
Abstract
Aims/hypothesis
This study was aimed at the elucidation of the pathogenesis of glucotoxicity, i.e. the mechanism whereby hyperglycaemia damages pancreatic beta cells. The identification of pathways in the process may help identify targets for beta cell-protective therapy. Carbohydrate response element-binding protein (ChREBP), a transcription factor that regulates the expression of multiple hyperglycaemia-induced genes, is produced in abundance in pancreatic beta cells. We hypothesise that ChREBP plays a pivotal role in mediating beta cell glucotoxicity.
Methods
We assessed the role of ChREBP in glucotoxicity in 832/13 beta cells, isolated mouse islets and human pancreas tissue sections using multiple complementary approaches under control and high-glucose-challenge conditions as well as in adeno-associated virus-induced beta cell-specific overexpression of Chrebp (also known as Mlxipl) in mice.
Results
Under both in vitro and in vivo conditions, ChREBP activates downstream target genes, including fatty acid synthase and thioredoxin-interacting protein, leading to lipid accumulation, increased oxidative stress, reduced insulin gene transcription/secretion and enhanced caspase activity and apoptosis, processes that collectively define glucotoxicity. Immunoreactive ChREBP is enriched in the nucleuses of beta cells in pancreatic tissue sections from diabetic individuals compared with non-diabetic individuals. Finally, we demonstrate that induced beta cell-specific Chrebp overexpression is sufficient to phenocopy the glucotoxicity manifestations of hyperglycaemia in mice in vivo.
Conclusions/interpretation
These data indicate that ChREBP is a key transcription factor that mediates many of the hyperglycaemia-induced activations in a gene expression programme that underlies beta cell glucotoxicity at the molecular, cellular and whole animal levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insulin deficiency, whether absolute or relative, leads to hyperglycaemia, the hallmark of type 1 and type 2 diabetes. Over time, hyperglycaemia leads to cellular damage and organ dysfunction. In the pancreas, chronic hyperglycaemia produces progressive deleterious effects on beta cells, a pathological process referred to as glucotoxicity [1–4], which is manifested by enhanced lipogenesis and intracellular lipid accumulation [5], increased oxidative stress [6, 7], followed by reduced insulin gene expression and/or secretion [2, 8], and increased apoptosis [7, 9]. A number of transcription factors, including sterol regulatory element-binding protein 1c (SREBP-1c) [10] and small heterodimer partner (SHP) [11], have been implicated in glucotoxicity.
Chrebp (also known as Mlxipl) is a glucose-responsive basic helix–loop–helix leucine zipper transcription factor that is present in multiple cell types, but its regulatory function on cell metabolism is thought to be especially important in the liver [12, 13], in which glucose controls carbohydrate response element-binding protein (ChREBP) functions via multiple mechanisms including increased expression and transactivation activity, phosphorylation-dependent upregulation of nuclear translocation and DNA binding [14–17]. ChREBP heterodimerises with max-like protein x (Mlx) and binds to a carbohydrate response element (ChoRE) in the promoters of genes for glycolytic and lipogenic enzymes, such as liver-type pyruvate kinase (L-PK), acetyl-coA carboxylase (ACC), and fatty acid synthase (FASN), and activates their transcription. These are the same genes that are stimulated by prolonged exposure of the beta cell line to high-glucose concentrations in vitro [5].
As in hepatocytes, Chrebp mRNA is also highly expressed in pancreatic islets and beta cell lines such as INS-1 and MIN6 cells; expression of Chrebp in INS-1 cells is dose-dependently stimulated by high glucose [18, 19]. In this study, we show that prolonged activation of ChREBP by high glucose leads to beta cell dysfunction and apoptosis. Use of multiple complementary approaches using mouse and human material supports the conclusion that ChREBP mediates many of the deleterious effects of high glucose on pancreatic beta cells in vitro and in vivo.
Methods
Animals
C57BL/6 and NONcNZO10/LtJ mice were purchased from Jackson Laboratory, Bar Harbor, ME, USA. All mice were maintained in the accredited pathogen-free Baylor College of Medicine (BCM) Transgenic Mice Facility on a 12 h light/dark cycle. All experiments were performed following approval of the protocol by the animal care research committee of BCM.
Plasmid construction
We subcloned mouse Chrebp∆1-196 (constitutively active [CA]-Chrebp) [17] into murine stem cell virus (MSCV)-based retroviral plasmid vector (pMSCV-puro; Clontech, Mountain View, CA, USA) or pcDNA3.1 expression vector (Invitrogen). Plasmids for RU486-inducible expression [20] and secreted alkaline phosphatase (SEAP) reporter driven by SR-α promoter (a fusion promoter which is composed of the SV40 early promoter and the R segment and part of the U5 sequence of the long terminal repeat derived from human T-cell leukaemia virus type I) were kindly provided by S. Tsai and D. Spencer (BCM, Houston, TX, USA), respectively. The 17 × 4tk inducible promoter and Chrebp∆1-196 were cloned into self-inactivating (SIN) retroviral vector with puromycin selection marker. The GLP65 transactivator, containing GAL4 DNA-binding domain, modified human progesterone receptor ligand-binding domain, and human p65 activation domain chimera, was cloned into pMSCV-hygro retroviral vector (Clontech). Rat insulin II (−696, +8) and Txnip (−200, +25) and mutated Txnip promoters were amplified by PCR from rat genomic DNA and cloned into pGLuc-Basic (New England Biolabs, Ipswich, MA, USA). Cytomegalovirus (CMV) promoter was transferred from pcDNA3.1 into pGLuc-Basic.
Cell culture
The 832/13 cells (a gift from C. Newgard, Duke University, Durham, NC, USA) were cultured as described previously [21].
Retrovirus preparation
Bosc23 cells were maintained as described previously [22]. We transiently transfected these cells with retroviral construct and pCL-Eco packaging vector. Retrovirus made with these constructs or empty vector were infected into target cells in the presence of polybrene. Infected cells were selected with puromycin or hygromycin.
Lipid accumulation assay
Oil Red O staining was performed as described previously [23].
