
Abstract
Aims/hypothesis
We have previously shown that NADPH oxidase (NOX) lies upstream of uncoupled endothelial nitric oxide synthase (eNOS), which is known to occur in diabetic endothelium. However, it remains unclear which specific NOX isoform(s) is responsible for eNOS uncoupling and endothelial dysfunction in diabetic mouse models. The aim of the present study was to test the hypothesis that one or more NOX isoform(s) mediate(s) diabetic uncoupling of eNOS, which has been shown to occur in patients with diabetes to contribute to endothelial dysfunction.
Methods
Diabetes was induced by streptozotocin administration. The N ω-nitro-l-arginine methyl ester (L-NAME)-sensitive superoxide production of aortic segments, reflective of eNOS uncoupling activity, was determined by electron spin resonance.
Results
The L-NAME-sensitive superoxide production was more than doubled in wild-type diabetic mice, implicating uncoupling of eNOS. This was abolished in diabetic p47 phox−/− (also known as Ncf1 −/−) mice, but preserved in Nox2 −/y (also known as Cybb −/−) mice made diabetic. The eNOS uncoupling activity was markedly attenuated in diabetic mice transfected with Nox1 or Nox1 organiser 1 (Noxo1) short interfering RNA (siRNA), and abolished in Nox1 −/y diabetic mice. Diabetes-induced impairment in endothelium-dependent vasorelaxation was also significantly attenuated in the Nox1 −/y mice made diabetic. By contrast, Nox4 siRNA, or inhibition of mitochondrial complex I or III with rotenone or siRNA, respectively, had no effect on diabetic uncoupling of eNOS. Overexpression of Dhfr, or oral administration of folic acid to improve dihydrofolate reductase (DHFR) function, recoupled eNOS in diabetes to improve endothelial function.
Conclusions/interpretation
Our data demonstrate for the first time that the p47phox and NOXO1-dependent activation of NOX1, but not that of NOX2, NOX4 or mitochondrion, mediates diabetic uncoupling of eNOS. NOX1-null mice are protected from diabetic endothelial dysfunction. Novel approaches to inhibit NOX1 and/or improve DHFR function, may prove to have therapeutic potential for diabetic endothelial dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accumulating evidence has indicated that oxidative stress contributes to diabetic cardiovascular complications. What remain unclear are the enzymatic sources of sustained reactive oxygen species (ROS) production in diabetic blood vessels. NADPH oxidase (NOX) is now recognised as an important enzymatic system responsible for vascular ROS production in diseases such as hypertension and atherosclerosis [1–3], as well as in both type 1 and type 2 diabetes mellitus [4–6]. We and others have shown that in the diabetic endothelium, uncoupled endothelial nitric oxide synthase (eNOS) represents the predominant source of superoxide [4, 5], following an initial activation of NOX that is angiotensin II-dependent. However, NOX remains active in the vascular smooth muscle cells [5, 7]. Indeed, angiotensin II uncoupling of eNOS requires upstream activation of NOX [8].
The next important question would be to identify which NOX isoform is specifically involved in eNOS uncoupling in diabetes, as different NOX isoforms have been shown to have differential roles in mediating vascular pathogenesis [9]. In addition, increased mitochondrial ROS production has been implicated in diabetic endothelium. The interrelationship between uncoupled eNOS and mitochondrial activation in diabetes is also unknown. Since, in patients with diabetes, eNOS uncoupling contributes to endothelial dysfunction [10, 11], the anticipated new findings would have therapeutic potential for treating vascular diseases in diabetic patients.
One or more functional NOX isoforms are produced in vascular cells. The NOX family is categorised into isoforms of the membrane-spanning catalytic subunit. Among family members of NOX1, NOX2 (previously known as gp91phox), NOX3, NOX4 and NOX5, NOX1 and 4 are produced in vascular smooth muscle and NOX1, 2, 4 and 5 are present in endothelial cells. NOX1, 2 and 4 bind p22phox for their activation, whereas NOX5 is believed to function independently of the traditional NOX regulatory subunits [12]. NOX1 is believed to interact with the p47phox homologue NOX organiser 1 (NOXO1) [13], the p67phox homologue NOX activator 1 and Rac1 [14]. However, p47phox binding to NOX1 is also functionally critical [15]. Both NOX1-null and p47phox-null mice have attenuated blood pressure in response to angiotensin II [16–18]. Phosphorylation and membrane translocation of p47phox is also known to activate NOX2.
