
Abstract
Aims/hypothesis
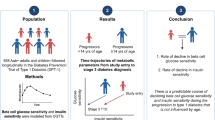
Few studies have explored the epidemiology of beta cell loss in youth with diabetes. This report describes the evolution and major determinants of beta cell function, assessed by fasting C-peptide (FCP), in the SEARCH for Diabetes in Youth study.
Methods
Participants were 1,277 youth with diabetes (948 positive for diabetes autoantibodies [DAs] and 329 negative for DAs), diagnosed when aged <20 years, who were followed from a median of 8 months post diagnosis, for approximately 30 months. We modelled the relationship between rate of change in log FCP and determinants of interest using repeated measures general linear models.
Results
Among DA-positive youth, there was a progressive decline in beta cell function of 4% per month, independent of demographics (age, sex, race/ethnicity), genetic susceptibility to autoimmunity (HLA risk), HbA1c and BMI z score, or presence of insulin resistance. Among DA-negative youth, there was marked heterogeneity in beta cell loss, reflecting an aetiologically mixed group. This group likely includes youths with undetected autoimmunity (whose decline is similar to that of DA-positive youth) and youth with non-autoimmune, insulin-resistant diabetes, with limited decline (~0.7% per month).
Conclusions/interpretation
SEARCH provides unique estimates of beta cell function decline in a large sample of youth with diabetes, indicating that autoimmunity is the major contributor. These data contribute to a better understanding of clinical evolution of beta cell function in youth with diabetes, provide strong support for the aetiological classification of diabetes type and may inform tertiary prevention efforts targeted at high-risk groups.
Similar content being viewed by others

Introduction
Diabetes mellitus is a spectrum of metabolic diseases with distinct aetiologies, characterised by a progressive decline in beta cell function and mass [1, 2]. An increasing body of evidence demonstrates the importance of preserving endogenous beta cell function in patients with diabetes, regardless of aetiology or presumed diabetes type [3, 4]. However, there is a paucity of data regarding the epidemiology of beta cell function, including the rate and major determinants of beta cell decline, especially among contemporary youth with diabetes [5]. Limited information has been provided by interventional studies in youth with autoimmune diabetes [6–9] and individuals with adult-onset diabetes [10]. Recent data suggest that there is a progressive deterioration of beta cell function both before and after the diagnosis of autoimmune diabetes [11, 12]; however, limited information is available regarding the factors that predict the rate of decline. Even less is known about the clinical evolution of beta cell function in youth without autoimmune diabetes. Moreover, most previous studies were designed to explore the effects of specific interventions, were usually much smaller and often included selected participants, making it difficult to draw conclusions applicable to the general population of youth with diabetes. Such data may be relevant for the aetiological classification of diabetes type.
SEARCH for Diabetes in Youth is a population-based, multicentre and multi-ethnic study of paediatric diabetes in the USA. With the aim of rigorously characterising the epidemiology of diabetes in youth, this study has carefully collected detailed clinical and pathophysiological data on a large number of youth with all types of diabetes. Moreover, SEARCH has recently developed operational definitions of diabetes types, as recommended by the ADA expert committee, using an aetiological approach based on assessment of autoimmunity and insulin resistance (IR), in which four aetiological subgroups were defined [13]. The purpose of this manuscript is to describe the presentation, short-term clinical evolution and major determinants of decline in beta cell function, as measured by fasting C-peptide (FCP), in a general population of youth with diabetes, using data from the prospective cohort of the SEARCH for Diabetes in Youth study.
Methods
Overview
Data for these analyses derive from the SEARCH for Diabetes in Youth prospective cohort study. A detailed description of SEARCH study methods has been published elsewhere [14]. Briefly, SEARCH has been conducting population-based ascertainment of youth and young adults with newly diagnosed (incident) diabetes diagnosed when <20 years of age, starting in 2002 and continuing through to the present. SEARCH recruited participants from four geographically defined populations in the USA (Ohio, Colorado, South Carolina and Washington) and also Indian Health Service beneficiaries from several American Indian populations and enrollees in several managed healthcare plans in California and Hawaii. Participants with newly diagnosed diabetes in 2002–2005 were invited to participate in a baseline research visit (median duration of diabetes 8 months, interquartile range [IQR] 4.5–14.5 months) and two follow-up exams centred around 12 and 24 months after their baseline visit. At each research visit fasting blood samples were obtained from metabolically stable participants (defined as no episode of diabetic ketoacidosis during the previous month), physical measurements were conducted and questionnaires were administered. The study was reviewed and approved by the local Institutional Review Board(s) that had jurisdiction over the local study population and all participants provided informed consent and/or assent.