Immunofluorescence
For immunofluorescence of 832/13 cells and primary islets, we used rabbit anti-ChREBP antibody (Cayman Chemical, Ann Arbor, MI, USA), Alexa568-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR, USA), guinea pig anti-insulin (Linco, St. Charles, MO, USA) and rabbit anti-cleaved poly (ADP-ribose) polymerase (PARP; Cell Signaling Technology) antibodies. Human pancreatic paraffin tissue sections were used from Biochain (Hayward, CA, USA). Images were taken using the LSM 510 META triple laser system (Zeiss, Thornwood, NY, USA).
Luciferase assay
Gaussia luciferase (GLuc) assay (New England Biolabs) and Phospha-Light (Applied Biosystems, Carlsbad, CA, USA) kits were used for luciferase and SEAP assay according to the manufacturers’ instructions. Results are shown as fold activation over reporter activity with empty vector or under low glucose concentration if not otherwise specified.
Quantitative RT-PCR
We extracted total RNA using the Total RNA Isolation Mini Kit (Agilent, Santa Clara, CA, USA) and treated it with DNase I (Sigma-Aldrich, St Louis, MO, USA). We then synthesised cDNA using Omniscript RT kit (Qiagen, Hilden, Germany) or SuperScript III (Invitrogen, Carlsbad, CA, USA). We performed PCR using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and monitored by Mx3000P Real-Time PCR System (Stratagene, Santa Clara, CA, USA). Expression of housekeeping genes was analysed by geNorm [24] and the expression of the two to three most stable genes was used to normalise the expression of genes of interest. Primer sequences used in this study are available upon request.
Apoptosis detection
We used the APOPercentage Assay (Biocolor, Carrickfergus, UK) according to the manufacturer’s instructions as an alternative method to quantify apoptosis.
Small interfering RNA transfection
Rat Wbscr14 (ChREBP) and non-targeting ON-TARGETplus SMARTpool small interfering RNA (siRNA; Dharmacon, Lafayette, CO, USA) were transfected into the cells at 18–24 h after seeding using DharmaFECT 2 transfection reagent (Dharmacon) according to the manufacturer’s instructions. Further analyses were performed in 24–72 h after transfection as indicated in each experiment.
2′,7′-Dichlorofluorescein diacetate oxidative stress assay
We quantified the accumulation of cellular reactive oxygen species by measuring 2′,7′-dichlorofluorescein diacetate (DCFH-DA) oxidation [22]. Cells were washed with PBS and then incubated with DCFH-DA in the dark for 30 min. The fluorescence was measured by a fluorometer using a SYBR Green filter.
Mouse pancreatic islets isolation
Islets were isolated from mice as previously described [25, 26]. Islets were trypsinised with 0.05% trypsin (wt/vol.) and maintained in RPMI medium supplemented with 5 mmol/l glucose and 10% FBS (vol./vol.) overnight. The islets were treated as indicated in each experiment.
Induction of hyperglycaemia in mice by subcutaneous glucose injections
We treated wild-type mice with 77 or 77 mmol/l NaCl + 66.67% glucose (5 g/kg body weight) by subcutaneous injection. We monitored blood glucose using a OneTouch Ultra2 glucometer (LifeScan, Milpitas, CA, USA) and periodically repeated injections to maintain blood glucose above 19.425 mmol/l for 24 h. Mice were fasted without water restriction during treatment.
Adeno-associated virus production and administration
The 293 Tcells were transfected with adeno-associated virus (AAV) shuttle vector, p5E18 and pAdΔF6 helper plasmid using calcium phosphate transfection method. We harvested these cells at 48–72 h after transfection and purified by deoxycholate digestion and iodixanol gradient. A total of 5 × 1012 viral genome particles were injected i.p. into 4-week-old mice.
Oral glucose tolerance test
We fasted all mice for 6 h and administered glucose solution by gavage (2 g/kg body weight) and collected tail blood at 0, 15, 30, 60, 120 and 180 min. We performed the hexokinase assay and insulin measurements (Ultrasensitive Mouse Insulin ELISA; Mercodia, Sweden) according to the manufacturer’s instructions.
Laser capture microdissection
Pancreas sections were embedded in optimal cutting temperature (OCT) compound, frozen on dry ice and stored at −80°C. We next performed cryosectioning (to 7 μm) and stained sections using the Histogene staining kit (Molecular Devices, Sunnyvale, CA, USA) in an RNase-free environment. Islets were isolated using the Veritas microdissection system (Molecular Devices).
Statistical analyses
All results are expressed as the mean ± SD. Sample groups were compared using the Student’s t test, with p < 0.05 considered statistically significant. Error bars indicate the SDs of three to four biological replicates.
Results
CA-Chrebp expression phenocopies glucotoxicity in beta cells
To test whether ChREBP activation mediates the glucotoxic effects of high glucose, we generated a CA-ChREBP construct that lacks the low-glucose inhibitory domain (LID; Fig. 1a). CA-ChREBP localises to the nucleus and activates target gene expression, independent of glucose concentration.

