
Abstract
Aims/hypothesis
Recent experiments in liver and adipocyte cell lines indicate that palmitate can induce endoplasmic reticulum (ER) stress. Since it has been shown that ER stress can interfere with insulin signalling, our hypothesis was that the deleterious action of palmitate on the insulin signalling pathway in muscle cells could also involve ER stress.
Methods
We used C2C12 and human myotubes that were treated either with palmitate or tunicamycin. Total lysates and RNA were prepared for western blotting or quantitative RT-PCR respectively. Glycogen synthesis was assessed by [14C]glucose incorporation.
Results
Incubation of myotubes with palmitate or tunicamycin inhibited insulin-stimulated protein kinase B (PKB)/ v-akt murine thymoma viral oncogene homologue 1 (Akt). In parallel, an increase in ER stress markers was observed. Pre-incubation with chemical chaperones that reduce ER stress only prevented tunicamycin but not palmitate-induced insulin resistance. We hypothesised that ER stress activation levels induced by palmitate may not be high enough to induce insulin resistance, in contrast with tunicamycin-induced ER stress. Indeed, tunicamycin induced a robust activation of the inositol-requiring enzyme 1 (IRE-1)/c-JUN NH2-terminal kinase (JNK) pathway, leading to serine phosphorylation of insulin receptor substrate 1 (IRS-1) and a decrease in IRS-1 tyrosine phosphorylation. In contrast, palmitate only induced a very weak activation of the IRE1/JNK pathway, with no IRS1 serine phosphorylation.
Conclusions/interpretation
These data show that insulin resistance induced by palmitate is not related to ER stress in muscle cells.
Similar content being viewed by others


Introduction
Insulin resistance observed in skeletal muscle is a main feature of obesity and type 2 diabetes. Elevated plasma concentrations of NEFA are consistently observed, leading to lipotoxicity through the over-accumulation of lipid derivatives such as diacylglycerol and ceramides in non-adipose tissues [1, 2]. Palmitate, a predominant saturated NEFA, acts indirectly on insulin signalling by inducing de novo ceramide synthesis and it is now clear that ceramides accumulate in the skeletal muscle of different rodent models of insulin resistance [3, 4], as well as in the muscles of obese insulin-resistant humans [5, 6]. Ceramides modulate diverse signalling pathways through the inhibition of pro-survival kinases such as protein kinase B (PKB)/v-akt murine thymoma viral oncogene homologue 1 (Akt) and the activation of stress-activated kinases such as c-Jun kinase (JNK) and protein kinase C zeta (PKCζ), as well as protein phosphatases such as protein phosphatase 2A (PP2A) and protein phosphatase 1 [7–9]. We, and others, have shown that palmitate-generated ceramides inhibit insulin signalling in muscle cells, specifically by targeting PKB/Akt [10–12], a key serine/threonine kinase implicated in the stimulation of glycogen synthesis and glucose transport by insulin in muscle [13]. Several studies in which ceramide biosynthesis was prevented pharmacologically or genetically have demonstrated that accumulation of ceramide inhibits insulin signalling and, therefore, induced insulin resistance [10, 14, 15].
Interesting observations indicate that saturated fatty acids may also act at the level of the endoplasmic reticulum (ER) by modulating the fusion/fission events of membranes [16, 17]. The result is a dilatation of the ER and the accumulation of unfolded or misfolded protein within the ER lumen. To deal with this stress, cells induce a response called unfolded protein response (UPR) [18]. Three main effectors are activated to restore ER function: protein kinase R-like ER protein kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme-1 (IRE-1) [19]. In the basal state, these factors are kept inactive through their association with a protein chaperone called binding immunoglobulin protein (BIP)/glucose regulated protein 78 (GRP78) in the ER. Following ER stress, BIP/GRP78 is displaced and interacts with unfolded proteins [19], allowing the activation of both PERK and IRE1 by trans-autophosphorylation, and ATF6 by proteolytic cleavage [19]. Once activated these pathways induce several genes and chaperones, such as activating transcription factor 4 (ATF4), CCAAT/enhancer binding protein [C/EBP] homologous protein (CHOP) and BIP/GRP-78, to restore ER homeostasis. The activation of PERK leads to the phosphorylation of eukaryotic initiation factor 2α (eiF2α) resulting in global attenuation of protein synthesis [19]. IRE-1 has an endonuclease activity that activates the transcription factor X box binding protein 1 (XBP-1) by removing a 26 bp segment of its mRNA [19]. Ultimately, when ER homeostasis cannot be restored, apoptosis is initiated [19].