Data collection
Study visits occurred after an 8 h overnight fast. Participants did not take diabetes medications on the morning of the visit and long-acting insulin was administered on the evening before the visit and then discontinued. Fasting blood samples were used to analyse diabetes autoantibodies (DAs), HbA1c, lipids and FCP and fasting plasma glucose (FPG). Assays were performed at the Northwest Lipid Metabolism and Diabetes Research Laboratories, University of Washington, the Central Laboratory for SEARCH. Glutamic acid decarboxylase-65 (GADA) and insulinoma-associated-2 (IA-2A) autoantibodies were analysed using a standardised protocol and a common serum calibrator developed by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)-sponsored harmonisation group [15]. The cut-off values for positivity were 33 NIDDKU/ml for GADA and 5 NIDDKU/ml for IA-2A [15]. HbA1c was measured in whole blood with an automated nonporous ion-exchange HPLC system (model G-7; Tosoh Bioscience, San Francisco, CA, USA). Measurements of plasma cholesterol, triacylglycerols and HDL-cholesterol were performed on a Roche Modular-P autoanalyzer (Roche Diagnostics, Indianapolis, IN, USA). Levels of FCP were determined by a two-site immunoenzymetric assay (Tosoh 1800; Tosoh Bioscience) with a sensitivity of 0.05 ng/ml. Human leucocyte antigen class II genotyping (HLA DR/DQ) was performed with a PCR-based sequence-specific oligonucleotide probe system in the laboratories of S. Gaur (University of Washington, Seattle, WA, USA) and H. Erlich (Roche Molecular Systems, Pleasanton, CA, USA) on all consenting participants [16].
Weight and height were measured using standardised procedures and used to calculate BMI. Waist circumference was measured using the National Health and Nutrition Examination Survey (NHANES) III protocol [17].
Insulin sensitivity (IS) was estimated using the following equation: log e IS = 4.64725 − 0.02032 (waist, cm) − 0.09779 (HbA1c, %) − 0.00235 (triacylglycerol, mg/dl; to convert triacylglycerol values from mg/dl to mmol/l, multiply by 0.0113). This equation was developed and validated in a previous study [18] using direct measurements of glucose disposal rate from euglycaemic–hyperinsulinaemic clamps. IR was defined as an IS value below the 25th percentile in the general US youth population (<8.15), as previously described [13].
Data on self-reported race and ethnicity were collected using 2000 US Census questions [19] and classified as Hispanic, non-Hispanic white (NHW), non-Hispanic black, American Indian and Asian/Pacific islander.
Determinants of interest
Autoimmunity was defined by positive titres for either GADA or IA-2A at baseline. Because many participants were treated with insulin, positive insulin autoantibodies were not used. Genetic susceptibility to autoimmunity was defined based on HLA DR/DQ. HLA genotypes were categorised as susceptible (DR3/4, DR4/4, DR4/8, DR4/1, DR 4/13, DR 3/3, DR 3/9, DR 4/9 and DR 9/9 genotypes) and not susceptible, including neutral and protective genotypes [13], as recommended by the Type 1 Diabetes Genetic Consortium, with modifications for the multi-ethnic population [16]. For these analyses race/ethnicity was categorised as NHW and other than non-Hispanic white, including all other racial/ethnic groups. BMI z scores from measured height and weight at baseline visit were derived for age and sex using the US Centers for Disease Control and Prevention National Center for Health Statistics growth charts [20]. Finally, SEARCH participants were also classified according to baseline aetiological category of diabetes type in four mutually exclusive groups as previously described [13]: autoimmune and insulin sensitive, autoimmune and insulin resistant, non-autoimmune and insulin sensitive, non-autoimmune and insulin resistant.
Study participants
The eligible study population consisted of all SEARCH participants who were diagnosed in 2002–2005, who had a baseline visit with non-missing FCP, DA and demographic data, plus at least another follow-up visit with non-missing FCP. There were 2,763 individuals that had a baseline SEARCH visit and 1,895 participants had non-missing FCP, DA and demographic data at baseline. An additional sample of 629 were dropped because they did not have at least one follow-up visit with non-missing FCP, resulting in 1,277 SEARCH individuals contributing to this analysis. Of the 1,277 participants included in these analyses, 634 contributed two visits (baseline plus one follow-up) and 643 contributed three visits (baseline plus two follow-up visits). There were 948 DA-positive participants (487 with double and 461 with single antibody positivity) and 329 DA-negative participants.
Statistical analyses
Statistical analyses were performed using SAS software (version 9.2; SAS Institute, Cary, NC, USA), with p < 0.05 indicating significance. Comparisons of baseline participant characteristics by baseline DA status were examined using χ2 tests, t tests and Wilcoxon tests. Beta cell function was estimated using FCP concentration [21].