ChREBP promotes lipid accumulation, downregulates insulin expression and impairs insulin secretion in 832/13 cells. Unless otherwise specified, the 832/13 cells were cultured in regular RPMI-1640 medium that contained 11 mmol/l glucose. a Schematic domain structure of CA-ChREBP and wild-type ChREBP. b Morphology of unstained 832/13 cells infected with CA-Chrebp-containing retrovirus or empty vector. c CA-ChREBP stimulates lipid accumulation in 832/13 cells in 11 mmol/l glucose. We stained 832/13 cells infected with CA-Chrebp-containing retrovirus or empty vector for neutral lipid (red dots) by Oil Red O. Histograms (d, e) represent the amount of stained intracellular lipid compared with 832/13 cells infected with empty vector. e CA-ChREBP stimulates Fasn transcript expression. We treated 832/13 cells infected with CA-Chrebp-containing retrovirus or empty vector for 18 h with 2.5 or 25 mmol/l d-glucose, extracted total RNA from the cells and performed qRT-PCR using primers specific for Fasn. The histograms show the means of relative RNA levels normalised to mRNA levels of Hmbs, Alas1 and Eef1g. White bars, glucose at 2.5 mmol/l; black bars, glucose at 25 mmol/l. f CA-ChREBP downregulates insulin transcript expression in 832/13 cells. We preincubated 832/13 cells in 2.5 mmol/l d-glucose and switched them to 25 mmol/l d-glucose. RNA was isolated from samples collected at 2, 8, 16, 24 h, and insulin 2 transcripts were quantified by qRT-PCR. g CA-ChREBP downregulates insulin promoter-driven luciferase expression in 832/13 cells. We co-transfected luciferase constructs driven by CMV promoter or rat insulin 2 promoter (Rip) with CA-Chrebp or empty vector in 832/13 cells. Luciferase activity was measured at 24 h after transfection and normalised to SEAP, and expressed as fold activation over the activity seen in cells transfected with secreted luciferase driven by CMV promoter construct and empty vector. White bars, empty vector; black bars CA-Chrebp. h CA-ChREBP impairs insulin secretion. We performed glucose-induced insulin secretion assay in 832/13 cells infected with CA-Chrebp-containing retrovirus or empty vector. * p < 0.05 compared with cells treated with empty vector. B, basic; HLH, helix–loop–helix; LZ, leucine zipper
The morphology of 832/13 cells, an INS-1-derived insulinoma line [27], harbouring a retrovirus expressing CA-Chrebp (Fig. 1b) is similar to that of uninfected 832/13 cells but displaying greatly increased lipid droplet accumulation (Fig. 1c). Quantitative analysis of 832/13 cells showed that CA-Chrebp transduction induced a ~15-fold increase in neutral lipids compared with transduction with an empty vector (Fig. 1d). Concomitantly, CA-ChREBP significantly stimulated the expression of Fasn (Fig. 1e), a lipogenic gene with a promoter that contains a known ChoRE, in both low- and high-glucose media, corroborated by the finding that Chrebp knockdown inhibits glucose-stimulated Fasn expression and lipid accumulation in beta cells [19]. Though CA-ChREBP is fully activated under low-glucose conditions, there is an augmentation of the effects seen with high glucose that may be secondary to both ChREBP-independent high-glucose effects and to the activation of endogenous ChREBP by high glucose.
Quantitative RT-PCR analysis showed that CA-ChREBP suppressed insulin mRNA expression in 832/13 cells (Fig. 1f). We next examined insulin promoter activity using a rat insulin promoter-luciferase reporter construct, co-transfected with CA-Chrebp or empty vector. These co-transfection experiments indicate that CA-Chrebp co-expression downregulated insulin promoter-driven luciferase reporter gene expression (Fig. 1g), but had no effect on CMV promoter activity. Therefore, CA-Chrebp expression is associated with downregulated insulin gene expression. CA-Chrebp expression also led to inhibition of glucose-stimulated insulin secretion (Fig. 1h), corroborating and extending the observations of high-glucose-stimulated insulin mRNA level in these cells (Fig. 1f).
As significant numbers of dysmorphic CA-Chrebp-expressing 832/13 cells were lost during each passage through cell death, we constructed an RU486-inducible CA-Chrebp vector (Fig. 2a) to further investigate the role of ChREBP activation on apoptotic beta cell death. This system uses a chimeric transactivator consisting of a mutated progesterone receptor–ligand binding domain fused at the N-terminus to the DNA binding domain of the yeast GAL4, and ligated at the C-terminus to part of the activation domain of the human p65 protein, a component of the nuclear factor of κ light polypeptide gene enhancer in B cells (NF-κB) complex. In the presence of the antiprogestin RU486, the chimeric regulator binds to the target gene (Chrebp) containing the 17-mer Gal4 binding site, resulting in an efficient ligand-inducible transactivation of the target gene [20]. Inducible CA-Chrebp-transduced 832/13 cells (iCA) displayed a low basal background expression due to a small leakage of the iCA construct (Fig. 2b). In these cells, addition of RU486 stimulated Chrebp mRNA approximately eightfold compared with vehicle treatment; it also significantly activated caspase-3/7 activity in iCA cells whether they were in low or high glucose, but not in wild-type 832/13 cells (Fig. 2c). Change in caspase activity was accompanied by a change in the number of apoptotic cells as determined by APOPercentage dye uptake (Fig. 2d, e). By this assay, in comparison with vehicle treatment, RU486 increased the number of apoptotic iCA cells by ~12-fold; it had no effect on non-transduced wild-type cells. Furthermore, addition of N-(2-quinolyl)valyl-aspartyl-(2,6-difluorophenoxy)methylketone (Q-VD-OPH), a broad-spectrum caspase inhibitor, but not vehicle, to iCA cells protected these cells against RU486-induced apoptosis (Fig. 2d, e), suggesting that ChREBP induces apoptosis predominantly through a caspase-dependent pathway. Furthermore, we found that siRNA treatment of these cells decreased ChREBP production by ~60% (see Fig. 3c) and partially attenuated the high-glucose-induced caspase-3/7 activity (Fig. 2f).