We have recently demonstrated that oral folic acid supplementation potently recouples eNOS in angiotensin II-induced hypertension via a novel mechanism of restoring endothelial dihydrofolate reductase (DHFR) production and activity [19]. A folic acid diet preserves tetrahydrobiopterin (H4B) and nitric oxide bioavailabilities to reduce oxidative stress in both angiotensin II-treated endothelial cells and mice [19]. We have also shown that overexpression of DHFR protects eNOS from being uncoupled by angiotensin II [8, 20]. Therefore in the present study we aimed to identify specific NOX isoform(s) responsible for diabetic uncoupling of eNOS; clarify interrelationships among NOX, mitochondrion and uncoupled eNOS; and discover novel therapeutics targeting uncoupled eNOS in diabetes.
Methods
Animals and induction of diabetes
Male C57BL/6 mice (3–4 months old) were purchased from Charles River Laboratories (Hollister, CA, USA) and used as wild-type (WT) controls. Age-matched NOX2-null (Nox2 −/y) and p47phox-null (p47 phox−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA), and bred and maintained at the University of California Los Angeles (UCLA). The NOX1-null (Nox1 −/y) founder mice were generously provided by Dr Karl-Heinz Krause from the University of Geneva [18]. To induce diabetes, the mice were injected with streptozotocin (STZ, 100 mg/kg) intravenously for three consecutive days [7]. Blood glucose was determined using a One Touch Ultra blood glucose meter (Lifescan, Milpitas, CA, USA) before STZ injection (day 0) and on the harvest day (day 7). On day 7, mice were killed and aortic tissues were collected for measurements of superoxide production in the presence or absence of N ω-nitro-l-arginine methyl ester (L-NAME). The Institutional Animal Care and Usage Committee at UCLA approved the use of animals and experimental procedures.
Electron spin resonance detection of superoxide production
Freshly isolated aortas were carefully and thoroughly cleaned of adhering fat in ice-cold modified Krebs/HEPES buffer and cut into 3 mm rings. The superoxide-specific spin trap methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH, 1 mmol/l; Axxora LLC, San Diego, USA) solution was freshly prepared in nitrogen gas-bubbled Krebs/HEPES buffer containing diethyldithiocarbamic acid (DETC, 5 μmol/l; Sigma-Aldrich, St Louis, MO, USA) and deferoxamine (25 μmol/l; Sigma-Aldrich). Aortic rings were mixed with spin trap solution and loaded into glass capillary for analysis of the superoxide signal using an e-Scan electron spin resonance spectrophotometer (Bruker BioSpin, Rheinstetten, Germany). Some aortic rings were preincubated with 100 μmol/l L-NAME or 50 μmol/l 1400W on ice for 1 h before the addition of spin trap solution. The electron spin resonance settings used were: centre field, 3,475; sweep width, 9 G; static field, 3,484.98; microwave frequency, 9.75 GHz; microwave power, 21.02 mW; modulation frequency, 86 kHz; modulation amplitude, 2.47 G; resolution in X, 512; and number of x-scans, 10.
In vivo RNA interference of NOX1, NOXO1, NOX4 and mitochondrial complex III
The short interfering RNA (siRNA) was prepared in cationic liposome-based in vivo transfection reagent (Altogen Biosystems, Las Vegas, NV, USA) for in vivo delivery. The siRNAs against Nox1, Nox4 or Noxo1 were obtained from Dharmacon (Chicago, IL, USA), Invitrogen (Grand Island, NY, USA) and Integrated DNA Technologies (Coralville, IA, USA) respectively. The sequences of target for Nox1, Nox4, Noxo1 and mitochondrial complex III domain Rieske iron-sulphur gene [21, 22] are presented in electronic supplementary material (ESM) Table 1. Briefly, 600 μg siRNA was dissolved in 50 μl PBS and mixed with 40 μl transfection reagent. After 15–20 min incubation at room temperature, 10 μl transfection enhancer reagent was added and incubated together for another 10 min at room temperature. The transfection mixture was then diluted with 150 μl 5% glucose and injected into mice via the tail vein. The mice were injected on days 1, 2, 4 and 6 and then killed.
Western blot analysis of aortic NOX1, NOX4, NOXO1, mitochondrial complex III, eNOS and DHFR protein levels
Freshly isolated aortas were powderised in liquid nitrogen before lysis with lysis buffer (50 mmol/l Tris pH 7.4, 2 mmol/l EDTA, 1 mmol/l EGTA) containing 1% Triton X-100, 0.1% sodium dodecyl sulphate (SDS), 50 mmol/l NaF, 10 mmol/l Na4P2O7, 1 mmol/l Na3VO4, 1 mmol/l dithiothreitol (DTT), 1 mmol/l phenylmethylfonylsulfonyl fluoride (PMSF) and 10 μl/ml protease inhibitor cocktail (Sigma-Aldrich). Forty microgram proteins were separated in 10% SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences, Pittsburgh, PA, USA). The membranes were then probed with NOX1 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), NOX4 (1:500; Abcam, Cambridge, MA, USA), NOXO1 (1:500, Rockland Immunochemicals, Gilbertsville, PA, USA), Rieske domain of mitochondrial complex III (1:500; MitoSciences, Eugene, OR, USA), eNOS (1:1,000; BD Biosciences, San Jose, CA, USA), DHFR (0:1,000; RDI, North Acton, MA, USA) or β-actin (1:5,000; Sigma) antibodies at 4°C overnight, and subsequently with peroxidase-conjugated secondary antibody for 1 h at room temperature. Proteins were visualised using electrochemiluminescent reagents (GE Healthcare Biosciences, Pittsburgh, PA, USA).
