosome, with 16,192 SNPs passing all filters.
Two analyses were done on stage 2 data. First, an enrichment analysis of SNPs selected from stage 1 based on nominal association (n = 1,681) was performed estimating the over-representation of low p values in analyses of type 2 diabetes, obesity and hypertension in stage 2 data. Second, we performed single SNP association analyses, including stage 2 genotyping data for all 15,989 stage 1 and stage 2 individuals, with 12 variables and SNPs for replication in stage 3 were selected based on these analyses. Three binary (type 2 diabetes, obesity, hypertension) and nine quantitative traits (BMI, waist circumference, systolic and diastolic BP, fasting levels of plasma glucose and serum insulin, cholesterol, HDL-cholesterol and triacylglycerol) were analysed. Association analysis of each SNP was performed using linear or logistic regression assuming an additive or log-additive model. Principal component analysis was performed using the covariance matrix [23] and the first principal component and sex were included in the model as covariates. All quantitative traits were rank normalised to a normal distribution before analysis. No inflations in test statistics for the 12 traits were observed after correction by genomic control (λGC 1.00–1.09, ESM Fig. 7).
SNP selection for stage 3
To follow up on the most promising associations from stage 2 association analyses we selected top hits for 12 different metabolic traits. SNPs were selected based on association for each trait (p < 10−3 for type 2 diabetes, obesity, BMI, waist circumference, fasting glucose, fasting insulin and p < 10−4 for all other traits). SNPs in linkage disequilibrium (LD) (r 2 > 0.2) with a known genome-wide significant associated lead SNP for the given metabolic trait were excluded. Phased data from the 1000 Genomes project were used to estimate LD. For the lipid traits we additionally defined a known associated locus as the region spanning 250 Kb up- and downstream of the known associated SNP. All SNPs within these regions were excluded for follow-up.
Stage 3 genotyping
Forty-five SNPs were selected for stage 3 replication and were genotyped in up to 63,896 individuals from seven centres (ESM Table 3). The trait-specific sample sizes are described in ESM Table 6.
Meta-analysis of stage 3 data and meta-analysis stage 2 and stage 3 results
First, association results from the seven centres of stage 3 were combined by meta-analysis to obtain an overall replication result. Second, the Danish discovery data from stage 2 were meta-analysed with the seven replication centres to obtain an overall combined result. The effect for each SNP was estimated by inclusion in a fixed-effects meta-analysis using METAL [24]. For quantifiable traits an overall z statistic relative to each reference allele was estimated based on p values and direction of effects adjusted for the number of individuals in each sample. For dichotomous traits the estimate was weighted according to the estimated SEs by using the inverse corresponding SE.
In meta-analysis of all data, we applied a Bonferroni correction for the number of SNPs and the number of traits analysed (p corrected = 0.05/(70,182 × 12) = 5.9 × 10−8). This correction is conservative as we did not take into account SNP or trait correlations. The corrected threshold is close to conventional genome-wide significance level (p = 5 × 10−8).
Gene expression analysis
Gene expression levels of CD300LG, COBLL1, MACF1, ACP1, ZFAND2B, GPSM1, PRRC2A and GRB14 were quantified by TaqMan real-time PCR (Applied Biosystems, Foster City, CA, USA) in a human tissue mRNA panel including aorta, leucocytes, total brain, hippocampus, hypothalamus, pituitary gland, colon, total small intestine, jejunum, ileum, adipose tissue, kidney, liver, pancreas, skeletal muscle and placenta (ClonTech Laboratories, Mountain View, CA, USA).
Further information
Additional description of methods can be found in the ESM Methods & Results.