ChREBP induces caspase-dependent apoptosis and oxidative stress. a Schematic diagram of RU486-inducible CA-Chrebp vectors. Expression of RU486 regulatable element (GLP65) is driven by CA (viral long terminal repeat) promoter. GLP65 homodimerises and binds to inducible promoter (17 × 4tk) in the presence of RU486, which leads to activation of CA-Chrebp transcription. b Overexpression of CA-Chrebp by the RU486-inducible system. We treated iCA cells for 24 h with RU486 10 nmol/l or vehicle (ethanol), and then isolated the RNA and performed qRT-PCR using CA-Chrebp-specific primers. The histograms are the means of relative RNA levels normalised to mRNA levels of Eef1g and expressed as fold activation over the activity seen in cells exposed to ethanol. White bars, glucose at 2.5 mmol/l; black bars, glucose at 25 mmol/l. c Activation of iCA cells leads to increased caspase-3/7 activity. We treated iCA and wild-type 832/13 cells with ethanol or RU486 10 nmol/l in 2.5 or 25 mmol/l d-glucose medium. Caspase-3/7 activity was measured after 48 h of treatment and normalised to wild-type cells exposed to ethanol and 2.5 mmol/l d-glucose. White bars, glucose at 2.5 mmol/l; black bars, glucose at 25 mmol/l. d, e Overexpression of CA-Chrebp induces apoptosis. We treated wild-type cells and iCA cells with ethanol or 10 nmol/l RU486 in the presence of DMSO or Q-VD-OPH 100 μmol/l for 48 h and stained them with APOPercentage dye. Apoptotic cells appear red in the image. Histograms (e) represent the amount of dye uptake by apoptotic cells compared with wild-type cells treated with ethanol. White bars, ethanol; black bars, RU486. f Chrebp silencing decreases glucose-induced caspase-3/7 activity. We transfected 832/13 cells with Chrebp siRNA, which led to ~60% reduction in ChREBP protein, incubated these cells in 25 mmol/l d-glucose for 48 h, and measured caspase-3/7 activity. g Activation of ChREBP increases oxidative stress to iCA cells. We treated iCA cells with ethanol or RU486 10 nmol/l for 18 h and incubated them with DCFH-DA to assay for accumulation of cellular reactive oxygen species. Intensity of fluorescence was measured. h Chrebp silencing reduces oxidative stress. We incubated 832/13 cells transfected with Chrebp siRNA in 25 mmol/l d-glucose for 48 h, and measured DCFH-DA oxidation. * p < 0.05 compared with cells treated with 2.5 mmol/l d-glucose or ethanol or control siRNA. ψ+, packaging signal; EtOH, ethanol; HygroR, hygromycin-resistance gene; LTR, long terminal repeat; PuroR, puromycin-resistance gene; PPGK, phosphoglycerate kinase promoter; WT, wild-type

ChREBP regulates Txnip expression. a Alignment of predicted ChoRE sequences present in Txnip promoters among vertebrates. Black, absolutely conserved nucleotide in E-boxes; grey, relatively conserved nucleotide between E-boxes, compared with human. b Induction of Chrebp stimulates Txnip transcripts in iCA cells. We preincubated iCA and wild-type cells for 16 h in 2.5 mmol/l d-glucose in the presence of RU486 10 nmol/l or ethanol, and then incubated them in 2.5 mmol/l or 25 mmol/l d-glucose for 8 h. We isolated the RNA and performed qRT-PCR using Txnip-specific primers. The histograms are the means of relative RNA levels normalised to Hmbs, Alas1 and Eef1g and expressed as fold activation over the activity seen in wild-type cells exposed to 2.5 mmol/l d-glucose and ethanol. c siRNA-mediated Chrebp mRNA silencing downregulates TXNIP protein production in 832/13 cells. We transfected 832/13 cells with Chrebp-targeting siRNA or control siRNA for 48 h and extracted total protein for western blotting using rabbit anti-ChREBP with goat anti-rabbit HRP-conjugated antibodies or monoclonal anti-TXNIP with goat anti-mouse horseradish peroxidase-conjugated antibodies. d Chrebp silencing diminishes glucose-induced Txnip mRNA expression. We transfected 832/13 cells with Chrebp-targeting siRNA or control siRNA for 48 h. We isolated the RNA and performed qRT-PCR using Txnip-specific primers. The histograms are expressed as fold activation over the activity seen in 832/13 cells transfected with control siRNA and exposed to 2.5 mmol/l d-glucose. e The effect of ChREBP on the Txnip promoter is localised to the ChoRE by serial mutagenesis. We co-transfected luciferase reporter driven by wild-type or mutated proximal rat Txnip promoter sequences with CA-Chrebp. Luciferase activity was measured at 24 h and normalised to SEAP, and expressed as fold activation over the activity seen in cells transfected with proximal rat Txnip promoter and empty vector. Numbers represent base pairs upstream of transcription start site provided by Ensembl (ENSRNOG00000021201). *p < 0.05 compared with cells treated with 2.5 mmol/l d-glucose or ethanol. In b and d: white bars, glucose at 2.5 mmol/l; black bars, glucose at 25 mmol/l. In e: white bars, empty vector; black bars, CA-Chrebp. EtOH, ethanol; Luc, luciferase; WT, wild-type
Chronic oxidative stress has been proposed as a major cause of pancreatic beta cell dysfunction and death, a pathogenic mechanism that is magnified by the very low levels of antioxidant enzyme activity in these cells [27]. We measured DCFH-DA oxidation [28] in iCA cells to determine whether ChREBP activation leads to an increase in cellular reactive oxygen species. Fluorometric measurements showed a doubling of the conversion of DCFH-DA to fluorescent dichlorofluorescein, indicating an increase in cellular reactive oxygen species in iCA cells within 18 h of RU486-induced ChREBP expression (Fig. 2g). On the other hand, siRNA knockdown of Chrebp led to partial amelioration of DCFH-DA oxidation stimulated by high glucose (25 mmol/l) in 832/13 cells (Fig. 2h), further supporting a direct role of ChREBP in regulating the accumulation of cellular reactive oxygen species.
Therefore, increased or constitutive Chrebp expression per se is sufficient to promote lipid accumulation, inhibit upregulation of insulin mRNA and secretion by high glucose, increase cellular reactive oxygen species, and induce caspase-dependent apoptosis, all of which are defects found in pancreatic beta cells exposed to prolonged high glucose (25 mmol/l).
ChREBP regulates Txnip mRNA and protein expression in pancreatic beta cells
Putative ChoRE sequences were recently identified in the promoter of Txnip, a critical factor for glucose-induced apoptosis in beta cells [29–31]. Sequence alignment of Txnip ChoRE from mammals to fish showed the element to be highly conserved (Fig. 3a), supporting a functionally important role for glucose-induced Txnip transcription in vertebrates. Although direct binding of ChREBP to the Txnip ChoRE sequence and regulation of Txnip promoter activity has been demonstrated [32], functional effects of this binding have not been reported.
We studied the impact of RU486-inducible CA-Chrebp expression on Txnip mRNA in 832/13 cells (Fig. 3b). Addition of RU486 had no effect on the basal Txnip mRNA expression in wild-type cells, but it increased basal Txnip mRNA expression in a low-glucose environment through induced expression of CA-Chrebp in iCA cells (Fig. 3b). To corroborate the role of ChREBP on Txnip expression, we transfected 832/13 cells with Chrebp siRNA and found that siRNA silencing of Chrebp attenuated the high-glucose-mediated upregulation of Txnip mRNA (Fig. 3d) as well as protein production (Fig. 3c). Therefore, results from both activation and silencing of Chrebp support a key role for this transcription factor in the expression of Txnip in 832/13 cells.