Folic acid and rotenone administration
Folic acid (15 mg/kg per day) or rotenone (80 mg/kg per day) was orally administrated to mice using customised food tablets [19]. In brief, standard mouse chow was grounded, mixed well with folic acid or rotenone, and food tablets were produced by an in-house customised press machine. All mice were fed ad libitum and tablets with folic acid or rotenone were replaced every 2 days. Folic acid or rotenone treatments were started 2 days before STZ injection.
HPLC detection of circulating rotenone levels
The circulating levels of rotenone in mice were determined by HPLC analysis as previously described [23]. Plasma was separated from 200 μl whole blood, and rotenone was extracted with the same volume of dichloromethane (Sigma-Aldrich) for 15 min. After centrifugation at 12,000 rpm for 10 min, the organic phase was evaporated using a centrifugal evaporator for 10 min. The extracted pellet was dissolved in 35 μl acetonitrile, and 15 μl of the sample was injected into a C18 column for analysis. The rotenone concentration was monitored at 214 nm. The mobile phase was 70% methanol with a flow rate of 1 ml/min. The retention time for the rotenone peak is 15.5 min.
HPLC detection of circulating 5-MTHF levels
The circulating levels of 5-methyltetrahydrofolic acid (5-MTHF) in mice were determined by HPLC analysis as described by Lu et al [24]. Whole blood was frozen and thawed to lyse erythrocytes and 300 μl ascorbic acid 57 mmol/l was added to 100 μl blood. After being vortexed for 30 s and incubated at 37°C for 60 min to accelerate the conversion from 5-MTHF polyglutamate to 5-MTHF monoglutamate, samples were diluted with 600 μl potassium phosphate dibasic buffer 0.2 mol/l containing 30 mmol/l mercaptoethanol (pH 8.5). Samples were heated at 100°C for 15 min for deproteination. After being vortexed for 30 s and centrifuged at 10,000 g for 15 min, the supernatant fraction was collected and used for HPLC analysis.
As for the standards, 2 mg/ml 5-MTHF was dissolved in 0.01 mol/l potassium phosphate dibasic containing 57 mmol/l ascorbic acid (pH 7.1), and its dilute was spiked into blood matrix. The concentration of 5-MTHF was monitored by fluorescent detection (295/360 nm). The mobile phase was composed of methanol and 0.6% acetic acid in water (14:86, vol./vol.) with a flow rate of 1 ml/min.
HPLC for aortic H4B content
Aortic content of H4B was determined by HPLC analysis as previously described [5, 19].
Assessment of endothelium-dependent vasorelaxation
Aortic ring was prepared in Krebs solution (119 mmol/l NaCl, 4.7 mmol/l KCl, 1.2 mmol/l MgSO4, 1 mmol/l KH2PO4, 2.5 mmol/l CaCl2, 25 mmol/l NaHCO3 and 5.6 mmol/l glucose) gassed with 5% CO2 in O2. Under the microscope a triangular vessel hook was placed through the lumen of a 3 mm vessel segment, which was then mounted into an organ bath (Myobath II; WPI, Sarasota, FL, USA). Aortic rings were stretched with 800 mg of tension and then equilibrated for 90 min. After equilibration, rings were precontracted with 1 μmol/l phenylephrine and then exposed to incremental concentrations of acetylcholine (10−9 to 10−6 mol/l). Data acquisition was performed using Data-Trax hardware and Lab-Trax software (WPI).
Statistical analysis
Statistical analyses were performed with one-way ANOVA. When ANOVA indicated significant differences, Tukey’s post hoc test was used for multiple comparisons. Statistical significance was set at p < 0.05. Relaxation data were analysed by two-way ANOVA, followed by Bonferroni’s post hoc test. All grouped data shown in the figures are presented as mean ± SEM.