Several studies carried out in liver and adipose tissue have shown that nutrient excess-induced chronic ER stress plays a role in the pathogenesis of diabetes, contributing to hepatic steatosis [20], beta cell loss and insulin resistance [21–23]. Insulin receptor substrate 1 (IRS-1), a docking protein downstream of the insulin receptor [24], is a target of prolonged ER stress through its inhibition by the IRE-1/c-Jun NH2-terminal kinase (JNK) pathway [22]. Reciprocally, prevention of ER stress by the use of chemical chaperones improves glucose homeostasis and insulin sensitivity [25–27].
Activation of ER stress markers in skeletal muscle is still contradictory. ER stress has also been shown to take place in mouse skeletal muscle in response to palmitate [28], and recent studies have shown that UPR takes place in skeletal muscles of diabetic patients [28] and in those of mice fed a high-fat diet [29]. On the other hand, other studies failed to detect ER stress in the skeletal muscles of obese mice [22], as well as of obese [30] and insulin-resistant [31] patients.
Because ceramide biosynthesis from palmitate occurs at the level of the ER [15], and since the activation of the ER can reciprocally induce ceramide biosynthesis in INS-1 cells [32], we questioned the relative roles of ER stress and ceramides in the deleterious action of palmitate on the insulin signalling pathway in muscle cells. We demonstrate that palmitate induces an ER stress in muscle cells, inhibits the insulin signalling pathway and alters glucose metabolism through the production of ceramides. However, inhibition of the palmitate-induced ER stress did not improve palmitate-induced insulin resistance in muscle cells, suggesting that the negative action of palmitate on insulin signalling through ceramide production is independent of ER stress induction.
Methods
Materials
All reagent-grade chemicals, insulin, palmitate, 4-phenyl butyric acid (PBA), and BSA were purchased from Sigma-Aldrich (St Louis, MO, USA). C2-ceramide and tauroursodeoxycholic acid (TUDCA) were obtained from Calbiochem (Darmstadt, Germany). Complete protein phosphatase inhibitor tablets were obtained from Boehringer-Roche Diagnostics (Mannheim, Germany). Antibodies against native PKB/Akt, 473Ser PKB/Akt, 183Thr /185Tyr JNK, β-actin, 980Thr PERK, 21/9Ser glycogen synthase kinase (GSK3)α/β and 51Ser eiF2α were from Cell Signaling (New England Biolabs, Boston, MA, USA), GRP-78 from Santa Cruz Biotechnology (Heidelberg, Germany), 724Ser IRE-1 from Novus Biologicals (Littleton, CO, USA), and 307Ser IRS-1 and 612Tyr IRS-1 from Upstate (Lake Placid, NY, USA). Horseradish peroxidase anti-rabbit, -mouse and -sheep/goat IgGs were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA) and the enhanced chemiluminescent substrate was from Thermo Scientific Pierce (Rockford, IL, USA). D-[U-14C]glucose was obtained from PerkinElmer (Courtaboeuf, France).
Cell culture
C2C12 myoblasts were maintained at 37°C in DMEM (Invitrogen, Paisley, UK) containing 10% (wt/vol.) fetal bovine serum. For differentiation into myotubes, the myoblasts were grown to confluency and the medium was replaced with DMEM containing 2% (wt/vol.) horse serum. Myotubes were used for experiments 4 days following differentiation.
Human skeletal muscle cells
Muscle biopsies were obtained in the context of approved preclinical and clinical trials [33], and via the Tissue Bank for Research (Myobank) of the French Association against Myopathies (AFM), in agreement with the French bioethical law (law no 94–654 of 29 July 1994, modified 22 January 2002) on informed consent. Donors were adults and had no clinical signs of muscular disease. Fresh muscle biopsies were minced and enzymatically dissociated with collagenase. Cell myoblasts were purified, grown and differentiated as myotubes as previously described [33].
Preparation of whole cell lysates
Cells were lysed following experimental manipulation (see figure legends) in an appropriate volume of lysis buffer [34].
Immunoblotting
Cell lysates were subjected to SDS/PAGE and immunoblotted as previously reported [34]. Nitrocellulose membranes were probed with various antibodies as described in the figure legends. Detection of primary antibodies was performed using appropriate peroxidase-conjugated IgGs and protein signals were visualised using enhanced chemiluminescence (Thermo Scientific Pierce) by exposure to Kodak autoradiographic film.
Fatty acid treatment
Cells were treated with palmitate conjugated with fatty acid free BSA as described [10]. Briefly, fatty acids were dissolved in ethanol and diluted 1:10 in DMEM containing 10% (wt/vol.) BSA. Final BSA concentration in the culture was 1.5%. Concentrations of palmitate are indicated in the figure legends.
Glycogen synthesis
C2C12 myotubes were pre-incubated with 0.75 mmol/l palmitate with or without 1 mmol/l TUDCA for 16 h. Five hours before the experiment, cells were deprived of serum in DMEM containing palmitate with or without TUDCA. Cells were then washed with PBS and incubated with or without 100 nmol/l insulin for 60 min in PBS containing D-[U-14C] glucose (3.7 × 10−3 MBq/ml). The incubation was terminated by three washes with ice-cold 0.9% (wt/vol.) NaCl prior to lysis in 60% (wt/vol.) KOH. Cellular glycogen was precipitated from lysates as described previously [35], and associated radioactivity was determined by liquid scintillation counting. Protein was determined using the Bradford method [36].