Using all baseline and follow-up data, we used mixed effects regression analysis to derive predicted FCP levels over time, based on fitted models that included up to three observed measures of FCP for each participant. These models adjust for the correlation between repeated observations in the same individual and have the advantage of handling a varying number of observations within individuals, thereby allowing for inclusion of the maximum number of data points [22]. Further, this method allows for a random intercept and slope in disease duration to account for within-individual dependence. These models incorporate duration of diabetes (actual time in months) as a continuous time-varying covariate and explore significant associations (statistical interactions) between determinants of interest (baseline DA status, age at diagnosis, sex, race/ethnicity, HLA DR/DQ susceptibility status, baseline BMI z score and HbA1c) and time (duration of diabetes) on FCP levels. All models are also adjusted for site- and time-varying FPG levels measured at each FCP measurement. Since baseline DA status emerged as the main determinant of FCP decline all analyses were stratified by baseline DA status. For DA-positive individuals, the effect of double vs single DA positivity at baseline on FCP decline over time was also explored, adjusting for all the covariates mentioned above. Age at diagnosis was modelled both continuously and categorically (<10 and ≥10 years, for illustrative purposes) with very similar results. To account for a skewed distribution of FCP levels, values were log-transformed for these analyses. Predicted FCP rate of change was expressed as per cent change in FCP level per month with 95% CIs.
Finally, we were also interested in modelling the rate of change in FCP levels as a function of baseline aetiological category of diabetes type. Therefore, a separate model was built to explore the association between the four-level aetiological diabetes type variable (autoimmune and insulin sensitive, autoimmune and insulin resistant, non-autoimmune and insulin sensitive, non-autoimmune and insulin resistant) and log-transformed FCP over time. This analysis excluded 66 participants due to missing data on variables that are part of the IS equation.
Results
Baseline characteristics of study participants, stratified by baseline DA status, are shown in Table 1. As expected, DA-positive participants were younger at diagnosis, more likely to be of male sex and NHW race/ethnicity, more likely to have susceptible HLA DR-DQ genotypes and had lower FCP levels and BMI z scores, but higher baseline FPG levels, compared with those who were DA negative. Table 2 presents the number of individuals participating at each of the three SEARCH visits, their average diabetes duration at each visit and mean FCP and FPG levels, by DA status.
Table 3 presents the associations between baseline FCP levels and determinants of interest, from multivariable analyses stratified by baseline DA status. In both DA-positive and DA-negative participants, younger age at diagnosis, NHW race/ethnicity, higher baseline HbA1c and lower baseline BMI z score were independently associated with significantly lower baseline FCP levels. In DA-positive participants only, male sex was also associated with lower baseline FCP levels. HLA DR/DQ susceptibility status was not significantly associated with FCP levels at baseline in either group.
Figure 1 shows predicted FCP levels over time among youth with positive DA at baseline, by age at diagnosis of diabetes (stratified as <10 and ≥10 years). The model showed that age at diagnosis, sex, race/ethnicity, HLA risk, baseline HbA1c and BMI z score were not associated with FCP decline over time (see electronic supplementary material [ESM] Table 1). The predicted rate of decline in FCP levels among youth positive for DA (shown in Fig. 1 for an average baseline HbA1c of 8%, BMI Z-score of 0.6 and an average time-varying FPG of 9.2 mmol/l) was 4.1% (95% CI 3.9, 4.3) per month, regardless of age at diagnosis, sex, race/ethnicity and HLA risk status. When baseline DA positivity status was added to the model, presence of two DAs vs one DA was the only variable significantly associated with faster decline in FCP levels (p < 0.0001): estimated 4.4% (95% CI 4.1, 4.7) per month in those with double DA positivity vs 3.7% (95% CI 3.4, 4.0) per month in those with single DA positivity (ESM Table 2).

Estimated mean FCP levels and rate of monthly decline among youth with positive DA status at baseline; (a) age at diagnosis of diabetes <10; (b) age at diagnosis ≥10. Black symbols, HLA susceptible; grey symbols, HLA not susceptible; diamonds, NHW race/ethnicity; triangles, other race. Multivariate mixed model predicting log-transformed FCP over time based on diabetes duration, age at diagnosis of diabetes, sex, race/ethnicity, HLA risk group, baseline HbA1c, BMI z score, site and time-varying fasting glucose (FPG). Estimates and rate of decline (95% CIs) are shown for an average baseline HbA1c of 8% (63.93 mmol/mol), BMI z score of 0.6 and an average time-dependent FPG of 9.2 mmol/l. The data under the graph present the number of FCP data points available in various time intervals
Figure 2 shows predicted FCP levels over time among youth with negative DA at baseline, by age at diagnosis of diabetes (<10 and ≥10 years). The model showed that age at diagnosis, race/ethnicity, HLA risk status, baseline HbA1c and BMI z score were independently associated with FCP decline over time (ESM Table 3). Predicted rates of decline are shown in Fig. 2 for an average baseline HbA1c of 7.5%, BMI z score of 1.3 and an average time-varying FPG of 7.3 mmol/l. The fastest rate of decline was predicted in youth of NHW race/ethnicity who had susceptible HLA DR/DQ genotypes (4.0% [95% CI 3.1, 4.9] per month in youth diagnosed at <10 years of age and 3.2% [95% CI 2.4, 3.9] in those diagnosed at age ≥10 years). These estimates are very similar to those predicted for youth who were DA positive at baseline, suggesting undetected autoimmunity in this group of SEARCH participants. Similarly, rates of decline that were consistently elevated were predicted among youth with susceptible HLA DR/DQ genotypes who were of other racial/ethnic groups than NHW (2.9% [95% CI 1.9, 4.0] per month among those with age at diagnosis <10 years and 2.1% [95% CI 1.2, 2.9] per month among those with older age at diagnosis). These data also suggest an undetected autoimmune process, given the presence of susceptible HLA markers. The slowest rates of decline were predicted among DA-negative youth of minority racial/ethnic groups who did not have susceptible HLA DR/DQ genotypes (1.4% [95% CI 0.6, 2.1] per month in those diagnosed at <10 years of age and 0.5% [95% CI 0.1, 1.0] per month in those diagnosed at an age older than 10 years). These participants seem to belong to a different aetiological group characterised by absence of DA and genetic markers of autoimmunity and also by a preponderance of minority racial/ethnic backgrounds. Interestingly, the group of NHW participants without susceptible HLA genotypes who were diagnosed at an age older than 10 years had an intermediate predicted rate of change, 2.5% (95% CI 1.7, 3.2) per month, likely marking an aetiologically mixed group of individuals with and without autoimmune diabetes.