Results
Whole exome sequencing (stage 1)
The workflow of the project is shown in Fig. 1. In stage 1, 2,000 individuals were exome sequenced to a median coverage per individual of 91% of the target region. Hereof, 986 metabolic cases and 988 controls with an average depth of 8× fulfilled quality filtering (ESM Fig. 2, ESM Table 4). In total, 70,182 low-frequency and common variants with an estimated MAF above 1% were identified (Table 1, ESM Table 5, ESM Fig. 8). In the initial association analyses with case–control status a general inflation of the test statistics was observed. Application of a stringent set of SNP filtering criteria to remove the bias and restriction of the association analysis to the 48,035 SNPs that fulfilled all filtering criteria resulted in a low inflation rate (λGC 1.05) (ESM Fig. 9). As expected no strong associations were found but instead, as part of the study design, a number of SNPs in the tail of the p value distribution were selected for genotyping in stage 2.
Association with metabolic traits in Danish individuals (stage 2)
To follow up on the outcome of exome sequencing-based SNP discoveries and association analysis in a larger sample set, 16,192 SNPs were analysed in 15,989 Danes. Of these SNPs, 54% were not present on any of the most commonly used GWAS arrays and 50% were not imputable from GWAS data using HapMap as reference panel.
Initially we performed an enrichment analysis for the SNPs nominally associated with case–control status in exome sequencing data (p < 0.05, n = 1,681). These analyses showed an excess of low p values in stage 2 association results for type 2 diabetes (ESM Figs 10, 11). The estimated fraction of associated SNPs was 3.1%, corresponding to 52 expected true associations among the 1,681 SNPs, yet some might be associated due to LD with the same causal variant. No excess of low p values were found in analyses of obesity and hypertension (ESM Figs 12, 13).
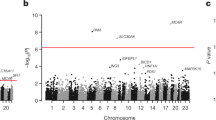
In further analyses of the 16,192 SNPs all 15,989 individuals were included to increase statistical power and SNPs for follow-up in stage 3 were prioritised from examinations of three binary traits (type 2 diabetes, obesity and hypertension) and nine quantifiable traits (BMI, waist circumference, systolic and diastolic BP, fasting levels of plasma glucose and serum insulin, total cholesterol, HDL-cholesterol and triacylglycerol). In analyses of the 12 metabolic phenotypes the strongest novel associations were demonstrated between the CD300LG R82C missense variant and fasting HDL-cholesterol (β = −0.18, p = 7.2 × 10−8) and the COBLL1 rs7607980 variant and type 2 diabetes (OR 0.80, p = 7.2 × 10−8). These were the only associations with p values below 10−6 while a number of potential associations with uncorrected p values below 10−4 were detected (Fig. 2, ESM Fig. 7).

Manhattan plots of 16,192 SNPs for 12 metabolic traits in up to 15,989 Danish individuals (stage 2). For each of the traits the −log10(p) was plotted against the chromosome position. SNPs that have been established as known genome-wide associated signals for each trait are marked in orange. The dotted line indicates Bonferroni correction significance threshold corrected for 16,192 SNPs and 12 traits. The association analyses were performed with logistic or linear regression adjusted for first principal component and sex. All p values were corrected by genomic control
SNPs showing potential novel association with one or more of the 12 traits were selected for replication in stage 3. This selection yielded 51 associations for eight traits covered by 45 unique SNPs (ESM Table 7). SNPs were selected from association results of type 2 diabetes (11 SNPs), obesity (nine SNPs), BMI (three SNPs), waist circumference (one SNP), fasting glucose (seven SNPs), fasting insulin (eight SNPs), fasting HDL-cholesterol (two SNPs) and fasting triacylglycerol (one SNP). A SNP in ELOVL3 showed potential association with both type 2 diabetes, fasting plasma glucose and waist circumference while two SNPs in ACP1 and SLC27A4 were selected from analyses of obesity, BMI and waist circumference (ESM Table 8). We did not identify any SNPs from association results of hypertension, systolic and diastolic BP and fasting total cholesterol that fulfilled the selection criteria.