Isolation of total RNA and quantitative RT-PCR
Total RNA was isolated as described previously [37]. We retro-transcribed 1 μg RNA using Superscript II (Invitrogen). Real-time quantitative RT-PCR (qRT-PCR) analyses were performed with 25 ng cDNA and 250 nmol/l sense and antisense primers (Eurogentec, Angers, France) in a final reaction volume of 25 μl by using the qPCR Core Kit (Eurogentec) and the MyiQ real-time PCR detection system (Bio-Rad). Specific primers were designed using Primer Express software (version 1.0; Applied Biosystems, Carlsbad, CA, USA). Relative quantification of each gene was calculated after normalisation to 18S ribosomal RNA by using the comparative Ct method. For analysis of Xbp1 mRNA splicing, 200 ng cDNA was amplified with specific primers for the rat Xbp1 gene (forward, 5′-CCATGGGAAGATGTTCTGGG-3′; reverse, 5′-ACACGCTTGGGGATGAATGC-3′). The conditions used for the PCR have been described previously [20]. PCR products were separated by electrophoresis on 2.5% agarose gel and visualised by ethidium bromide staining.
Statistics
Results are expressed as mean ± SEM. Statistical significance was assessed using one-way ANOVA followed by a Bartlett test. A p value <0.05 was considered significant.
Results
Palmitate activates ER stress and alters insulin signalling in C2C12 myotubes
We treated C2C12 myotubes with increasing concentrations of palmitate (0.25–0.75 mmol/l) for 16 h, prior to an acute 10 min insulin challenge. Figure 1a shows that, in the absence of palmitate, an acute insulin challenge induces a significant increase in PKB/Akt phosphorylation on its serine 473 residue. One physiological target of PKB/Akt, GSK3α/β, was also phosphorylated (Fig. 1a). Both PKB/Akt and GSK3α/β insulin-induced phosphorylation were dose-dependently inhibited by palmitate with the highest effect for a concentration of 0.5 mmol/l (Fig. 1a). A concentration of palmitate of 0.75 mmol/l (which remains in the physiological range) was chosen for all further experiments since higher concentrations yielded similar results (including on ER stress, see below) but were slightly toxic for the cells (results not shown). We next investigated the kinetics of palmitate-induced loss of PKB/Akt phosphorylation. Figure 1b shows that PKB/Akt phosphorylation on serine 473 was reduced in a time-dependent manner, and nearly undetectable following a 16 h incubation of cells with palmitate.

Palmitate induces both an alteration in insulin signalling and an ER stress in C2C12 myotubes. a C2C12 myotubes were incubated with various concentrations of palmitate for 16 h followed by 100 nmol/l insulin for the last 10 min. Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt, native PKB/Akt, 21Ser GSK3α, 9Ser GSK3β, GRP-78 and 51Ser eiF2α. b C2C12 myotubes were incubated with 0.75 mmol/l palmitate from 1 h to 16 h followed by 100 nmol/l insulin for the last 10 min before being lysed and immunoblotted with antibodies against 473Ser PKB/Akt, native PKB/Akt, GRP-78 and 51Ser eiF2α. c C2C12 myotubes were incubated with 0.75 mmol/l palmitate for 16 h before total RNA extraction. Unspliced (u) and spliced (s) forms of Xbp1 mRNA were measured by RT-PCR. Representative immunoblots from at least three independent experiments are shown. Protein sizes (kDa) are shown on the left hand size of the immunoblots
Interestingly, palmitate also triggered a UPR in C2C12 myotubes (Fig. 1). An increase in ER stress markers was observed in response to palmitate, i.e. phosphorylation of the translation initiation factor eIF2α (Fig. 1 a,b) splicing of Xbp1 mRNA (Fig. 1c), and activation by two- to fourfold of several ER stress chaperones and foldases, such as the protein content of GRP78 (Fig. 1a, b), and Chop (also known as Ddit3), Atf4, oxygen regulated protein 150 (Orp150 [also known as Hyou1]) and Trb3 (also known as Trib3) mRNA (Fig. 3b).