Estimated mean FCP levels and rate of monthly decline among youth with negative DA status at baseline; (a) age at diagnosis of diabetes <10; (b) age at diagnosis ≥10. Black symbols, HLA susceptible; grey symbols, HLA not susceptible; diamonds, NHW race/ethnicity; triangles, other race. Multivariate mixed model predicting log-transformed FCP over time based on diabetes duration, age at diagnosis of diabetes, sex, race/ethnicity, HLA risk group, baseline HbA1c, BMI z score, site and time-dependent fasting glucose (FPG) levels. Estimates and rates of decline (95% CIs) are shown for an average baseline HbA1c of 7.5% (58.47 mmol/mol) and BMI z score of 1.3 and an average time-dependent FPG of 7.3 mmol/l. The data under the graph present the number of FCP data points available in various time intervals
Finally, predicted rates of decline in FCP levels by baseline aetiological category of diabetes type are presented in Table 4. Overall, baseline category of diabetes type was significantly associated with FCP decline (p < 0.0001). The fastest rates of decline were predicted among youth with autoimmunity, regardless of whether they were insulin sensitive (4.0% [95% CI 3.8, 4.3] per month) or insulin resistant (4.2% (95% CI 3.8, 4.7) per month). An intermediate rate of decline (2.4% [95% CI 1.9, 3.0] per month) was predicted for youth without evidence of autoimmunity who were insulin sensitive, while the predicted rate of decline among youth with non-autoimmune, insulin-resistant diabetes was significantly slower (0.7% [95% CI 0.3, 1.2] per month).
Discussion
Our study provides unique quantitative estimates of decline in beta cell function over the first 30 months following diagnosis of diabetes in a large, diverse sample of youth in the USA. SEARCH provides strong evidence that autoimmunity is the main driver of beta cell loss even after diabetes diagnosis. Among DA-positive youth, there is a progressive decline in beta cell function, independent of demographic factors, genetic susceptibility to autoimmunity (HLA risk), HbA1c levels and BMI z score. Among DA-negative youth, there is marked heterogeneity in beta cell loss, reflecting an aetiologically mixed group. Our data contribute to a better understanding of the clinical progression of diabetes in youth in the first years following diagnosis.
In youth with autoimmune diabetes there was an estimated decline in beta cell function of ~4% per month, or approximately 40% per year. Similarly, in a series of 204 consecutive patients with type 1 diabetes, Snorgaard et al [23] reported that FCP levels (on average 0.5 ng/ml at diagnosis) declined in a homogeneous manner at a rate of 0.24 ng/ml annually. Other studies have estimated the decline in beta cell function during the first year after diagnosis of type 1 diabetes, with variable results. Most of the recent studies are based on data provided by the control arm of intervention studies, include small numbers of highly selected participants and use stimulated C-peptide as the measure of beta cell function. While some report modest reduction in beta cell function at 1 year [24, 25], others report a 50% decline in the first year post-diagnosis [1, 26–28], consistent with our data in an unselected population.
There are limited data on factors influencing beta cell function in youth with autoimmune diabetes [12, 29–35]. We found that younger age, male sex, NHW race/ethnicity, higher HbA1c and lower BMI z score were each associated with lower FCP levels at baseline (Table 3). The Diabetes Prevention Trial-Type 1 (DPT-1) study has demonstrated that C-peptide levels at diagnosis are much higher in individuals diagnosed through surveillance of those at high risk [36] compared with those diagnosed in the community, with signs and symptoms of hyperglycaemia. Our findings that higher HbA1c and lower BMI z score are associated with lower baseline FCP levels are consistent with DPT-1 findings, suggesting a more severe or longer preclinical course in those with less residual beta cell function at presentation. Consistent with our data, several reports have shown that younger age is associated with lower C-peptide levels at diagnosis with type 1 diabetes [8, 9, 36]. This may suggest a more aggressive preclinical course or an overall lower beta cell mass in younger children [6], while in older patients greater IR may lead to a clinical presentation at an earlier preclinical stage of disease [21, 37].