The presence of two E-box motifs with proper spacing in ChoRE is critical for determining glucose regulation [33]. We note, however, that ChREBP may not be the only glucose-responsive ChoRE-binding protein [34]. To dissect the role of an intact ChoRE in the activation of the Txnip promoter by ChREBP, we constructed luciferase reporter constructs driven by the Txnip promoter containing the native ChoRE or different mutated ChoREs (Fig. 3e). In comparing the response of these different constructs with CA-Chrebp, we found that mutations in one of the two E-boxes of ChoRE, deletion of a single nucleotide between the E-boxes, or deletion of the entire ChoRE sequence on the proximal Txnip promoter, leads to complete loss of activation by CA-ChREBP. These data suggest that all elements of ChoRE sequence in the Txnip promoter are essential for ChREBP activation.
Hyperglycaemia activates ChREBP in human pancreatic beta cells
To determine if ChREBP plays a similar role in human beta cells, we examined tissue sections of the pancreas of type 2 diabetic individuals and non-diabetic controls. By immunofluorescence, pancreatic beta cells of diabetic individuals produced increased levels of immunoreactive ChREBP and thioredoxin-interacting protein (TXNIP) (and reduced levels of insulin) compared with those of non-diabetics. There was also markedly increased overlap in ChREBP and nuclear (DAPI) staining in sections from diabetic individuals (Fig. 4a). These observations are corroborated by confocal microscopy, which revealed in greater morphological detail an intense overall ChREBP staining with substantially more ChREBP translocation in the nuclei of beta cells in diabetic than in non-diabetic individuals (Fig. 4b). The difference in the distribution of ChREBP was observed in pancreatic sections from all individuals examined (three with diabetes and three without), consistent with a correlation with the diabetic state and not individual variation.

Activation of ChREBP in pancreatic beta cells is associated with Txnip upregulation in vivo. a Diabetic human beta cells produce high levels of ChREBP and TXNIP protein. We stained for nucleus (blue), ChREBP (red), insulin (purple) and TXNIP (green) in non-diabetic and diabetic pancreas paraffin sections and imaged by fluorescence microscopy. b More nuclear ChREBP is present in diabetic beta cells. We labelled insulin (blue), ChREBP (green), nucleus (red) and imaged by confocal microscopy to confirm nuclear localisation of ChREBP (yellow colour in merged picture) in non-diabetic and diabetic pancreas. White arrows indicate the presence of nuclear ChREBP in the beta cells from the pancreatic sections of diabetic patients. c Txnip expression is upregulated in isolated islets. We performed qRT-PCR to detect Txnip expression in isolated islets treated with 5 or 25 mmol/l d-glucose for 48 h and normalised to Eef1g expression. d High glucose induces caspase-3/7 activity in isolated islets. We incubated isolated islets in medium containing 5 or 25 mmol/l d-glucose for 72 h and measured caspase-3/7 activity. e Glucose-induced hyperglycaemia activates ChREBP in vivo. Pancreas paraffin sections from mice injected with 77 mmol/l NaCl only or glucose in 77 mmol/l NaCl solution for 24 h were stained for nucleus (red), ChREBP (green) or insulin (blue) and imaged by confocal microscopy. White arrows indicate the presence of nuclear ChREBP in the beta cells from the pancreatic sections of mice with glucose-induced hyperglycaemia. f Upregulation of Txnip transcripts by glucose-induced hyperglycaemia. We isolated pancreatic islets from mice injected with glucose or 77 mmol/l NaCl by the LCM technique and extracted RNA. We performed qRT-PCR to quantify Txnip expression, which is normalised by expression of Rn18s. g TXNIP protein production is also induced in glucose-injected mice. We stained pancreas paraffin sections from 77 mmol/l NaCl- or glucose-injected mice for insulin (purple) or TXNIP (green). * p < 0.05 compared with cells treated with 5 mmol d-glucose. Scale bar, 5 μm
High glucose activates ChREBP in isolated islets as well as mouse pancreatic beta cells in vivo
To exclude confounding factors that might have influenced the observations in human pancreatic beta cells, we isolated pancreatic islets from non-diabetic mice and incubated them in culture medium with different concentrations of glucose for 48 h. By immunofluorescence we found that, compared with 5 mmol/l glucose, exposure to 25 mmol/l glucose led to enhanced nuclear localisation of ChREBP in insulin-producing cells in these islets (electronic supplementary material [ESM] Fig. 1). By quantitative (q)RT-PCR, incubation in high glucose was found to lead to an approximate threefold increased Txnip mRNA level compared with low glucose (Fig. 4c). Furthermore, incubation in 25 mmol/l glucose led to an approximate threefold increase in caspase-3/7 activity compared with 5 mmol/l glucose (Fig. 4d).
Finally, to determine if ChREBP activation is regulated by hyperglycaemia in pancreatic islets in vivo, we induced steady hyperglycaemia over a 24 h period in non-diabetic mice by repeated subcutaneous glucose injections, at the end of which we collected their pancreases for analysis. Immunofluorescent staining revealed that pancreatic beta cells of glucose-injected mice displayed enhanced ChREBP nuclear localisation compared with controls injected with 77 mmol/l NaCl solution (Fig. 4e). In addition, RNA isolated by laser capture microdissection followed by qRT-PCR showed that high glucose induced the expression of Txnip mRNA (Fig. 4f). Moreover, by immunofluorescence the hyperglycaemia also induced the appearance of TXNIP in the insulin-producing cells of the pancreas of these mice (Fig. 4g). Therefore, as in 832/13 cells exposed to high glucose in vitro, hyperglycaemia leads to translocation of ChREBP into the nucleus, and activation of Txnip transcription and protein accumulation in pancreatic beta cells in mice in vivo.