Ceramides mediate the action of palmitate on both PKB/Akt and the UPR
We then tested whether ER stress was mediated by the endogenous production of ceramides from palmitate. We used myriocin, a commonly used inhibitor of serine palmitoyl transferase, the key enzyme that commits palmitate to the de novo synthesis of ceramides. Figure 2 shows that pretreatment of C2C12 muscle cells with myriocin protected against the palmitate-induced inhibition of PKB/Akt. In parallel, myriocin partially prevented the palmitate-induced phosphorylation of eiF2α in these cells, suggesting that ceramides mediated in part the activation of UPR induced by palmitate. This is confirmed in Fig. 2b where C2C12 cells were treated directly with C2-ceramides. Similarly to palmitate, C2-ceramides (short-chain ceramides) induced the phosphorylation of eiF2α on serine 51 (Fig. 2b), the splicing of Xbp1 mRNA (Fig. 2c) and a concomitant inhibition of the insulin-induced phosphorylation of PKB/Akt (Fig. 2b).

Implication of ceramides in the palmitate-induced activation of ER stress. a C2C12 myotubes were incubated with 0.75 mmol/l palmitate with or without 10 μmol/l myriocin for 16 h followed by 100 nmol/l insulin for the last 10 min. Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt, native PKB/Akt, and 51Ser eiF2α. Histograms show densitometric quantification of both 473Ser PKB/Akt and 51Ser eiF2α. Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to cells treated with insulin; significant change (‡ p < 0.05) relative to cells treated with both palmitate and insulin. b C2C12 myotubes were incubated with 100 μmol/l C2-ceramide for 2 h followed by 100 nmol/l insulin for the last 10 min. Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt, native PKB/Akt, and 51Ser eiF2α. Representative immunoblots from at least three independent experiments are shown. Protein sizes (kDa) are shown on the left hand size of the immunoblots. c C2C12 myotubes were incubated with 100 μmol/l C2-ceramide for 16 h before total RNA extraction. Unspliced (u) and spliced (s) forms of Xbp1 mRNA were measured by RT-PCR
ER stress does not mediate palmitate-induced alterations in insulin signalling in C2C12 and human muscle cells
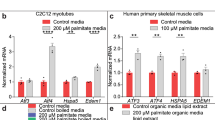
Since there is a temporal correlation between ER stress activation and alterations in insulin signalling induced by palmitate in C2C12 myotubes, we then examined whether inhibition of ER stress could restore insulin sensitivity in C2C12 myotubes. Cells were treated with TUDCA, a chemical chaperone that inhibits ER stress, for 1 h [22, 25], before adding palmitate for a further 16 h. TUDCA significantly decreases palmitate-induced Xbp1 splicing (Fig. 6b), as well as the expression of Atf4, Chop, Orp150 and Trb3 (Fig. 3b), indicating reduced ER stress in these cells. However, TUDCA pretreatment did not improve PKB/Akt phosphorylation in the presence of palmitate (Fig. 3a). Similarly to what was observed with palmitate, pre-incubation of C2C12 cells with TUDCA did not prevent the inhibition of insulin-stimulation of PKB/Akt by C2-ceramide (data not shown). Interestingly, as shown in Fig. 4, glycogen synthesis reflected the pattern observed for insulin signalling, that is, activation by insulin and inhibition in the presence of insulin and palmitate, which still prevails in the presence of TUDCA.

Inhibition of ER stress does not prevent palmitate-induced insulin resistance in C2C12 and human myotubes. a, b C2C12 myotubes were incubated with 0.75 mmol/l palmitate with or without 1 mmol/l TUDCA for 16 h followed by 100 nmol/l insulin for the last 10 min. a Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt, native PKB/Akt, GRP-78, and 51Ser eiF2α. b Quantification of Atf4, Chop, Trb3 and Orp150 mRNA levels was performed by qRT-PCR. Results are mean ± SEM (n = 3). Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to palmitate-treated cells. c, d C2C12 myotubes were incubated with 0.75 mmol/l palmitate with or without 5 mmol/l PBA for 16 h followed by 100 nmol/l insulin for the last 10 min. c Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt and native PKB/Akt. d Quantification of Atf4, Chop, Trb3 and Orp150 mRNA levels was performed by qRT-PCR. Results are mean ± SEM (n = 3). Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to palmitate-treated cells. e, f Human myotubes were incubated with 0.75 mmol/l palmitate with or without 1 mmol/l TUDCA for 16 h followed by 100 nmol/l insulin for the last 10 min. e Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt and native PKB/Akt. f Quantification of ATF4, CHOP, TRB3 and GRP78 (also known as HSPA5) mRNA levels was assessed by qRT-PCR. Results are mean ± SEM (n = 3). Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to palmitate-treated cells. Representative immunoblots from at least three independent experiments are shown. Protein sizes are shown on the left hand size of the immunoblots

Palmitate-induced inhibition of glycogen synthesis is not reversed by ER stress inhibition in C2C12 myotubes. C2C12 myotubes were incubated with 0.75 mmol/l palmitate with or without 1 mmol/l TUDCA for 16 h. Glycogen synthesis was assessed after treating cells with 100 nmol/l insulin for 1 h. Results are mean ± SEM (n = 3). Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to insulin-treated cells; significant change (‡ p < 0.05) relative to insulin- and TUDCA-treated cells
We repeated the experiments on insulin signalling in C2C12 cells using another ER stress inhibitor, namely 4-phenylbutyric acid (PBA) [25]. As shown with TUDCA, PBA reversed palmitate-induced ER stress but insulin signalling was not improved (Fig. 3c, d).