Although age, sex, race/ethnicity, HbA1c and BMI z score were each associated with lower FCP levels at baseline (Table 3), none of them were significantly associated with the subsequent rate of decline (Fig. 1). Based on our data, on average, residual beta cell function at any point in time is a function of baseline C-peptide levels and disease duration. Others have also reported that baseline C-peptide level is the strongest predictor of decline in beta cell function in youth [21] and young adults [38] with autoimmune diabetes.
One of our most novel findings is that markers of obesity and IR do not significantly influence the decline in FCP levels after diagnosis of autoimmune diabetes. Investigators and clinicians have previously described a group of youth with autoimmunity and obesity as having ‘hybrid’ or ‘double’ diabetes [39], suggesting a mixed aetiology and, likely, a more aggressive clinical evolution. The aetiological classification of diabetes type proposed by SEARCH [13] and findings from other recent studies [40] provide evidence that rather than being a distinct aetiological category this group represents the upper tail of the distribution of IR and obesity among youth with autoimmune diabetes. We now provide novel evidence that the rate of FCP decline in youth with autoimmune, insulin-resistant diabetes is not significantly different from that observed in youth with autoimmune, insulin-sensitive diabetes (4.2% [95% CI 3.8, 4.7] per month vs 4.0% [95% CI 3.8, 4.3] per month, p = 0.5, Table 4). This was also suggested by baseline BMI z score not being significantly associated with decline in FCP levels in DA-positive youth. These findings lend additional support to the notion that type 1 diabetes is a distinct aetiological category and that the main driver of beta cell loss is autoimmunity, regardless of prevailing IR that may occur simultaneously. These findings also have important clinical implications indicating the importance of a correct diagnosis of autoimmune type 1 diabetes in obese youth, and thus adequate insulin treatment.
Less is known about the clinical history of beta cell function in youth with non-autoimmune diabetes. Although small studies suggested that youth with type 2 diabetes have some degree of beta cell dysfunction at diagnosis [41, 42], there are no large prospective studies of the natural evolution and determinants of beta cell function in youth with type 2 diabetes. The estimated rate of decline among SEARCH youth who had non-autoimmune, insulin-resistant diabetes was 0.7% per month, or approximately 8% per year, in the first 30 months following diagnosis (Table 4). This is consistent with studies in adults showing a 7% decline annually [3, 43]. However, a recent study has suggested no signs of progressive beta cell damage during 20 years of prospective follow-up of autoantibody-negative, adult-onset diabetes [44], which contrasts with the generally held view of progressive beta cell loss in type 2 diabetes. There is an obvious need for long-term follow-up studies of individuals with youth-onset, non-autoimmune, type 2 diabetes.
Our study has some limitations and unique strengths. Because SEARCH is a large epidemiological study, we used fasting, rather than stimulated, C-peptide levels to assess baseline and change in beta cell function over time. FCP level is easy to obtain and correlates well with stimulated C-peptide [45–47], although it may be insufficient to detect subtle effects of therapy. In addition, we used a standard protocol to assess beta cell function, with all measurements conducted in the fasting state, in the absence of hypo-or hyperglycaemia, which are known to affect FCP levels [48]. Only a fraction of participants had FCP measures in the first 6 months after diagnosis and, therefore, during this period the rate of decline in FCP may have been estimated with less precision. Only two autoantibodies were measured in SEARCH. Thus, individuals who may have autoimmune markers other than GADA or IA-2A were misclassified as having non-autoimmune diabetes. However, the predicted rates of change in FCP levels based on models that included other characteristics, such as HLA DR/DQ susceptibility genotypes, race/ethnicity and age at diagnosis, identified groups of participants with likely undetected autoimmune diabetes. In addition, SEARCH is planning to assess ZnT8 autoantibodies in stored samples once assays are standardised. We also do not have follow-up data on all SEARCH participants. However, minimal differences existed between those who did (n = 1,277) and did not participate (n = 629) in the follow-up assessment. In particular, the proportion of DA-positive and DA-negative participants, the racial/ethnic distribution, the mean age and FCP levels according to DA status were not substantially different. Finally, our data are not the result of a randomised clinical trial, which is much better suited to address specific questions regarding the role of glycaemic control or that of specific therapies, on beta cell decline in youth with diabetes. Our study also has very important strengths. This is a unique cohort of youth with diabetes ascertained without regard to presumed diabetes type, family history or related factors, such as age, treatment or race/ethnicity, which has permitted us to accurately quantify progression of beta cell damage in a large sample of youth with diabetes.