ChREBP is sufficient for recapitulating the downstream effects of pancreatic beta cell glucotoxicity in mouse models
To mimic the effects of high-glucose-induced ChREBP activation in pancreatic beta cells in vivo, we used gene delivery by AAV. AAV-mediated gene transfer provides robust and stable expression of transgene in multiple tissues. Double-stranded (ds)AAV serotype 8 has been used successfully for delivering genes to pancreatic islets in mice by i.p. injection [35]. However, the packaging limit of dsAAV is about 2.5 kb, which is too short for the beta cell-specific Chrebp expression cassette. We optimised conditions for i.p. injection of single-stranded (ss)AAV8 using a viral construct that contains secreted luciferase driven by the rat insulin promoter (Fig. 5a). We monitored plasma luciferase as an indicator of overall transgene expression in beta cells and found that i.p. injection of AAV8 in 3–4-week-old mice with a high dose of virus provided better results than injection in newborn mice, with the peak expression at 5–6 week after injection (data not shown). We next constructed another AAV vector which contains Rip-driven CA-Chrebp (Fig. 5a).

Chrebp overexpression in beta cells induces diabetes in mice and oxidative beta cell damage in vivo. a Schematic diagram of AAV vectors. CA-Chrebp expression is driven by rat insulin 2 promoter (Rip) for beta cell-specific expression. Control vector contains GLuc and enhanced green fluorescent protein (eGFP) cDNA. b,c Beta cell-specific overexpression of CA-Chrebp induces beta cell damage and diabetes in diabetic-prone mice. Male NONcNZO10/LtJ mice were injected i.p. with CA-Chrebp-containing AAV (n = 3) or control vector (n = 3). Tail blood glucose (b) and body weight (c) were measured every week. d–g Mice injected with CA-Chrebp-containing AAV display beta cell dysfunction. Oral glucose tolerance tests were performed at 4 weeks after AAV injection. Tail blood was collected at 0, 15, 30, 60, 120 and 180 min after gavage and measured for glucose (d) and insulin (f). Area under the curve of glucose (120 min) (e) and insulin/glucose (60 min) (g) were calculated and reported as percentages compared with control groups. * p < 0.05 compared with control group. Diamonds, Rip-GLuc-IRES-eGFP; squares, Rip-CA-Chrebp; IRES, internal ribosomal entry site; ITR, inverted terminal repeat
We chose the diabetes-prone NONcNZO10/LtJ mouse strain, which was developed to model human type 2 diabetes [36]. Male NONcNZO10/LtJ mice develop hyperglycaemia after high-fat feeding to attain a specified body weight during peripuberty and afterwards. We injected i.p. AAV8 expressing either Rip-driven CA-Chrebp or Rip-driven secreted luciferase into chow-fed NONcNZO10/LtJ mice and monitored their fasting blood glucose and body weight. We found that, compared with those injected with AAV8-luciferase, mice injected with AAV8 expressing CA-Chrebp developed progressive hyperglycaemia (Fig. 5b) with arrested body weight gain over time (Fig. 5c). Oral glucose tolerance tests demonstrated abnormally elevated blood glucose excursions (Fig. 5d,e) associated with reduced serum insulin levels (Fig. 5f) in these mice. The insulin/glucose ratio, a measure of beta cell function, was reduced by 57% compared with controls (Fig. 5g). We killed these mice 6 weeks after gene transfer and collected their pancreases. Localisation of transgene in pancreatic beta cells was confirmed by immunofluorescence (Fig. 6b). We found that the control vector-injected animals had normal islet morphology, whereas the CA-Chrebp-AAV8-injected mice displayed markedly disorganised islet architecture (Fig. 6a). Suppressed endogenous insulin protein and increased Txnip expression were observed in CA-Chrebp overexpressing mouse islets by immunostaining (Fig. 6c,d). Furthermore, levels of 8-hydroxyguanosine (8OHG) and cleaved PARP, markers for oxidative stress and apoptosis [37], respectively, were markedly increased in these mice (Fig. e,f). These results indicate that overexpression of CA-Chrebp in vivo in beta cells is sufficient to promote oxidative stress, beta cell dysfunction and apoptosis, as well as suppress insulin production, culminating in a diabetic phenotype.

CA-Chrebp-containing AAV reduces insulin expression and induces oxidative damage and apoptosis. Mice were killed and pancreases collected at 6.5 weeks after AAV injection. Pancreas sections were processed. a Haematoxylin and eosin staining of Rip-GLuc-IRES-eGFP and Rip-CA-Chrebp. b Immunofluorescent staining for insulin and luciferase in control group. Immunofluorescent staining for insulin (c), TXNIP (d), 8OHG (e) and PARP (f)
Discussion
The concept of glucotoxicity, i.e. that long-term hyperglycaemia damages beta cells, was recognised over 25 years ago [1]. There is now compelling evidence that glucotoxicity is responsible, at least in part, for the time-dependent deterioration of beta cell function [2, 4] and loss of beta cell mass [38, 39] in patients with type 2 diabetes. Over the years, a number of biochemical pathways have been implicated in the process [4, 40, 41]. Experiments presented here suggest that ChREBP, a hyperglycaemia-responsive gene product that regulates multiple downstream effector genes, is a mediator of beta cell glucotoxicity via activation of a transcription programme that culminates in beta cell dysfunction and apoptosis.