In order to increase the physiological relevance of these findings, we repeated these experiments in cultured human myotubes. In human myotubes, palmitate induces an ER stress (Fig. 3e) and impairs insulin signalling. As is the case in rodent cells, TUDCA, despite alleviating the ER stress, did not restore insulin signalling (Fig. 3e, f).
Tunicamycin-induced ER stress alters insulin signalling in C2C12 myotubes
Overall, our data suggest that the induction of ER stress by palmitate does not play a role in the onset of insulin resistance in muscle cells. This is in contradiction with what has been observed in other cell types, such as hepatocytes and adipocytes [19, 20, 22], where the induction of ER stress by chemical inducers was followed by an insulin resistance state. We then hypothesised that the level of activation of the UPR by palmitate was lower than by chemical inducers. To test this hypothesis, we treated C2C12 muscle cells with tunicamycin, an agent that inhibits protein glycosylation leading to a strong ER stress [20, 22]. A 16 h incubation of the cells with tunicamycin provoked the phosphorylation of IRE-1 and complete splicing of Xbp1 (Fig. 5b). Simultaneously, ER stress genes were activated from 25- to 120-fold by tunicamycin (Fig. 5c). In these conditions, tunicamycin inhibited the phosphorylation of PKB/Akt induced by insulin (Fig. 5a). Interestingly, TUDCA pretreatment partially reversed IRE-1 phosphorylation and Xbp1 mRNA splicing (Fig. 5b), as well as ER stress gene activation (Fig. 5c), and prevented the inhibitory effects of tunicamycin on PKB/Akt phosphorylation (Fig. 5a). This suggests that the action of tunicamycin on insulin signalling is mediated by an ER stress-related pathway.

TUDCA reverses tunicamycin-induced insulin signalling alterations in C2C12 myotubes. a C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin with or without 1 mmol/l TUDCA for 16 h followed by 100 nmol/l insulin for the last 10 min. Cell lysates were immunoblotted with antibodies against 473Ser PKB/Akt and β-actin. Histogram shows densitometric quantification of 473Ser PKB/Akt from three independent experiments. Significant change (*p < 0.05) relative to untreated control. b C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin with or without 1 mmol/l TUDCA for 16 h before RNA extraction or being lysed. Cell lysates were immunoblotted with antibodies against 724Ser IRE-1 and β-actin. Unspliced (u) and spliced (s) forms of Xbp1 mRNA were measured by RT-PCR. c C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin with or without 1 mmol/l TUDCA for 16 h before total RNA extraction. Quantification of Chop, Edem (also known as Edem1), Orp150 and Trb3 mRNA levels were assessed by qRT-PCR. Results are mean ± SEM (n = 3). Significant change (*p < 0.05) relative to untreated control; significant change († p < 0.05) relative to tunicamycin-treated cells. Protein sizes (kDa) are shown on the left hand size of the immunoblots
Tunicamycin activates the IRE-1/JNK pathway to a greater extent than palmitate
It has been proposed in liver cells that ER stress induces an insulin resistance state through the stimulation of the IRE-1 pathway, leading to the activation of the stress-activated kinase JNK [22]. A strong phosphorylation of IRE-1 on its serine 724 residue was observed in the presence of tunicamycin whereas palmitate had a mild effect (Fig. 6a). By contrast, PERK phosphorylation was equally stimulated in response to palmitate and tunicamycin (Fig. 6a).