Results
p47phox-dependent NOX mediates diabetic uncoupling of eNOS
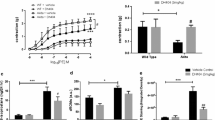
To examine if the specific NOX isoform(s) mediating eNOS uncoupling in diabetes is p47phox-dependent, we subjected WT and p47 phox−/− mice to STZ induction of diabetes. Plasma blood glucose was significantly increased to 27.44 ± 1.9 mmol/l in the WT diabetic mice (from 8.63 ± 0.7 mmol/l on day 0). As shown in Fig. 1a, neither knockout of p47phox nor knockout of NOX2 (using NOX2-null mice; see below) affected STZ-induced hyperglycaemia. Of interest, blood glucose levels were lower in the p47 phox−/− mice at baseline. L-NAME-sensitive aortic superoxide production, reflective of eNOS uncoupling activity, was more than doubled in the diabetic mice. This is consistent with previous observations [4, 5, 7]. On the other hand, 1400W-dependent superoxide production was not different in the WT and diabetic aortas, as shown in Fig. 1b. Importantly, the eNOS uncoupling activity was substantially attenuated in the p47 phox−/− mice made diabetic (Fig. 1c), implicating an intermediate role of p47phox-dependent NOX(s) in diabetic uncoupling of eNOS.

p47phox-dependent NOX, but not NOX2, mediates diabetic uncoupling of eNOS. WT, p47 phox−/− or Nox2 −/y mice were made diabetic by STZ injection (100 mg/kg per day for 3 days). On day 7, blood glucose levels were determined and aortas were harvested for detection of L-NAME-sensitive superoxide production (reflective of eNOS uncoupling activity) using electron spin resonance. a Blood glucose levels. b Superoxide level inhibited by NOS inhibitor L-NAME (left panel) and iNOS inhibitor 1400W (right panel, c). d eNOS uncoupling activity in p47 phox−/− mice. e eNOS uncoupling activity in Nox2 −/y mice. *p < 0.05, **p < 0.01, ***p < 0.001 vs sham mice; †† p < 0.01 vs STZ-diabetic WT mice (n = 9–13)
NOX2 does not contribute to eNOS uncoupling in diabetes
As a cytosolic regulatory subunit, p47phox binds NOX1 and NOX2 to modulate their enzymatic activities. p47phox has been known to be important for vascular NOX activity despite identification of the new homologue NOXO1. NOX2 knockout mice were used to examine a potential role of NOX2 in diabetic uncoupling of eNOS. As shown in Fig. 1d, L-NAME-sensitive superoxide production was not different in WT and Nox2 −/y mice made diabetic. This result indicates that NOX2 does not contribute to eNOS uncoupling in diabetes.
NOX1, rather than NOX4, mediates diabetic uncoupling of eNOS
We next employed in vivo RNA interference to examine the potential roles of NOX1 and NOX4 in diabetic uncoupling of eNOS. As shown in Fig. 2a, NOX1 and NOX4 protein levels were effectively reduced by repetitive in vivo siRNA transfection (see Methods section). The blood glucose levels, however, were not affected by Nox1 or Nox4 siRNA transfection (Fig. 2b). In Nox1 siRNA-transfected diabetic mice, eNOS-derived superoxide production was significantly decreased compared with scrambled siRNA-transfected diabetic mice (percentage of control, 161.5 ± 15.39 vs 227.3 ± 25.89; Fig. 2c). By contrast, Nox4 siRNA transfection had no effect on eNOS uncoupling activity (Fig. 2d). Transfection of mice with NOXO1 siRNA, however, resulted in a similar attenuation of eNOS uncoupling activity as was found with Nox1 siRNA transfection (Fig. 3a). Of note, Noxo1 siRNA markedly diminished aortic NOXO1 protein content (Fig. 3b), without affecting blood glucose levels (Fig. 3c). These results implicate the roles of NOXO1 and NOX1, but not that of NOX4, in mediating diabetic uncoupling of eNOS.