Replication of selected associations in European samples (stage 3)
The 45 SNPs covering the 51 potential associations discovered in stage 2 were genotyped in up to 63,896 Europeans for replication in stage 3 (Fig. 1, ESM Table 6). Meta-analysis of stage 3 replication data showed nominal replication (p < 0.05) for the same trait in a consistent direction for seven of the 51 selected associations (ESM Table 9). In meta-analysis of Danish stage 2 data and stage 3 replication data, three SNPs were associated at p < 5.9 × 10−8 (Table 2, ESM Figs 14, 15). A low-frequency (MAF 3.5%) non-synonymous (R82C) polymorphism in CD300LG was associated with lower fasting levels of serum HDL-cholesterol while two common (MAF 12.5% and 23.4%, respectively) non-synonymous polymorphisms in COBLL1 and MACF1 were associated with type 2 diabetes (Table 2). All three replicated SNPs in CD300LG, COBLL1 and MACF1 were selected for stage 2 based on their annotation (missense) and the COBLL1 variant was also selected based on its stage 1 association p value. The effect of the minor allele of CD300LG R82C on fasting HDL-cholesterol in repeated analyses in replication cohorts without rank normalisation of HDL-cholesterol was 0.051–0.072 mmol/l. Further in-silico replication data for COBLL1 and MACF1 were obtained from existing GWAS meta-analysis data [2] (ESM Table 10) while no previous association data exists for the CD300LG variant. In additional analyses of other metabolic traits, the CD300LG variant also associated (p < 0.001) with increased fasting serum triacylglycerol while the MACF1 rs2296172 variant also associated with decreased fasting HDL-cholesterol (ESM Table 11). No secondary associations were seen for the COBLL1 rs7607980 variant. Analyses of gene expression in a tissue panel showed that CD300LG is expressed in adipose tissue, skeletal muscle and placenta (ESM Fig. 16). Data showed that COBLL1 is expressed in pancreatic islets and kidney and to some degree in skeletal muscle, liver and adipose tissue while MACF1 is expressed in more tissues including pancreas and skeletal muscle.
Discussion
To discover novel associations between coding polymorphisms with a MAF above 1% and common metabolic traits we sequenced the exomes of 1,974 Danes to a depth of 8× and subsequently performed a two-stage follow-up in 15,989 Danes and in a further 63,896 Europeans. We identified a low-frequency amino-acid polymorphism in CD300LG associated with fasting HDL-cholesterol and two common amino-acid polymorphisms in COBLL1 and MACF1 associated with type 2 diabetes.
While the outcome of this comprehensive study may seem modest it remains a first-line report of challenges with large-scale next-generation sequencing studies of complex traits. Strengths of the study include the thorough replication in European samples, bringing high confidence in the reported associations, yet notable drawbacks are related to the early-stage exome capture technology and sequencing with a relatively low depth; together with bias in the sequencing data, in part coming from lack of sample randomisation, leading to the inability to assess the impact of rare variation alone or as gene-based combinations.
The effect of CD300LG R82C on fasting HDL-cholesterol was higher than all but one of the GWAS-identified HDL-cholesterol-associated variants [10]. CD300LG is a type I membrane glycoprotein that contains a single immunoglobulin V-like domain [25, 26]. The protein has been proposed to serve multiple functions, including endocytosis of various immunoglobulins [25] and mediation of L-selectin-dependent lymphocyte rolling [26], and has been shown to bind a broad range of polar lipids [27]. In-silico prediction by PolyPhen and SIFT indicated that the non-conservative R82C substitution is damaging to protein function suggesting that R82C could be the functional variant in this locus. Obviously, functional studies are needed to provide further evidence of the role of this variant.