Tunicamycin inhibits insulin signalling via the activation of the IRE-1/JNK pathway in C2C12 myotubes. a C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin, or 0.75 mmol/l palmitate for 16 h, or 100 nmol/l insulin for 10 min. Cell lysates were immunoblotted with antibodies against 980Thr PERK, 724Ser IRE-1, and native IRE-1. Histogram shows densitometric quantification of 724Ser IRE-1. Significant change (*p < 0.05) relative to untreated control. b C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin or 0.75 mmol/l palmitate with or without 1 mmol/l TUDCA for 16 h before total RNA extraction. Unspliced (u) and spliced (s) forms of Xbp1 mRNA were measured by RT-PCR. c C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin or 0.75 mmol/l palmitate for 16 h before being lysed. Cell lysates were immunoblotted with antibodies against 183Thr/185Tyr JNK and native JNK. d C2C12 myotubes were incubated with 0.5 μg/ml tunicamycin or 0.75 mmol/l palmitate for 16 h in the presence of insulin (100 nmol/l) for the last 10 min before being lysed. Cell lysates were immunoblotted with antibodies against 307Ser IRS-1, 612Tyr IRS-1 and native IRS-1. Representative immunoblots from at least three independent experiments are shown. Protein sizes (kDa) are shown on the left hand size of the immunoblots
Considering the events downstream of IRE-1 activation, the level of Xbp1 splicing reflected the higher phosphorylation of IRE-1 by tunicamycin, compared with what was observed with palmitate (Fig. 6b) and phosphorylation of JNK was more prominent in cells treated with tunicamycin than with palmitate (Fig. 6c). It has been shown in liver cells that the insulin signalling pathway was affected after tunicamycin treatment through the inhibition of the docking protein IRS-1 when phosphorylated on serine residues by JNK [22]. Tunicamycin-treated C2C12 myotubes displayed a significant increase in phosphorylation of IRS-1 on serine 307 and a drastic decrease in insulin-induced IRS-1 tyrosine phosphorylation (Fig. 6d). In contrast, and as already described [12], palmitate-treated C2C12 cells did not exhibit any serine phosphorylation of IRS-1 (Fig. 6d) and IRS-1 tyrosine phosphorylation was not affected. This confirms the lack of activation of the IRE-1/JNK pathway by palmitate in these cells and corroborates a more downstream action of palmitate on the insulin signalling pathway at the level of PKB/Akt.
Discussion
Palmitate induces concomitantly an ER stress and insulin resistance in muscle cells
Accumulation of unfolded proteins in the ER lumen induces an adaptive response called UPR in order to decrease protein synthesis and to restore ER folding capacities [19]. However, in situations where ER stress is activated chronically, a decline of insulin action appears in liver and adipose tissue, thus contributing to the development of type 2 diabetes [22]. Although a direct role of ER stress in the pathogenesis of diabetes and obesity has been demonstrated in both liver and adipose cells [19, 20, 22, 23, 25], much less information is available in skeletal muscle. Studies have shown the appearance of ER stress markers in response to high-fat diet in mouse skeletal muscles [29] or in diabetic patients [28], but very recently, the involvement of ER stress in human skeletal muscle at the onset of glucose intolerance has been questioned [30, 31]. In the present study, we examined the direct effects of NEFA (i.e. palmitate) on both muscle cell ER homeostasis and insulin sensitivity. Our data demonstrate that palmitate concomitantly disrupts ER homeostasis and alters insulin signalling and glucose metabolism, but that palmitate-induced ER stress is not responsible for the decrease in insulin sensitivity.
Palmitate-induced ER stress is linked to ceramide production
Interestingly, and in contrast with what had been shown in INS-1 cells [32] and H4IIE liver cells [38], de novo ceramide synthesis from palmitate was required for fatty acid-induced ER stress in C2C12 myotubes. Indeed, we show that ceramides alone can induce an ER stress and that inhibition of ceramide synthesis in the presence of palmitate prevents ER stress in muscle cells. The action of ceramides on ER stress has also been described in various cell types such as prostate epithelial cells [39], mouse T hybridoma cells [40], pancreatic beta cells [41] and human glioma cells [42]. However, the mechanism by which ceramides can induce an ER stress remains largely unknown.
Tunicamycin-induced ER stress promotes an insulin resistance in muscle cells
As the activation of ER stress itself can be involved in the development of insulin resistance, we have reproduced a situation of high ER stress without any added NEFA that could be a substrate for ceramide production. Our data indicate that activation of ER stress by tunicamycin downregulates the insulin signalling pathway in C2C12 myotubes, an effect which was reversed in the presence of a chemical chaperone that reduced the intensity of ER stress. Tunicamycin strongly activates the IRE-1/JNK pathway, leading to serine phosphorylation of IRS-1 and a blockade of insulin signalling at this step. Insulin resistance induced by tunicamycin occurred independently of any ceramide production, since tunicamycin action was not prevented when cells were pre-incubated with myriocin (data not shown).
Palmitate-induced insulin resistance is not linked to the concomitant palmitate-induced ER stress in muscle cells
Surprisingly, and despite the fact that a tunicamycin-induced ER stress was able to inhibit insulin signalling (see above), ER stress induced by palmitate does not play a role in the induction of insulin resistance. Indeed, lowering the ER stress with two chemical chaperones (TUDCA and PBA) did not improve insulin sensitivity or glucose metabolism in rodent and human muscle cells, in contrast with what was observed when ER stress was induced by tunicamycin.