NOX1, but not NOX4, mediates diabetic uncoupling of eNOS. C57BL/6 mice were made diabetic by STZ injection (100 mg/kg per day for 3 days) and transfected with control, Nox1 or Nox4 siRNA. On day 7, blood glucose levels were determined and aortas were harvested for L-NAME-sensitive superoxide detection using electron spin resonance. a Protein contents of NOX1 and NOX4 in siRNA-transfected diabetic mouse aortas. b Blood glucose levels. c, d eNOS uncoupling activity in scrambled siRNA, Nox1 or Nox4 siRNA-transfected diabetic mice. *p < 0.05, **p < 0.01, ***p < 0.001 vs sham mice; † p < 0.05 vs untransfected diabetic mice (n = 6–7). White bars, sham mice; black bars, STZ-diabetic mice. RNAi, RNA interference; Sc, scrambled

NOXO1-dependent NOX1 mediates diabetic uncoupling of eNOS. C57BL/6 and NOX1−/y mice were made diabetic by STZ injection (100 mg/kg per day for 3 days) and transfected with control or Noxo1 siRNA as described in the Methods section. On day 7, blood glucose levels were determined and aortas were harvested for L-NAME-sensitive superoxide detection using electron spin resonance. a eNOS uncoupling activity in control siRNA or Noxo1 siRNA-transfected diabetic mice. b Protein content of NOXO1 in siRNA-transfected diabetic mouse aortas. c Blood glucose levels. *p < 0.05 vs sham mice; † p < 0.05 vs control siRNA-transfected diabetic mice (n = 4–6). White bars, sham mice; black bars, STZ-diabetic mice; RNAi, RNA interference. WT and Nox1 −/y mice were made diabetic by STZ injection (100 mg/kg per day for 3 days). d eNOS uncoupling activity in WT and Nox1 −/y mice. e Vasorelaxation in response to acetylcholine in precontracted aortic rings. f Blood glucose levels. *p < 0.05, **p < 0.01, ***p < 0.001 vs WT mice; ††† p < 0.001 vs Nox1 −/y mice (n = 6–7). Black line, STZ-diabetic mice; light grey line, Nox1 −/y/STZ-diabetic mice; medium grey line, Nox1 −/y mice; dark grey line, control mice. g Western blot for NOX1. h Upregulation of NOX1 protein production in STZ-diabetic mice. i quantitative grouped data for NOX1 protein content. **p < 0.01 vs WT mice (n = 3)
In additional experiments we employed NOX1-null mice (Nox1 −/y, Fig. 3g). The eNOS uncoupling activity was almost completely abolished in Nox1 −/y mice made diabetic (Fig. 3d), further confirming a specific and intermediate role of NOX1 in mediating eNOS uncoupling in diabetes. In addition, the impairment in endothelium-dependent vasorelaxation was also markedly improved in the diabetic Nox1 −/y mice (Fig. 3e). Blood glucose levels were not different in diabetic WT and Nox1 −/y mice (Fig. 3f). Indeed, NOX1 protein content in diabetes was upregulated more than threefold compared with nondiabetic WT mice (Fig. 3h–i). As shown in Fig. 4a, b, eNOS level was not different between WT and diabetic mice. Consistent with our previous findings, DHFR levels and aortic H4B contents were markedly attenuated in diabetic mice [5], which were significantly restored in Nox1−/y mice (Fig. 4c, d).

NOX1-mediated eNOS uncoupling in diabetes is accompanied by deficiencies in H4B and DHFR. WT and Nox1 −/y mice were made diabetic by STZ injection (100 mg/kg/day for 3 days). On day 7, aortas were harvested for analysis of eNOS level, DHFR level and H4B content. a Representative western blots for eNOS and DHFR. b Quantified grouped data for eNOS protein content. c Quantified grouped data for DHFR protein content. d HPLC analysis of aortic H4B content. *p < 0.05, **p < 0.01 vs WT mice; † p < 0.05 vs Nox1 −/y mice (n = 4)
Mitochondrial superoxide does not contribute to diabetic uncoupling of eNOS
In view of the observations that mitochondrial production of superoxide is elevated in diabetic endothelium, and that hydrogen peroxide functions as an intermediate for eNOS uncoupling in response to pathological agonists such as angiotensin II [8], which is also found to mediate diabetic uncoupling of eNOS [5], we investigated a potential role of mitochondrial ROS in provoking eNOS uncoupling. Inhibitions of mitochondrial complexes in vivo were achieved by RNA interference targeting complex III iron–sulphur centre or a diet containing complex I inhibitor rotenone. Protein abundance of Rieske iron–sulphur was significantly attenuated by siRNA transfection (Fig. 5a). In parallel experiments, plasma rotenone levels from mice fed the rotenone diet were determined by HPLC analysis. As shown in Fig. 5b, rotenone levels were increased 1 h after feeding, which remained elevated above 0.1 μmol/l throughout the entire study period. However, neither rotenone nor complex III RNA interference attenuated eNOS uncoupling activity in diabetic mice (Fig. 5c). Blood glucose levels were unaffected by these treatments (Fig. 5d). These data indicate that mitochondrial superoxide does not contribute to diabetic uncoupling of eNOS. Whether or not uncoupled eNOS induces mitochondrial dysfunction in diabetes, however, remains to be further investigated.