The variants N939D in COBLL1 and M2290V in MACF1 were associated with type 2 diabetes, yet non-coding SNPs in these loci have previously been associated with other metabolic phenotypes [3, 6, 10, 28]. In the COBLL1 locus (ESM Fig. 17) the intergenic rs10195252 is reported to associate with fasting triacylglycerol [10] and waist-to-hip ratio in women while the intergenic rs3923113 was reported to associate with type 2 diabetes in a GWAS in individuals of South Asian ancestry [3]. COBLL1 N939D found here is in partial LD with these variants (HapMap release 27: r 2 = 0.18 and r 2 = 0.20, respectively) and conditional analysis showed that COBLL1 N939D carries the effect on type 2 diabetes when conditioning on rs10195252 or rs3923113. Two SNPs in the region have been implicated in the regulation of fasting circulating levels of triacylglycerol and HDL-cholesterol [10]. COBLL1 N939D is in high LD (HapMap release 27: r 2 = 0.98) with the HDL-cholesterol-associated variant [10], and we confirmed the association with HDL-cholesterol for this locus. COBLL1 N939D is also in high LD (HapMap release 27: r 2 = 0.97) with rs12328675 reported to associate with fasting triacylglycerol; however, we observed no association with fasting triacylglycerol levels. The M2290V variant in MACF1 was shown to increase the risk of type 2 diabetes (Table 2) and subsequent analyses of related metabolic phenotypes showed that the same allele also decreased fasting serum HDL-cholesterol levels. PABPC4 rs4660293, which is correlated with MACF1 rs2296172 (HapMap CEU release 27: r 2 = 0.64) has previously been reported to associate with HDL-cholesterol [10]. The biological functions of the associated variants in COBLL1 and MACF1 are unknown; variants in the COBLL1 locus may, however, influence expression of nearby GRB14 to change insulin sensitivity [6, 29].
In the present sequencing-based study initiated in early 2008 we applied exome sequencing to a depth of 8× in 1,974 individuals to discover variants associated with metabolic traits. This and other reports [17, 18] constitute the first indications that exome sequencing is a useful tool in complex traits genetics. Yet, for studying low-frequency and common variation not captured by the standard GWAS design the most cost-effective design for the near future may be to impute variants in standard SNP chip genotyped samples based on whole-genome sequence reference panels such as data from the 1000 Genomes Project [30]. In this context, studies based on SNP chip genotyping and imputation based on a local genome-wide sequencing reference set have lately been published [12]. Interestingly, a recently published report suggested that extremely low-pass whole-genome sequencing (0.1–0.5×) and imputation from 1000 Genomes Project reference panel is more cost-efficient than array genotyping for the study of variants with MAF above 1% [31]. While these approaches may work for low-frequency and common variation, the study of rare variation (MAF <0.5–1%) necessitates resequencing to capture the spectrum of variation. As highlighted by restraints in the present study, issues of sequencing depth, sample size and unbiased data generation are of foremost importance. Deep unbiased exome sequencing will also allow for burden test analyses of the combined impact on phenotype of multiple rare and low-frequency variants in a given locus or in other functional units such as a biologically relevant pathway [32].
In conclusion, we performed medium-depth exome sequencing in 2,000 individuals with follow-up in up to 76,071 Europeans and discovered three amino-acid polymorphisms with a frequency above 1% associated with specific metabolic phenotypes. Therefore, low-frequency and common coding polymorphisms with impact on metabolic traits do exist but they do not seem to be widespread. This study serves as an indication of the utility of exome sequencing in complex metabolic traits.