In summary, SEARCH provides unique quantitative estimates of beta cell function decline over the first 30 months following diagnosis in a large sample of youth with diabetes in the USA. Our data provide strong evidence that autoimmunity is the main driver of beta cell loss even after diabetes diagnosis. We also provide novel evidence that markers of obesity and IR do not influence the decline in beta cell function in youth with autoimmune diabetes. Finally, youth with non-autoimmune, insulin-resistant diabetes have a much slower progression of beta cell dysfunction. These data contribute to a better understanding of clinical evolution of beta cell function in youth with diabetes, provide strong support for the aetiological classification of diabetes type and may inform tertiary prevention efforts targeted at high-risk groups.
Abbreviations
- DA:
-
Diabetes autoantibodies
- DPT-1:
-
Diabetes Prevention Trial-Type 1
- FCP:
-
Fasting C-peptide
- FPG:
-
Fasting plasma glucose
- GADA:
-
Glutamic acid decarboxylase-65 autoantibody
- HLA DR/DQ:
-
Human leucocyte antigen DR/DQ genotypes
- IA-2A:
-
Insulinoma-associated-2 autoantibody
- IQR:
-
Interquartile range
- IR:
-
Insulin resistance
- IS:
-
Insulin sensitivity
- NHW:
-
Non-Hispanic white
- NIDDK:
-
National Institute of Diabetes and Digestive and Kidney Diseases
References
Steele C, Hagopian WA, Gitelman S et al (2004) Insulin secretion in type 1 diabetes. Diabetes 53:426–433
Turner RC, Cull CA, Frighi V, Holman RR (1999) Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 281:2005–2012
Kahn SE, Montgomery B, Howell W et al (2001) Importance of early phase insulin secretion to intravenous glucose tolerance in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 86:5824–5829
Steffes MW, Sibley S, Jackson M, Thomas W et al (2003) Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 26:832–836
Dahlquist G, Blom L, Persson B, Wallensteen M, Wall S (1988) The epidemiology of lost residual beta-cell function in short term diabetic children. Acta Paediatr Scand 77:852–859
Gottsater A, Landin-Olsson M, Lernmark A, Fernlund P, Sundkvist G (1994) Islet cell antibodies are associated with beta-cell failure also in obese adult onset diabetic patients. Acta Diabetologia 31:226–231
Wallensteen M, Dahlquist G, Persson B et al (1988) Factors influencing the magnitude, duration, and rate of fall of B cell function in type 1 (insulin-dependent) diabetic children followed for two years from their clinical diagnosis. Diabetologia 31:664–669
Bonfati R, Bazzigaluppi E, Calori G et al (1998) Parameters associated with residual insulin secretion during the first year of disease in children and adolescents with type 1 diabetes mellitus. Diabet Med 15:844–850
Karjalainen J, Salmela P, Ilonen J, Surcel HM, Knip M (1989) A comparison of childhood and adult type I diabetes mellitus. N Engl J Med 320:881–886
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Sosenko JM, Palmer JP, Rafkin-Mervis L et al (2008) Glucose and C-peptide changes in the perionset period of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 31:2188–2192
Greenbaum CJ, Anderson AM, Dolan LM et al (2009) Preservation of beta-cell function in autoantibody-positive youth with diabetes. Diabetes Care 32:1839–1844
Dabelea D, Pihoker C, Talton JW et al (2011) Etiological approach to characterization of diabetes type: the SEARCH for Diabetes in Youth Study. Diabetes Care 34:1628–1633
The SEARCH Study Group (2004) SEARCH for Diabetes in Youth: a multicenter study of the prevalence, incidence and classification of diabetes mellitus in youth. Control Clin Trials 25:458–471
Bonifacio E, Yu L, Williams AK et al (2010) Harmonization of glutamic acid decarboxylase and islet antigen-2 autoantibody assays for national institute of diabetes and digestive and kidney diseases consortia. J Clin Endocrinol Metab 95:3360–3367
Erlich H, Valdes AM, Noble J et al (2008) HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57:1084–1092
Fernandez JR, Redden DT, Pietrobelli A, Allison DB (2004) Waist circumference percentiles in nationally representative samples of African-American, European-American, and Mexican-American children and adolescents. J Pediatr 145:439–444
Dabelea D, D’Agostino RB Jr, Mason CC et al (2011) Development, validation and use of an insulin sensitivity score in youths with diabetes: the SEARCH for Diabetes in Youth study. Diabetologia 54:78–86
Ingram DD, Parker JD, Schenker N et al. (2003) United States Census 2000 population with bridged race categories. Vital Health Stat 21–55
Kuczmarski RJ, Ogden CL, Guo SS et al (2002) 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11:1–190
Palmer JP, Fleming GA, Greenbaum CJ et al (2004) C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21–22 October 2001. Diabetes 53:250–264
Brown H, Prescott R (2011) Applied mixed models in medicine. Wiley, New York
Snorgaard O, Lassen LH, Binder C (1992) Homogeneity in pattern of decline of beta-cell function in IDDM. Prospective study of 204 consecutive cases followed for 7.4 yr. Diabetes Care 15:1009–1013