Mitochondrion does not contribute to diabetic uncoupling of eNOS. C57BL/6 mice were fed the mitochondrial complex I inhibitor rotenone for 2 days prior to being made diabetic by STZ injection (100 mg/kg per day for 3 days), and throughout the study period of 7 days. Some diabetic mice were transfected with complex III siRNA as described in the Methods section. a Expression of Rieske subunit of mitochondrial complex III. b Circulating levels of rotenone in mice as determined by HPLC analysis (see Methods section). c L-NAME-sensitive superoxide production (eNOS uncoupling activity). d Blood glucose levels. *p < 0.05 vs sham mice (n = 6). White bars, sham mice; black bars, STZ-diabetic mice; Mito III, mitochondrial complex III; RNAi, RNA interference
Recoupling of eNOS in diabetes by restoration of DHFR function
We have previously shown that diabetic uncoupling of eNOS is accompanied by a deficiency in the H4B salvage enzyme DHFR [5]. Without affecting blood glucose levels (Fig. 6a), overproduction of DHFR was induced by in vivo transfection of pcDNA3.1-Dhfr expression vector (Fig. 6b), which turned out to be highly effective in attenuating eNOS uncoupling activity in diabetic mice (percentage of control, 187.7 ± 44.4 to 97.38 ± 40.21; Fig. 6c). The impairment in endothelium-dependent vasorelaxation in diabetes was also corrected by DHFR overproduction (Fig. 6d). Folic acid supplementation recouples eNOS in angiotensin II-induced hypertensive mice via restoration of endothelial DHFR content and activity [19, 20]. Interestingly, oral administration of folic acid also completely attenuated eNOS uncoupling in diabetic mice (Fig. 6e), though without lowering blood glucose (Fig. 6f). The plasma levels of 5-MTHF, one of the metabolites of folic acid, were unaffected in folic acid-supplemented mice (Fig. 6g). This is consistent with our previous findings that folic acid restoration of eNOS function is independent of 5-MTHF in angiotensin II-induced hypertension [19]. Of note, endothelium-dependent vasorelaxation was improved in folic acid-treated diabetic mice (Fig. 6h), which was accompanied by restoration of DHFR protein abundance and aortic H4B content (ESM Fig. 1a–c). Taken together, these data indicate that approaches effective in restoring DHFR function may serve as powerful therapeutic tools to eliminate eNOS uncoupling and endothelial dysfunction in diabetes.

Recoupling of eNOS in diabetes via restoration of DHFR. C57BL/6 mice were made diabetic by STZ injection (100 mg/kg per day for 3 days) and transfected with empty vector or pcDNA3.1-Dhfr plasmid (a–d). Oral administration with folic acid (15 mg/kg per day) was started 2 days prior to induction of diabetes with STZ (100 mg/kg per day for 3 days), and mice were fed with folic acid-containing food throughout the study period of 7 days (e–h). On day 7, blood glucose levels were determined and aortas were harvested for superoxide production using electron spin resonance. a Blood glucose levels. *p < 0.05 vs sham b DHFR levels in diabetic aortas. *p < 0.05, ***p < 0.001 vs sham c L-NAME-sensitive superoxide production (uncoupling activity of eNOS). *p < 0.05 vs sham mice; † p < 0.05 vs untransfected diabetic mice. d Vasorelaxation in response to acetylcholine in precontracted aortic rings. **p < 0.01, ***p < 0.001 vs sham mice; †† p < 0.01, ††† p < 0.001 vs Dhfr overexpressed sham mice (n = 5). Black line, STZ-diabetic mice; light grey line, Dhfr/STZ-diabetic mice; dark grey line, sham mice; medium grey line, Dhfr-overexpressed mice. e L-NAME-sensitive superoxide production (eNOS uncoupling activity). *p < 0.05 vs sham mice; † p < 0.05 vs control diet-treated diabetic mice (n = 4). f Blood glucose levels. *p < 0.05 vs sham. g Circulating levels of 5-MTHF as determined by HPLC analysis (see Methods section) in mice fed a standard diet or a diet supplemented with folic acid (15 mg/kg per day). h Vasorelaxation in response to acetylcholine in precontracted aortic rings. **p < 0.01, ***p < 0.001 vs sham mice; † p < 0.05, ††† p < 0.001 vs folic acid-treated sham mice; ‡ p < 0.05 vs folic acid-treated diabetic mice (n = 4–6). Black line, STZ-diabetic mice; medium grey line, folic acid-treated STZ diabetic mice; light grey line, folic acid diet-fed mice; dark grey line, WT mice; white bars, sham mice; black bars, STZ-diabetic mice
Discussion
The major novel findings of the present study are: (1) diabetic uncoupling of eNOS is mediated by p47phox and NOXO1-dependent NOX1 activation; (2) NOX1 deficiency prevents uncoupling of eNOS and endothelial dysfunction in diabetes; (3) neither NOX2 nor NOX4 is involved in mediating diabetic uncoupling of eNOS; (4) mitochondrion does not lie upstream of uncoupled eNOS in diabetes; (5) approaches restoring DHFR function via direct overexpression of Dhfr or folic acid administration are vasoprotective in diabetes by the mechanism of recoupling eNOS.