Abbreviations
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- SNP:
-
Single-nucleotide polymorphism
References
Manolio TA, Brooks LD, Collins FS (2008) A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118:1590–1605
Voight BF, Scott LJ, Steinthorsdottir V et al (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42:579–589
Kooner JS, Saleheen D, Sim X et al (2011) Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 43:984–989
Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116
Grarup N, Sparsø T, Hansen T (2010) Physiologic characterization of type 2 diabetes-related loci. Curr Diab Rep 10:485–497
Heid IM, Jackson AU, Randall JC et al (2010) Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 42:949–960
Speliotes EK, Willer CJ, Berndt SI et al (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42:937–948
The International Consortium for Blood Pressure Genome-Wide Association Studies (2011) Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478:103–109
Wain LV, Verwoert GC, O'Reilly PF et al (2011) Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 43:1005–1011
Teslovich TM, Musunuru K, Smith AV et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Manolio TA, Collins FS, Cox NJ et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Holm H, Gudbjartsson DF, Sulem P et al (2011) A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet 43:316–320
Albert TJ, Molla MN, Muzny DM et al (2007) Direct selection of human genomic loci by microarray hybridization. Nat Methods 4:903–905
Li Y, Vinckenbosch N, Tian G et al (2010) Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 42:969–972
Ng SB, Buckingham KJ, Lee C et al (2010) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42:30–35
Ng SB, Turner EH, Robertson PD et al (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461:272–276
Sanders SJ, Murtha MT, Gupta AR et al (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–241
O'Roak BJ, Deriziotis P, Lee C et al (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43:585–589
Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38:209–213
World Health Organization Study Group (1999) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Tech. Rep. Ser. WHO/NCD/NCS/99, 2nd edn. World Health Organization, Geneva
Li R, Li Y, Fang X et al (2009) SNP detection for massively parallel whole-genome resequencing. Genome Res 19:1124–1132
Kim SY, Lohmueller KE, Albrechtsen A et al (2011) Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinforma 12:231
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191
Takatsu H, Hase K, Ohmae M et al (2006) CD300 antigen like family member G: a novel Ig receptor like protein exclusively expressed on capillary endothelium. Biochem Biophys Res Commun 348:183–191
Umemoto E, Tanaka T, Kanda H et al (2006) Nepmucin, a novel HEV sialomucin, mediates L-selectin-dependent lymphocyte rolling and promotes lymphocyte adhesion under flow. J Exp Med 203:1603–1614
Cannon JP, O'Driscoll M, Litman GW (2012) Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 64:39–47
Dehghan A, Je D, Barbalic M et al (2011) Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels/clinical perspective. Circulation 123:731–738
Cooney GJ, Lyons RJ, Crew AJ et al (2004) Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23:582–593
1000 Genomes Project Consortium, Durbin RM, Abecasis GR et al (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Pasaniuc B, Rohland N, McLaren PJ et al (2012) Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet 44:631–635
Bansal V, Libiger O, Torkamani A, Schork NJ (2010) Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet 11:773–785
Acknowledgements
The authors thank M. Boehnke (University of Michigan, Ann Arbor, Michigan, USA) for valuable comments on the manuscript. The authors wish to thank staff at Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, Denmark: A. Forman, T. Lorentzen, B. Andreasen and G. J. Klavsen for technical assistance and A. L. Nielsen, G. Lademann and M. M. H. Kristensen for management assistance.
Funding
This project was funded by the Lundbeck Foundation and produced by The Lundbeck Foundation Centre for Applied Medical Genomics in Personalised Disease Prediction, Prevention and Care (LuCamp, www.lucamp.org). The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). Further funding came from the Danish Council for Independent Research (Medical Sciences).
The Inter99 was initiated by Torben Jørgensen (PI), Knut Borch-Johnsen (co-PI), Hans Ibsen and Troels F. Thomsen. The steering committee comprises the former two and Charlotta Pisinger. The study was financially supported by research grants from the Danish Research Council, the Danish Centre for Health Technology Assessment, Novo Nordisk Inc., Research Foundation of Copenhagen County, Ministry of Internal Affairs and Health, the Danish Heart Foundation, the Danish Pharmaceutical Association, the Augustinus Foundation, the Ib Henriksen Foundation, the Becket Foundation, and the Danish Diabetes Association.
The Health2006 was initiated by Allan Linneberg (PI) and Torben Jørgensen (co-PI). The study was financially supported by grants from the Velux Foundation, The Danish Medical Research Council, Danish Agency for Science, Technology and Innovation, The Aase and Ejner Danielsens Foundation, ALK-Abelló, (Hørsholm, Denmark) and Research Centre for Prevention and Health (the Capital Region of Denmark).