ER stress induced by palmitate is not strong enough to induce insulin resistance
We then tried to understand why palmitate-induced ER stress does not play a role in the initiation of insulin resistance in muscle cells. One potential answer was that palmitate could modulate only selective genes of the UPR and/or that palmitate did not induce ER stress to a sufficiently high level to activate the whole UPR system. We confirmed both of these assumptions. We showed that the magnitude of activation of ER stress genes was much higher in cells treated with tunicamycin than with palmitate (compare Figs 3, 5). Importantly, a 50% decrease in tunicamycin-induced stress markers in the presence of TUDCA is sufficient to prevent the ER stress-mediated tunicamycin action on insulin signalling. This demonstrates that ER stress activation has to be strong to play a deleterious action on insulin signalling. In particular, the IRE-1/JNK pathway was poorly activated after palmitate treatment, but fully activated with tunicamycin. The modest activation of this pathway by palmitate (or directly with ceramides) explains why, in contrast with tunicamycin, palmitate and ceramides have no negative impact on IRS-1 in muscle cells and act only at the level of PKB/Akt, as described previously [8, 10, 12]. A difference in ER stress activation between palmitate and chemical agents such as tunicamycin had already been observed in H4IIE liver cells [38]. Indeed, in that study, palmitate induced the UPR only moderately, with no real action on the IRE-1/XBP-1 pathway, compared with what was observed in cells treated with tunicamycin [38].
We must point out that the cellular models used in this study to investigate an individual fatty acid remains far from an in vivo situation where a mixture of fatty acids is always chronically present. In previous studies, it has been shown that unsaturated fatty acids such as oleate could prevent both palmitate-induced ER stress-mediated apoptosis and insulin resistance in liver and muscle cells by diverting palmitate from producing ceramide to neutral lipid storage [43, 44]. Thus, the balance between saturated and unsaturated fatty acids remains very important for the study of their impact on glucose metabolism.
During the course of our study, an article has challenged our results. Peng et al. have shown that palmitate-induced insulin resistance was mediated by ER stress in C2C12 cells [45]. It is hard to explain the discrepancy between this study and our data, but potential explanations could be first, that they were using undifferentiated C2C12 myoblasts for their study and second, that added palmitate was not complexed to albumin leading to supraphysiological free palmitate concentrations.
Altogether, these data show that insulin resistance induced by palmitate is not related to ER stress in rodent and human muscle cells. As clearly demonstrated in the literature, the main site of action of palmitate-derived ceramides in downregulating the insulin signalling pathway is PKB/Akt (Fig. 7). On the other hand, we have shown that a marked activation of the UPR can also induce insulin resistance at the level of IRS-1 through the activation of the IRE-1/JNK pathway in muscle cells (Fig. 7). Thus, we cannot exclude the possibility that chronic exposure of the muscle cells to saturated fatty acids, as in the case of obesity, may induce a more significant ER stress than that seen in our conditions, therefore affecting the insulin pathway.

Model recapitulating mechanisms by which ER stress could be involved in the development of insulin resistance in muscle cells. Binding of insulin to its receptor at the plasma membrane induces the activation of the insulin signalling pathway leading to the activation of major cellular events such as glucose transport and glycogen synthesis. Palmitate accumulation into muscle cells is concomitant with the biosynthesis of ceramides at the level of the ER. Increased levels of ceramides inside cells inhibits the stimulation of PKB/Akt by insulin through PKCζ/PP2A activation, and concomitantly induces mild ER stress that is not involved in the PKB/Akt inhibition process. By contrast, induction of a strong pharmacological ER stress with tunicamycin induces a full UPR response, including the activation of the IRE-1/JNK pathway and the inhibition of IRS-1 activity, contributing to the loss of insulin sensitivity. PI3K, phosphatidylinositol 3-kinase
Abbreviations
- Akt:
-
v-akt murine thymoma viral oncogene homologue 1
- ATF4:
-
Activating transcription factor 4
- ATF6:
-
Activating transcription factor 6
- BIP:
-
Binding immunoglobulin protein
- CHOP:
-
CCAAT/enhancer binding protein (C/EBP) homologous protein
- eiF2α:
-
Eukaryotic initiation factor 2α
- ER:
-
Endoplasmic reticulum
- GSK3:
-
Glycogen synthase kinase
- GRP78:
-
Glucose regulated protein 78
- IRE-1:
-
Inositol-requiring enzyme-1
- IRS-1:
-
Insulin receptor substrate-1
- JNK:
-
c-Jun NH2-terminal kinase
- PBA:
-
4-Phenyl butyric acid
- PERK:
-
Protein kinase R-like ER protein kinase
- PKB:
-
Protein kinase B
- PKCζ:
-
Protein kinase C ζ
- PP2A:
-
Protein phosphatase 2A
- TUDCA:
-
Tauroursodeoxycholic acid
- UPR:
-
Unfolded protein response
- XBP-1:
-
X box binding protein 1
References
Turban S, Hajduch E (2011) Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett 585:269–274
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Turinsky J, Bayly BP, O’Sullivan DM (1990) 1,2-Diacylglycerol and ceramide levels in rat skeletal muscle and liver in vivo. Studies with insulin, exercise, muscle denervation, and vasopressin. J Biol Chem 265:7933–7938
Kim JK, Fillmore JJ, Chen Y et al (2001) Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A 98:7522–7527