Keymeulen B, Vandemeulebroucke E, Ziegler AG et al (2005) Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352:2598–2608
Cook JJ, Hudson I, Harrison LC et al (1989) Double-blind controlled trial of azathioprine in children with newly diagnosed type I diabetes. Diabetes 38:779–783
Brown RJ, Sinaii N, Rother KI (2008) Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care 31:1403–1404
Herold KC, Gitelman SE, Masharani U et al (2005) A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 54:1763–1769
Herold KC, Hagopian W, Auger JA et al (2002) Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 346:1692–1698
Picardi A, Visalli N, Lauria A et al (2006) Metabolic factors affecting residual beta cell function assessed by C-peptide secretion in patients with newly diagnosed type 1 diabetes. Horm Metab Res 38:668–672
Decochez K, Keymeulen B, Somers G et al (2000) Use of an islet cell antibody assay to identify type 1 diabetic patients with rapid decrease in C-peptide levels after clinical onset. Belgian Diabetes Registry. Diabetes Care 23:1072–1078
Sabbah E, Savola K, Kulmala P et al (1999) Diabetes-associated autoantibodies in relation to clinical characteristics and natural course in children with newly diagnosed type 1 diabetes. The Childhood Diabetes in Finland Study Group. J Clin Endocrinol Metab 84:1534–1539
Kordonouri O, Danne T, Enders I, Weber B (1998) Does the long-term clinical course of type I diabetes mellitus differ in patients with prepubertal and pubertal onset? Results of the Berlin Retinopathy Study. Eur J Pediatr 157:202–207
Pozzilli P, Mesturino CA, Crino A, Gross TM, Jeng LM, Visalli N (2001) Is the process of beta-cell destruction in type 1 diabetes at time of diagnosis more extensive in females than in males? Eur J Endocrinol 145:757–761
Fernandez CM, Montana E, Camps I et al (1996) Ketoacidosis at diagnosis is predictive of lower residual beta-cell function and poor metabolic control in type 1 diabetes. Diabetes Metab 22:349–355
Montana E, Fernandez-Castaner M, Rosel P, Gomez J, Soler J (1991) Age, sex and ICA influence on beta-cell secretion during the first year after the diagnosis of type 1 diabetes mellitus. Diabetes Metab 17:460–468
Sosenko JM, Palmer JP, Greenbaum CJ et al (2006) Patterns of metabolic progression to type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 29:643–649
Sherry NA, Tsai EB, Herold KC (2005) Natural history of beta-cell function in type 1 diabetes. Diabetes 54(Suppl 2):S32–S39
Torn C, Landin-Olsson M, Lernmark A et al (2000) Prognostic factors for the course of beta cell function in autoimmune diabetes. J Clin Endocrinol Metab 85:4619–4623
Libman IM, Pietropaolo M, Arslanian SA, LaPorte RE, Becker DJ (2003) Evidence for heterogeneous pathogenesis of insulin-treated diabetes in Black and White children. Diabetes Care 26:2876–2882
Tfayli H, Bacha F, Gungor N, Arslanian S (2009) Phenotypic type 2 diabetes in obese youth: insulin sensitivity and secretion in islet cell antibody-negative versus -positive patients. Diabetes 58:738–744
Druet C, Tubiana-Rufi N, Chevenne D, Rigal O, Polak M, Levy-Marchal C (2006) Characterization of insulin secretion and resistance in type 2 diabetes of adolescents. J Clin Endocrinol Metab 91:401–404
Elder DA, Prigeon RL, Wadwa RP, Dolan LM, D’Alessio DA (2006) Beta-cell function, insulin sensitivity, and glucose tolerance in obese diabetic and nondiabetic adolescents and young adults. J Clin Endocrinol Metab 91:185–191
Matthews DR, Cull CA, Stratton IM, Holman RR, Turner RC (1998) UKPDS 26: sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabetes Med 15:297–303
Ekholm E, Gottsater A, Dahlin LB, Sundkvist G (2011) No signs of progressive beta cell damage during 20 years of prospective follow-up of autoantibody-negative diabetes. Acta Diabetol 49:57–62
Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR (2001) Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet 358:1749–1753
Pozzilli P, Pitocco D, Visalli N et al (2000) No effect of oral insulin on residual beta-cell function in recent-onset type I diabetes (the IMDIAB VII). IMDIAB Group. Diabetologia 43:1000–1004
Chaillous L, Lefevre H, Thivolet C et al (2000) Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: a multicentre randomised controlled trial. Diabete Insuline Orale group. Lancet 356:545–549
Greenbaum CJ, Harrison LC (2003) Guidelines for intervention trials in subjects with newly diagnosed type 1 diabetes. Diabetes 52:1059–1065
Acknowledgements
The SEARCH for Diabetes in Youth study is indebted to the many young individuals, their families and their healthcare providers, whose participation made this study possible. The writing group for this manuscript wishes to acknowledge the contributions of the following individuals to the SEARCH for Diabetes in Youth study:
California: J. M. Lawrence, K. Reynolds, J.-W. Hsu, M. H. Black, K. Holmquist and H. S. Takhar for the Department of Research and Evaluation and A. K. Kershnar for the Department of Pediatrics, Kaiser Permanente Southern California; and D. J. Pettitt for the Sansum Diabetes Research Institute.