In Finland this work has been supported by the following grants to M. Laakso: Academy of Finland, the Finnish Diabetes Research Foundation, the Finnish Cardiovascular Research Foundation, and EVO grant from the Kuopio University Hospital (5263).
In the UK the Collection of the UK type 2 diabetes cases was supported by Diabetes UK, BDA Research and the UK Medical Research Council (Biomedical Collections Strategic Grant G0000649). The UK Type 2 Diabetes Genetics Consortium collection was supported by the Wellcome Trust (Biomedical Collections Grant GR072960). We acknowledge use of DNA from The UK Blood Services collection of Common Controls (UKBS-CC collection), funded by the Wellcome Trust grant 076113/C/04/Z and by NIHR programme grant to NHSBT (RP-PG-0310-1002). The collection was established as part of the Wellcome Trust Case Control Consortium (WTCCC). For the 1958 Birth Cohort, venous blood collection was funded by the Medical Research Council grant G0000934 (awarded under the Health of the Public initiative), peripheral blood lymphocyte preparation by Juvenile Diabetes Research Foundation/Wellcome Trust and the cell-line production, DNA extraction and processing by the Wellcome Trust grant 06854/Z/02/Z. The genotyping was supported by the Wellcome Trust (083270) and EU (ENGAGE: HEALTH-F4-2007- 201413). A. P. Morris is a Wellcome Trust Senior Fellow (081682/Z/06/Z) and M. McCarthy receives funding from the Oxford NIHR Biomedical Research Centre. We acknowledge the contribution of M. Sampson.
In the Netherlands the work in this study was financially supported by the Dutch Diabetes Research Foundation grant 2006.00.060 and Biobanking and Biomolecular Research Infrastructure the Netherlands (BBMRI-NL).
The D.E.S.I.R. cohort was supported by co-operative contracts between Inserm, CNAMTS, Novartis, Lilly and sanofi-aventis, by Inserm (Réseaux en Santé Publique, Interactions entre les determinants de la santé, Cohortes Santé TGIR 2008), by the Association Diabète Risque Vasculaire, the Fédération Française de Cardiologie, La Fondation de France, ALFEDIAM, ONIVINS, Société Francophone du Diabète; Ardix Medical, Bayer Diagnostics, Becton Dickinson, Cardionics, Lilly, Merck Santé, Novo Nordisk, Pierre Fabre, Roche, sanofi-aventis, Topcon.
Work in Sweden was supported by grants from the Swedish Research Foundation (Dnr-349-2006-6589, 2009-1039, 521-2010-3490) and Knut & Alice Wallenberg Foundation.
Work in Finland was supported by grants from the Sigrid Juselius Foundation, Folkhälsan Research Foundation and the Finnish Medical Society.
The Gene-Lifestyle interactions And Complex traits Involved in Elevated Disease Risk (GLACIER) study is nested within the Northern Swedish Health and Disease Study cohort and the Västerbotten Intervention Programme (VIP). The research programme was approved by the Ethical Review Board in Umeå, Sweden. We are indebted to the study participants who dedicated their time and samples to these studies. We also thank the VIP and Umeå Medical Biobank staff for biomedical data collection and preparation. We specifically thank Å. Ågren (Umeå Medical Biobank) for data organisation, and K. Enqvist and T. Johansson (Västerbottens County Council) for expert technical assistance with DNA preparation. The GLACIER Study was funded by project grants from Novo Nordisk (P. W. Franks [PWF]), the Swedish Heart-Lung Foundation (PWF), the Swedish Diabetes Association (to PWF), Påhlssons Foundation (PWF), the Swedish Research Council (PWF), Umeå University Career Development Award (PWF) and The Heart Foundation of Northern Sweden (PWF).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
All authors substantially contributed to conception and design, acquisition of data, or analysis and interpretation of data used in the study.
OP, A. Albrechtsen, NG, YL, TS, JH, TH, JW and RN conceived, coordinated and executed the present study.