Haus JM, Kashyap SR, Kasumov T et al (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58:337–343
Adams JM, Pratipanawatr T, Berria R et al (2004) Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53:25–31
Blouin CM, Prado C, Takane KK et al (2010) Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 59:600–610
Hajduch E, Balendran A, Batty IH et al (2001) Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia 44:173–183
Nikolova-Karakashian MN, Rozenova KA (2010) Ceramide in stress response. Adv Exp Med Biol 688:86–108
Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS (2004) Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J 382:619–629
Schmitz-Peiffer C, Craig DL, Biden TJ (1999) Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274:24202–24210
Chavez JA, Knotts TA, Wang LP et al (2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 278:10297–10303
Hajduch E, Litherland GJ, Hundal HS (2001) Protein kinase B (PKB/Akt)—a key regulator of glucose transport? FEBS Lett 492:199–203
Summers SA (2006) Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res 45:42–72
Kitatani K, Idkowiak-Baldys J, Hannun YA (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20:1010–1018
Turner MD (2004) Fatty acyl CoA-mediated inhibition of endoplasmic reticulum assembly. Biochim Biophys Acta 1693:1–4
Haque ME, Lentz BR (2004) Roles of curvature and hydrophobic interstice energy in fusion: studies of lipid perturbant effects. Biochemistry 43:3507–3517
Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47:2726–2737
Flamment M, Kammoun HL, Hainault I, Ferre P, Foufelle F (2010) Endoplasmic reticulum stress: a new actor in the development of hepatic steatosis. Curr Opin Lipidol 21:239–246
Kammoun HL, Chabanon H, Hainault I et al (2009) GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest 119:1201–1215
Ozawa K, Miyazaki M, Matsuhisa M et al (2005) The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes 54:657–663
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Gregor MF, Hotamisligil GS (2007) Thematic review series: adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res 48:1905–1914
Litherland GJ, Hajduch E, Hundal HS (2001) Intracellular signalling mechanisms regulating glucose transport in insulin-sensitive tissues (review). Mol Membr Biol 18:195–204
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Jiao P, Ma J, Feng B et al (2011) FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKbeta pathways. Obesity (Silver Spring) 19:483–491
Lee YY, Hong SH, Lee YJ et al (2010) Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem Biophys Res Commun 397:735–739
Peter A, Weigert C, Staiger H et al (2009) Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes 58:1757–1765
Deldicque L, Cani PD, Philp A et al (2010) The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab 299:E695–E705
Sharma NK, Das SK, Mondal AK et al (2008) Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab 93:4532–4541
Deldicque L, van Proeyen K, Francaux M, Hespel P (2010) The unfolded protein response in human skeletal muscle is not involved in the onset of glucose tolerance impairment induced by a fat-rich diet. Eur J Appl Physiol 111:1553–1558
Lei X, Zhang S, Bohrer A, Ramanadham S (2008) Calcium-independent phospholipase A2 (iPLA2 beta)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem 283:34819–34832
Vilquin JT, Marolleau JP, Sacconi S et al (2005) Normal growth and regenerating ability of myoblasts from unaffected muscles of facioscapulohumeral muscular dystrophy patients. Gene Ther 12:1651–1662
Hajduch E, Alessi DR, Hemmings BA, Hundal HS (1998) Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes 47:1006–1013
Blair AS, Hajduch E, Litherland GJ, Hundal HS (1999) Regulation of glucose transport and glycogen synthesis in L6 muscle cells during oxidative stress. Evidence for cross-talk between the insulin and SAPK2/p38 mitogen-activated protein kinase signaling pathways. J Biol Chem 274:36293–36299
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Foretz M, Guichard C, Ferre P, Foufelle F (1999) Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A 96:12737–12742
Wei Y, Wang D, Topczewski F, Pagliassotti MJ (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291:E275–E281
Sauane M, Su ZZ, Dash R et al (2010) Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J Cell Physiol 222:546–555
Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS (2008) Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 111:4365–4374
Boslem E, Macintosh G, Preston AM et al (2011) A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J 435:267–276
Salazar M, Carracedo A, Salanueva IJ et al (2009) Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 119:1359–1372
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 52:726–733
Listenberger LL, Han X, Lewis SE et al (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082
Peng G, Li L, Liu Y et al (2011) Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 152:2206–2218
Acknowledgements
We are grateful to P. Guicheney for helpful advice, to X. Le Liepvre for technical help and to the tissue bank of the Association Française des Myopathies for human biopsy samples. R. Hage Hassan was supported by the French Ministry of Research.
Contribution statement
All authors were responsible for the conception and design of the study, analysis and interpretation of the data, drafting and revising the manuscript. All authors approved the final version to be published.
Duality of interest
All authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hage Hassan, R., Hainault, I., Vilquin, JT. et al. Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia 55, 204–214 (2012). https://doi.org/10.1007/s00125-011-2328-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2328-9