Our findings demonstrate an upstream role of NOX1 in eNOS uncoupling in diabetes. It was previously shown that production of NOX1, but not that of NOX4, was upregulated in STZ-induced diabetic rats [25]. Utilising RNA interference in vivo and mice genetically deficient in NOX isoforms and subunits, our present study systematically evaluated NOX–eNOS interaction in diabetes and identified a specific role of NOX1 in mediating diabetic uncoupling of eNOS and consequent impairment of endothelial function. This novel role of NOX1 in diabetes shares similarities with its role in angiotensin II-induced hypertension [17, 18, 26], where it mediates blood pressure regulation. Indeed, we have previously shown that angiotensin II also mediates diabetic uncoupling of eNOS [5]. In line with these findings, angiotensin II is known to upregulate NOX1 level in vascular smooth muscle cells [27]. Therefore NOX1 activation in endothelial cells, vascular smooth muscle cells or both cell types can lead to high levels of superoxide production, H4B deficiency and eNOS uncoupling in diabetes.
Silencing Nox4 with siRNA or knockout of NOX2 in mice was not effective in preventing eNOS uncoupling in diabetes. These data additionally support the conclusion that NOX1, rather than NOX2 or NOX4, mediates diabetic uncoupling of eNOS. Although NOX5 is produced in human endothelial cells and has been found to be involved in human coronary artery disease [28], it is not present in rodents [29].
The p47phox homologue, NOXO1, has been shown to bind NOX1 to modulate its enzymatic activity [13]. It was also reported to interact with NOX3 [14], which is, however, not produced in vascular cells. When initially identified, NOXO1 was believed to bind vascular NOX in replacement of neutrophil p47phox. Nonetheless, both NOX1-null and p47phox-null mice have attenuated blood pressure and aortic superoxide production in response to angiotensin II [17, 18, 26, 30], implicating roles of p47phox in modulating vascular NOX and pathogenesis. Likewise, our data showed that inhibition of either p47phox or NOXO1 attenuates eNOS uncoupling in diabetes, while Noxo1 siRNA was less effective than that of Nox1 siRNA, suggesting that co-inhibition of p47phox and NOXO1 might be more potent than the effect of Noxo1 siRNA alone.
p47phox also binds to NOX2. Uncoupling of eNOS, however, remained intact in the Nox2 −/− mice made diabetic. This indicates that eNOS uncoupling is attributed to p47phox-dependent activation of NOX1 rather than NOX2. Similarly, angiotensin II-induced eNOS uncoupling was not affected in Nox2 −/− mice (data not shown), further confirming that NOX2 activation was not involved. It has been known that p47phox anchors to plasma membrane through its PX (phox) domain while it binds to p22phox via its bis SH3 domain, once these domains become accessible after phosphorylation-induced conformational change of the autoinhibitory loop. The PX domain of NOXO1 has been reported to allow membrane targeting and binding of p22phox, along with the SH3 domain, without agonist stimulation. This is because NOXO1 lacks autoinhibitory loop and phosphorylation sites [14]. Given previous reports, NOXO1 binding has been suggested to be involved in constitutive activation of NOX1 to produce low levels of superoxide, unlike the role of p47phox, which is supposed to be agonist-dependent. Indeed, substitution of NOXO1 to p47phox induced a loss of constitutive activation in vitro [13]. On the other hand, NOX1 can be an agonist specifically activated by angiotensin II and platelet-derived growth factor, and p47phox-dependent NOX1 activation has been documented in vascular smooth muscle cells [27]. We propose that in diabetes NOXO1 is bound to the NOX1/p22phox complex at baseline, and, when angiotensin II is elevated, p47phox is phosphorylated and recruited to the complex to fully activate NOX1, leading to initial superoxide production, consequent oxidation of H4B and DHFR deficiency, and ultimate uncoupling of eNOS. This proposed mechanism of action is illustrated in Fig. 7.

Proposed mechanisms of action for an intermediate role of p47phox and NOXO1-dependent NOX1 activation in diabetic uncoupling of eNOS. NOXA1, NOX activator 1
Our data have excluded an intermediate role of mitochondrial ROS in diabetic uncoupling of eNOS. Previous findings have shown that cytoplasmic hydrogen peroxide is capable of inducing respiratory enzyme deficiency and ROS production [31, 32]. The opposite might also be true that mitochondrial dysfunction occurs downstream of uncoupled eNOS. Indeed, our data indicate that inhibition of mitochondrial complex I or III had no effect on diabetic uncoupling of eNOS. The detailed mechanisms as to whether and how uncoupled eNOS contributes to mitochondrial dysfunction in diabetes, however, are beyond the scope of the current study. In experimental models with triggered eNOS uncoupling, mitochondrial dysfunction developed in the heart, leading to altered contractile and morphological properties [33].
In conclusion, our data demonstrate for the first time that activation of p47phox and NOXO1-dependent NOX1 mediates eNOS uncoupling and endothelial dysfunction in diabetes. The other NOX isoforms, NOX2, 3, 4 and 5, are, however, not involved. Mitochondrion is also found not to contribute to diabetic uncoupling of eNOS. These findings provide innovative insights into oxidase interactions in the setting of diabetes, which are important in understanding sustained oxidative stress and endothelial dysfunction in diabetic patients. We have also shown that oral administration of folic acid, or Dhfr overexpression, potently recouples eNOS in diabetes via restoration of DHFR function. Therefore novel approaches to improve DHFR function and/or inhibit NOX1 may prove to be beneficial in treating diabetic endothelial dysfunction.