Colorado: D. Dabelea, R. F. Hamman and L. Testaverde for the Department of Epidemiology, Colorado School of Public Health, University of Colorado Denver; G. J. Klingensmith, M. J. Rewers, D. Maahs and P. Wadwa for the Barbara Davis Center for Childhood Diabetes; S. Daniels and K. Nadeau, Department of Pediatrics and Children’s Hospital; C. A. Bloch for the Pediatric Endocrine Associates; C. Sorrelman and A. Beartrack for the Navajo Area Indian Health Prevention Program.
Hawaii: B. L. Rodriguez, B. Waitzfelder, W. Fujimoto, J. D. Curb, F. Kennedy, G. Uramoto, S. Waxman, T. Hillier and R. Chung for Kuakini Medical Center.
Ohio: L. M. Dolan, M. Seid, E. Urbina and D. A. Standiford for the Cincinnati Children’s Hospital Medical Center.
Carolina: E. J. Mayer-Davis and J. Thomas for the University of North Carolina, Chapel Hill; A. Merchant, A. D. Liese, R. R. Moran, G. Gaillard-McBride, D. Lawler and M. Jackson for the University of South Carolina; D. Bowlby for the Medical University of South Carolina; J. Amrhein for Greenville Hospital Systems; P. Clark for McLeod Pediatric Subspecialists; M. Parker for Pediatric Endocrinology & Diabetes Specialists, Charlotte, North Carolina.
Washington: C. Pihoker, M. Afkarian, A. Badaru, L. Gilliam, I. Hirsch, L. L. Liu, J. Neff, and J. Yi-Frazier for the University of Washington; B. Loots, R. O’Connor, S. Kearns, S. Semana and M. Yaptangco for Seattle Children’s Hospital; and C. Greenbaum for Benaroya Research Institute.
Centers for Disease Control and Prevention: G. Imperatore, D. E. Williams, H. S. Kahn, B. Moore G. W. Edward and S. H. Saydah.
National Institute of Diabetes and Digestive and Kidney Diseases, NIH: B. Linder.
Central Laboratory (University of Washington): S. M. Marcovina, V. P. Gaur and K. Gadbois
Coordinating Center (Wake Forest University School of Medicine): R. Bell, R. D’Agostino, Jr, B. Beech, D. Case, T. Morgan, M. J. Naughton, S. Moxley, G. Hargis, D. Kronner, A. Anderson, C. Davis, J. Andrews and J. W. Talton.
Funding
SEARCH for Diabetes in Youth is funded by the Centers for Disease Control and Prevention (PA Nos 00097, DP-05-069 and DP-10-001) and supported by the National Institute of Diabetes and Digestive and Kidney Diseases.
Site contract Nos: Kaiser Permanente Southern California (U48/CCU919219, U01 DP000246 and U18DP002714), University of Colorado Denver (U48/CCU819241-3, U01 DP000247 and U18DP000247-06A1), Kuakini Medical Center (U58CCU919256 and U01 DP000245), Children’s Hospital Medical Center (Cincinnati) (U48/CCU519239, U01 DP000248 and 1U18DP002709), University of North Carolina at Chapel Hill (U48/CCU419249, U01 DP000254 and U18DP002708-01), University of Washington School of Medicine (U58/CCU019235-4, U01 DP000244 and U18DP002710-01), Wake Forest University School of Medicine (U48/CCU919219, U01 DP000250 and 200-2010-35171).
The authors wish to acknowledge the involvement of General Clinical Research Centers (GCRC) at the South Carolina Clinical & Translational Research (SCTR) Institute, at the Medical University of South Carolina (NIH/NCRR Grant No. UL1RR029882); Children’s Hospital and Regional Medical Center (Grant No. M01RR00037); Colorado Pediatric General Clinical Research Center (Grant No. M01 RR00069) and the Barbara Davis Center at the University of Colorado at Denver (DERC NIH P30 DK57516); and the Institutional Clinical and Translational Science Award (CTSA), NIH/NCRR at the University of Cincinnati (Grant No. 1UL1RR026314-01).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DD designed the study, obtained funding, researched the data, wrote the manuscript and takes full responsibility for the contents of the article. EJM-D, LMD, CP, SM, RD’A Jr, RFH, CG, WF, BL and GI designed the study, obtained funding, contributed to discussion and edited the manuscript. JSA analysed the data and edited the manuscript. The final version of this manuscript has been approved by all authors. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention and the NIDDK.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 14 kb)
ESM Table 2
(PDF 32 kb)
ESM Table 3
(PDF 32 kb)
Rights and permissions
About this article
Cite this article
Dabelea, D., Mayer-Davis, E.J., Andrews, J.S. et al. Clinical evolution of beta cell function in youth with diabetes: the SEARCH for Diabetes in Youth study. Diabetologia 55, 3359–3368 (2012). https://doi.org/10.1007/s00125-012-2719-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2719-6