High glucose not only induces ChREBP nuclear translocation, it also upregulates its transactivation activity. ChREBP requires high glucose for full activation [17]; therefore, wild-type ChREBP overproduction may not effectively uncover all of its functions, especially under low-glucose conditions. On the other hand, it is difficult to differentiate ChREBP function from other high-glucose effects in wild-type Chrebp-overexpressing cells under high-glucose conditions. It is thus not unexpected that Wang et al found that wild-type Chrebp overexpression in INS-1 cells only slightly upregulated downstream target genes and failed to affect apoptosis [10]. Da Silva Xavier et al reported that ChREBP directly binds to the Fasn promoter in MIN6 cells and Chrebp silencing causes reduction of intracellular triacylglycerol and potentiates glucose-stimulated insulin secretion [19]. Similarly, we found that Chrebp knockdown reduces cellular triacylglycerol content and a dominant negative form of ChREBP increases glucose-induced insulin secretion in 832/13 cells (data not shown). Using CA-ChREBP, we demonstrated further that ChREBP induces Fasn transcription and lipid accumulation, while CA-ChREBP downregulates insulin promoter activity and inhibits high-glucose-induced stimulation of insulin gene expression in 832/13 cells. Suppression of insulin gene transcription is likely an indirect effect of ChREBP by repression of mRNA expression of Pdx-1 (also known as Pdx1) and MafA [42], key transcription factors for insulin gene transcription. Increased oxidative stress in these cells could also underlie this effect of ChREBP, because chronic oxidative stress has been shown to interfere with the availability of pancreatic and duodenal homeobox 1 (PDX-1) and v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian) (MAFA) [43, 44]. Interestingly, Noordeen et al. showed that ChREBP may also contribute to beta cell dysfunction by repression of Arnt (also known as Hif-1β) mRNA expression in 832/13 cells [45]. Taken together, these data suggest that activation of ChREBP is sufficient for high-glucose-induced lipid accumulation and beta cell dysfunction.
CA-Chrebp-overexpressing cells display increased caspase-dependent apoptosis and oxidative stress. Lipid overload in pancreatic beta cells could partially explain these phenomena [46]. Txnip is a direct ChREBP target gene [47] that may mediate part of these effects of ChREBP. TXNIP binds to and inhibits the activity of thioredoxin, a key reactive oxygen species scavenger, and upregulated Txnip expression increases oxidative stress [47]. Txnip overexpression has been shown to lead to induction of cleaved caspase-3 and apoptosis in INS-1 cells [29, 48]. Moreover, high-glucose-induced apoptosis is abolished in TXNIP-deficient islets [31].
Induced overexpression of CA-Chrebp using an RU486-inducible construct strongly stimulates Txnip transcription and activates apoptosis. However, low-level Txnip expression resulting from a leaky ChREBP in non-induced cells does not produce detectable effects on apoptosis. Although we cannot exclude a minimal effect not detected by the assay, we believe that a threshold level of downstream activation may be required to produce significant phenotypes. We found that Chrebp silencing not only diminishes Txnip expression, but also reduces caspase-3/7 activity and DCFH-DA oxidation.
Beta cell overexpression of CA-Chrebp in NONcNZO10/LtJ mice, using AAV to deliver CA-Chrebp, led to impaired insulin secretion, progressive hyperglycaemia and weight loss with reduced insulin expression and secretory response, as well as evidence of increased oxidative damage and apoptosis, indicating that CA-Chrebp overexpression is sufficient to recapitulate the major manifestations associated with glucotoxicity in vivo.
Though the level of activation achieved in our experiments with CA-ChREBP appears to be higher than that achieved by high glucose alone, the results from these experiments and our conclusion that ChREBP mediates the glucotoxicity effects on beta cells are validated by other experiments we have presented. These findings were corroborated by studies in mice wherein first, exposure of isolated mouse islets to high and low glucose ex vivo, and second, induction of hyperglycaemia in vivo recapitulated these changes in the pancreatic beta cells, supporting a role for ChREBP and its downstream target, Txnip, in the beta cell response to prolonged high glucose concentration. Our data also indicate that these findings may be applicable in humans as immunofluorescence examination of pancreas tissue sections from patients with type 2 diabetics and from non-diabetic controls revealed that hyperglycaemia induced: (1) increased ChREBP protein in beta cells; (2) nuclear localisation of ChREBP; and (3) enhanced Txnip expression in the beta cells of people with diabetes.
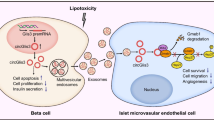
In summary (Fig. 7), this study demonstrates that activated ChREBP is a mediator of glucotoxicity by upregulating downstream target genes Fasn and Txnip, leading to lipid accumulation, increased oxidative stress, stimulated caspase activity and apoptosis, downregulated insulin gene transcription and impaired insulin secretion. Experiments in vitro, ex vivo and in vivo from mouse models and in diabetic and non-diabetic human pancreatic islet tissue sections support a pivotal role for ChREBP in beta cell glucotoxicity.

Model of ChREBP-mediated beta cell glucotoxicity
Abbreviations
- AAV:
-
Adeno-associated virus
- BCM:
-
Baylor College of Medicine
- ChoRE:
-
Carbohydrate response element
- ChREBP:
-
Carbohydrate response element-binding protein
- CMV:
-
Cytomegalovirus
- DCFH-DA:
-
2′,7′-Dichlorofluorescein diacetate
- ds:
-
Double-stranded
- FASN:
-
Fatty acid synthase
- GLuc:
-
Gaussia luciferase
- iCA:
-
Inducible CA-Chrebp-transduced 832/13 cells
- LID:
-
Low-glucose inhibitory domain
- 8OHG:
-
8-Hydroxyguanosine
- PARP:
-
Poly (ADP-ribose) polymerase
- pMSCV:
-
Murine stem cell virus (MSCV)-based retroviral plasmid
- qRT-PCR:
-
Quantitative RT-PCR
- SEAP:
-
Secreted alkaline phosphatase
- siRNA:
-
Small interfering RNA
- ss:
-
Single-stranded
- TXNIP:
-
Thioredoxin-interacting protein
References
Unger RH, Grundy S (1985) Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: implications for the management of diabetes. Diabetologia 28:119–121
Leahy JL, Bonner-Weir S, Weir GC (1992) Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 15:442–455
Del PS (2009) Role of glucotoxicity and lipotoxicity in the pathophysiology of type 2 diabetes mellitus and emerging treatment strategies. Diabet Med 26:1185–1192
Poitout V, Robertson RP (2008) Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 29:351–366
Roche E, Farfari S, Witters LA et al (1998) Long-term exposure of beta-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 47:1086–1094