Abbreviations
- DHFR:
-
Dihydrofolate reductase
- eNOS:
-
Endothelial nitric oxide synthase
- H4B:
-
Tetrahydrobiopterin
- L-NAME:
-
N ω-nitro-l-arginine methyl ester
- 5-MTHF:
-
5-Methyltetrahydrofolic acid
- NOX:
-
NADPH oxidase
- NOXO1:
-
NADPH oxidase organiser 1
- ROS:
-
Reactive oxygen species
- siRNA:
-
Short interfering RNA
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
Harrison DG (1997) Endothelial function and oxidant stress. Clin Cardiol 20:II-11–II-17
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Cai H, Griendling KK, Harrison DG (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24:471–478
Hink U, Li H, Mollnau H et al (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88:E14–E22
Oak JH, Cai H (2007) Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 56:118–126
San Martin A, Du P, Dikalova A et al (2007) Reactive oxygen species-selective regulation of aortic inflammatory gene expression in type 2 diabetes. Am J Physiol Heart Circ Physiol 292:H2073–H2082
Oak JH, Youn JY, Cai H (2009) Aminoguanidine inhibits aortic hydrogen peroxide production, VSMC NOX activity and hypercontractility in diabetic mice. Cardiovasc Diabetol 8:65
Chalupsky K, Cai H (2005) Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA 102:9056–9061
Rivera J, Sobey CG, Walduck AK, Drummond GR (2010) Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep 15:50–63
Heitzer T, Brockhoff C, Mayer B et al (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res 86:E36–E41
Guzik TJ, Mussa S, Gastaldi D et al (2002) Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105:1656–1662
Lassegue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30:653–661
Banfi B, Clark RA, Steger K, Krause KH (2003) Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278:3510–3513
Cheng G, Lambeth JD (2004) NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J Biol Chem 279:4737–4742
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Landmesser U, Cai H, Dikalov S et al (2002) Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40:511–515
Matsuno K, Yamada H, Iwata K et al (2005) Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112:2677–2685
Gavazzi G, Banfi B, Deffert C et al (2006) Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580:497–504
Gao L, Chalupsky K, Stefani E, Cai H (2009) Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice. A novel HPLC-based fluorescent assay for DHFR activity. J Mol Cell Cardiol 47:752–760
Gao L, Siu KL, Chalupsky K et al (2011) Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension 59:158–166
Brunelle JK, Bell EL, Quesada NM et al (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1:409–414
Bell EL, Klimova TA, Eisenbart J et al (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177:1029–1036
Marella M, Seo BB, Nakamaru-Ogiso E, Greenamyre JT, Matsuno-Yagi A, Yagi T (2008) Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS One 3:e1433
Lu W, Li H, Zhang Y, Ang CY (2002) Rapid method for the determination of total 5-methyltetrahydrofolate in blood by liquid chromatography with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci 766:331–337
Wendt MC, Daiber A, Kleschyov AL et al (2005) Differential effects of diabetes on the expression of the gp91phox homologues nox1 and nox4. Free Radic Biol Med 39:381–391
Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, Krause KH (2007) NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension 50:189–196
Lassegue B, Sorescu D, Szocs K et al (2001) Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88:888–894
Guzik TJ, Chen W, Gongora MC et al (2008) Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol 52:1803–1809
Fulton DJR (2009) Nox5 and the regulation of cellular function. Antioxid Redox Signal 11:2443–2452
Landmesser U, Dikalov S, Price SR et al (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111:1201–1209
Cai H (2005) NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 96:818–822
Lanza IR, Nair KS (2010) Mitochondrial function as a determinant of life span. Pflugers Arch 459:277–289
Ceylan-Isik AF, Guo KK, Carlson EC et al (2009) Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: role of mitochondrial biogenesis. Hypertension 53:1023–1031
Funding
This study was supported by the National Heart, Lung, and Blood Institute (NHLBI) Grants HL077440 (HC), HL088975 (HC), HL101228 (PP, JNW, HC), and an American Diabetes Association Award 7-08-RA-23 (HC).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
HC contributed to the conception and experimental design of the study. JYY and LG contributed to the performance of the experiments. All authors contributed to the analysis and interpretation of data, as well as drafting of the article. JYY and HC revised the article critically for important intellectual content. All authors have approved the final version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Target sequences for siRNAs (PDF 6 kb)
ESM Fig. 1
(PDF 20 kb)
Rights and permissions
About this article
Cite this article
Youn, J.Y., Gao, L. & Cai, H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55, 2069–2079 (2012). https://doi.org/10.1007/s00125-012-2557-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2557-6