NG, TS, JH, M. Lajer, AAN, PR, LT, CC, IB, TL, DRW, AL, T. Jørgensen, TH and OP recruited and phenotyped study participants enrolled in exome sequencing (stage 1) and large-scale follow-up genotyping (stage 2).
SC, GC, JMD, ASFD, MD, TF, TMF, GH, ATH, GAH, JK, OL, M. Marre, ADM., GN, PN, CNAP, FR, OR, TT, EV, TVV, MW, LY, PWF, BB, PF, MIM, M. Laakso and LG recruited and phenotyped study participants used in replication study (stage 3).
A. Albrechtsen, YL, TS, GT, T. Jiang, SYK, TK, RW, HJ, HZ, XJ, HL, XL, TM, XM, BM, MT, BW, HW, FX, CY, XZ, JZ, QZ, HZ, YZ, JW, RN and OP performed exome sequencing and the related data analysis.
A. Albrechtsen, NG, TS, GT, HC, Q. Li, CN, LS, KB, YC, YG, KH, SH, XJ, JMJ, WL, Q. Liao, XL, MPM, YW, HW, XZ, QZ, WZ, HZ, JW, RN and OP performed stage 2 genotyping and statistical analyses.
A. Albrechtsen, NG, TS, APM, CL, SC, A. Stančáková, AJB, CJG, GH, NWR, FR, NR, OR, PES, MS, NVL, TVV, LMT, PWF, PF, M. Laakso and OP performed stage 3 genotyping and statistical analyses and meta-analyses.
JM, RRM, TWS, and MAV performed biological studies.
The overall analysis group consisted of A. Albrechtsen, NG, TS, SYK, TK, Q. Li, LS, YC, YG, QL and RN.
A. Albrechtsen, NG, TS, TH and OP wrote the initial manuscript and all authors contributed substantially to data interpretation and paper review and approved the final manuscript.
OP, GA, A. Astrup, LB, KK, TWS, TL, DRW, AL, T. Jørgensen, TH, JW and RN are founders of the Lundbeck Foundation Centre for Applied Medical Genomics in Personalised Disease Prediction, Prevention and Care and designed the experimental protocols of the consortium.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Additional information
A. Albrechtsen N. Grarup, Y. Li, T. Sparsø and G. Tian contributed equally to this study.
Members of the D.E.S.I.R. Study Group and the DIAGRAM Consortium are listed in the electronic supplementary material (ESM).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods and Results
(PDF 562 kb)
ESM Fig. 1
(PDF 200 kb)
ESM Fig. 2
(PDF 263 kb)
ESM Fig. 3
(PDF 223 kb)
ESM Fig. 4
(PDF 228 kb)
ESM Fig. 5
(PDF 339 kb)
ESM Fig. 6
(PDF 224 kb)
ESM Fig. 7
(PDF 474 kb)
ESM Fig. 8
(PDF 241 kb)
ESM Fig. 9
(PDF 330 kb)
ESM Fig. 10
(PDF 334 kb)
ESM Fig. 11
(PDF 473 kb)
ESM Fig. 12
(PDF 481 kb)
ESM Fig. 13
(PDF 473 kb)
ESM Fig. 14
(PDF 341 kb)
ESM Fig. 15
(PDF 332 kb)
ESM Fig. 16
(PDF 391 kb)
ESM Fig. 17
(PDF 420 kb)
ESM Table 1
(PDF 200 kb)
ESM Table 2
(PDF 199 kb)
ESM Table 3
(XLS 72 kb)
ESM Table 4
(PDF 197 kb)
ESM Table 5
(PDF 198 kb)
ESM Table 6
(PDF 317 kb)
ESM Table 7
(PDF 321 kb)
ESM Table 8
(PDF 233 kb)
ESM Table 9
(PDF 348 kb)
ESM Table 10
(PDF 290 kb)
ESM Table 11
(PDF 296 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Albrechtsen, A., Grarup, N., Li, Y. et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 56, 298–310 (2013). https://doi.org/10.1007/s00125-012-2756-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2756-1