Kaneto H, Katakami N, Kawamori D et al (2007) Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid Redox Signal 9:355–366
Piro S, Anello M, Di PC et al (2002) Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism 51:1340–1347
Briaud I, Rouault C, Reach G, Poitout V (1999) Long-term exposure of isolated rat islets of Langerhans to supraphysiologic glucose concentrations decreases insulin mRNA levels. Metabolism 48:319–323
Efanova IB, Zaitsev SV, Zhivotovsky B et al (1998) Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular Ca2+ concentration. J Biol Chem 273:33501–33507
Wang H, Kouri G, Wollheim CB (2005) ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J Cell Sci 118:3905–3915
Park KG, Lee KM, Seo HY et al (2007) Glucotoxicity in the INS-1 rat insulinoma cell line is mediated by the orphan nuclear receptor small heterodimer partner. Diabetes 56:431–437
Uyeda K, Repa JJ (2006) Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab 4:107–110
Postic C, Dentin R, Denechaud PD, Girard J (2007) ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 27:179–192
Yamashita H, Takenoshita M, Sakurai M et al (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA 98:9116–9121
Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K (2001) Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci USA 98:13710–13715
Tsatsos NG, Towle HC (2006) Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochem Biophys Res Commun 340:449–456
Li MV, Chang B, Imamura M, Poungvarin N, Chan L (2006) Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 55:1179–1189
Wang H, Wollheim CB (2002) ChREBP rather than USF2 regulates glucose stimulation of endogenous L-pyruvate kinase expression in insulin-secreting cells. J Biol Chem 277:32746–32752
da Silva XG, Rutter GA, Diraison F, Andreolas C, Leclerc I (2006) ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic beta-cells. J Lipid Res 47:2482–2491
Burcin MM, Schiedner G, Kochanek S, Tsai SY, O’Malley BW (1999) Adenovirus-mediated regulable target gene expression in vivo. Proc Natl Acad Sci USA 96:355–360
Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49:424–430
Onishi M, Kinoshita S, Morikawa Y et al (1996) Applications of retrovirus-mediated expression cloning. Exp Hematol 24:324–329
Ramirez-Zacarias JL, Castro-Munozledo F, Kuri-Harcuch W (1992) Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry 97:493–497
Vandesompele J, de PK, Pattyn F et al (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034
Yechoor V, Liu V, Espiritu C et al (2009) Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell 16:358–373
Lee J, Kim MS, Li R et al (2011) Loss of Bmal1 leads to uncoupling and impaired glucose-stimulated insulin secretion in beta-cells. Islets 3:381–388
Robertson RP, Harmon JS (2006) Diabetes, glucose toxicity, and oxidative stress: a case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med 41:177–184
Jakubowski W, Bartosz G (2000) 2,7-dichlorofluorescin oxidation and reactive oxygen species: what does it measure? Cell Biol Int 24:757–760
Minn AH, Hafele C, Shalev A (2005) Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 146:2397–2405
Muoio DM (2007) TXNIP links redox circuitry to glucose control. Cell Metab 5:412–414
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A (2008) Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57:938–944
Cha-Molstad H, Saxena G, Chen J, Shalev A (2009) Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem 284:16898–16905
Shih HM, Liu Z, Towle HC (1995) Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J Biol Chem 270:21991–21997
Yamada K, Kawata H, Shou Z, Mizutani T, Noguchi T, Miyamoto K (2003) Insulin induces the expression of the SHARP-2/Stra13/DEC1 gene via a phosphoinositide 3-kinase pathway. J Biol Chem 278:30719–30724
Wang Z, Zhu T, Rehman KK et al (2006) Widespread and stable pancreatic gene transfer by adeno-associated virus vectors via different routes. Diabetes 55:875–884
Cho YR, Kim HJ, Park SY et al (2007) Hyperglycemia, maturity-onset obesity, and insulin resistance in NONcNZO10/LtJ males, a new mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab 293:E327–E336
Oliver FJ, de la RG, Rolli V, Ruiz-Ruiz MC, de MG, Murcia JM (1998) Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem 273:33533–33539
Maedler K, Spinas GA, Lehmann R et al (2001) Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 50:1683–1690
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Leahy JL (2005) Pathogenesis of type 2 diabetes mellitus. Arch Med Res 36:197–209
Maedler K (2008) Beta cells in type 2 diabetes—a crucial contribution to pathogenesis. Diabetes Obes Metab 10:408–420
da SX, Sun G, Qian Q, Rutter GA, Leclerc I (2010) ChREBP regulates Pdx-1 and other glucose-sensitive genes in pancreatic beta-cells. Biochem Biophys Res Commun 402:252–257
Kaneto H, Kawamori D, Matsuoka TA, Kajimoto Y, Yamasaki Y (2005) Oxidative stress and pancreatic beta-cell dysfunction. Am J Ther 12:529–533
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280:11107–11113
Noordeen NA, Khera TK, Sun G et al (2010) Carbohydrate-responsive element-binding protein (ChREBP) is a negative regulator of ARNT/HIF-1beta gene expression in pancreatic islet beta-cells. Diabetes 59:153–160
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem 279:30369–30374
Minn AH, Pise-Masison CA, Radonovich M et al (2005) Gene expression profiling in INS-1 cells overexpressing thioredoxin-interacting protein. Biochem Biophys Res Commun 336:770–778
Acknowledgements
We thank C. Newgard for 832/13 cells, S. Tsai for the RU486-inducible system, D. Spencer for the SEAP reporter construct, D. Moore, P. Barger and A. Rajun for comments, and M.-J. Tsai, M. Imamura and A. Antar for critical readings of the manuscript.
Funding
This work was supported by National Institutes of Health Grants HL R01-51586 to L. Chan, and by the Diabetes & Endocrinology Research Center (P30-DK079638) at BCM. L. Chan also received support from the Betty Rutherford Chair for Diabetes Research from St Luke’s Episcopal Hospital, the T.T. & W.F. Chao Global Foundation.
Contribution statement
NP, JKL, VKY, MVL, TA, PS, JJT, PKS, KO and LC contributed to the conception and design, or analysis and interpretation of data. NP, JKL, VKY, MVL, TA, PS, JJT, PKS, KO and LC contributed to drafting the article or revising it critically for important intellectual content. All authors approved the final version.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 184 kb)
Rights and permissions
About this article
Cite this article
Poungvarin, N., Lee, J.K., Yechoor, V.K. et al. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 55, 1783–1796 (2012). https://doi.org/10.1007/s00125-012-2506-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2506-4