
Abstract
Aim/hypothesis
To assess whether low-dose glucocorticoid treatment induces adverse metabolic effects, as is evident for high glucocorticoid doses.
Methods
In a randomised placebo-controlled double-blind (participants and the investigators who performed the studies and assessed the outcomes were blinded) dose–response intervention study, 32 healthy men (age 22 ± 3 years; BMI 22.4 ± 1.7 kg/m2) were allocated to prednisolone 7.5 mg once daily (n = 12), prednisolone 30 mg once daily (n = 12), or placebo (n = 8) for 2 weeks using block randomisation. Main outcome measures were glucose, lipid and protein metabolism, measured by stable isotopes, before and at 2 weeks of treatment, in the fasted state and during a two-step hyperinsulinaemic clamp conducted in the Clinical Research Unit of the Academic Medical Centre, Amsterdam, the Netherlands
Results
Prednisolone, compared with placebo, dose dependently and significantly increased fasting plasma glucose levels, whereas only prednisolone 30 mg increased fasting insulin levels (29 ± 15 pmol/l). Prednisolone 7.5 mg and prednisolone 30 mg decreased the ability of insulin to suppress endogenous glucose production (by 17 ± 6% and 46 ± 7%, respectively, vs placebo). Peripheral glucose uptake was not reduced by prednisolone 7.5 mg, but was decreased by prednisolone 30 mg by 34 ± 6% (p < 0.0001). Compared with placebo, prednisolone treatment tended to decrease lipolysis in the fasted state (p = 0.062), but both prednisolone 7.5 mg and prednisolone 30 mg decreased insulin-mediated suppression of lipolysis by 11 ± 5% and 34 ± 6%, respectively. Finally, prednisolone treatment increased whole-body proteolysis during hyperinsulinaemia, which tended to be driven by prednisolone 30 mg (5 ± 2%; p = 0.06). No side effects were reported by the study participants. All participants completed the study and were analysed.
Conclusions/interpretation
Not only at high doses but also at low doses, glucocorticoid therapy impaired intermediary metabolism by interfering with the metabolic actions of insulin on liver and adipose tissue. These data indicate that even low-dose glucocorticoids may impair glucose tolerance when administered chronically.
Trial registration:
ISRCTN83991850
Funding:
The study was funded by the Dutch Top Institute Pharma T1-106.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucocorticoids (GCs) are the cornerstone in the treatment of numerous chronic autoimmune and inflammatory diseases, including rheumatoid arthritis [1], polymyalgia rheumatica [2] and giant cell arteritis [3], systemic lupus erythematosus [4] and inflammatory bowel diseases [5]. GC treatment is accompanied by significant metabolic adverse effects, including insulin resistance, glucose intolerance and diabetes, visceral adiposity, dyslipidaemia and skeletal muscle atrophy [6]. The development of these adverse metabolic effects is dependent on the administered dose [7], and has mainly been studied with high doses. As such, daily administration of 36 mg prednisolone equivalent impaired glucose tolerance by reducing hepatic [8] and peripheral insulin sensitivity [9]. Furthermore, 30 mg prednisolone daily stimulated adipose tissue, but not whole-body, lipolysis [10], and induced dyslipidaemia [11]. In addition, 60 mg prednisolone daily was shown to stimulate breakdown of skeletal muscle tissue [12]. Finally, chronic exposure to markedly increased endogenous GC levels, as shown in patients with Cushing’s syndrome, unfavourably affected body fat distribution by promoting visceral fat accumulation and hepatic steatosis [13].
However, most patients treated with GCs for prolonged time periods receive, following an induction dose, GC dosages in the lower range: ≤7.5 mg prednisolone equivalent daily. Although the efficacy of these doses on disease activity in various conditions is well established [1–4, 7], the extent to which these low dosages induce adverse metabolic effects is unclear. In large retrospective case–control studies, it was demonstrated that low-dose GC treatment increased the odds for the initiation of blood-glucose lowering therapy by 63% [14] and increased the risk of developing diabetes [15]. In contrast, in clinical trials with chronic use of low-dose GC treatment, incident diabetes is reported less frequently [16]. The main limitation of these data is that no distinction can be made between disease-induced and age-related abnormalities on the one hand, and the metabolic abnormalities induced by low-dose prednisolone treatment on the other. To date, no studies have assessed the effects of low-dose GC treatment on the three most important pathways of intermediary metabolism. In addition, previous studies addressing the metabolic adverse effects of GC treatment have only studied the (hyper)acute effects and typically administered a single dose.
Thus, in the present study, we studied the effects of 2 week low-dose prednisolone treatment (7.5 mg daily) on glucose, lipid and protein metabolism using stable isotopes in healthy men, and included a typical induction dosage of prednisolone (i.e. 30 mg daily), which is known to impair intermediary metabolism, and a matching placebo treatment. Through combined infusion of tracers for glucose and protein metabolism, as well as lipolysis, we assessed differences in the sensitivity of metabolic fluxes to the dose-dependent effects of GC treatment.
Methods
Participants
Thirty-two healthy white men were recruited via local advertisements. All participants were in good health as confirmed by medical history, physical examination, screening blood tests and a 75 g 2 h OGTT, performed at screening visit. Inclusion criteria included: age 18–35 years; BMI 20–25 kg/m2; and normoglycaemia as defined by fasting plasma glucose (FPG) < 5.6 mmol/l and 2 h glucose < 7.8 mmol/l during OGTT. Exclusion criteria were the presence of any disease, use of any medication, first-degree relative with type 2 diabetes, smoking, shift work, a history of GC use, excessive sport activities (i.e. more often than twice/week) and changes in weight in the 3 months prior to study participation. The study was approved by an independent ethics committee and the study was conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent before participation.
Study design
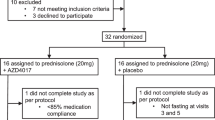
The study was a randomised double-blind placebo-controlled dose-response intervention study. Following assessment of eligibility and baseline measurements, participants were randomised to receive either prednisolone 7.5 mg once daily (n = 12), prednisolone 30 mg once daily (n = 12), or placebo (n = 8) treatment for a period of 14 days using block randomisation, as carried out by the department of experimental pharmacology of the VU University Medical Centre. On day −2 and day 13 of treatment, body composition, body fat distribution and liver fat content were quantified. On day −1 and on day 14 of treatment, glucose kinetics, lipolysis and proteolysis were measured in the basal state and during a two-step hyperinsulinaemic–euglycaemic clamp using stable isotopes (electronic supplementary material [ESM] Fig. 1a, b). All measurements were conducted following a 12 h overnight fast with the individuals in the semi-supine position. Participants refrained from drinking alcohol for a period of 24 h before the study days and did not perform strenuous exercise for a period of 48 h before the study days. During all visits, including a follow-up visit at day 7 of treatment, safety and tolerability were assessed. A patient flow diagram is shown in ESM Fig. 2.
Assessment of body composition/body fat distribution
Body composition was measured by dual energy X-ray absorptiometry (DEXA) scans (Delphi A; Hologic, Waltham, MA, USA). Magnetic resonance imaging (MRI), for determination of visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) area at the level of L3-L4, and proton magnetic resonance spectroscopy (1H-MRS), to quantify liver fat content, were performed using a 1.5 T MRI scanner (Sonata; Siemens, Erlangen, Germany), as described previously by Tushuizen et al. [17, 18]. All magnetic resonance examinations (DVR) and quantification of abdominal fat compartments (MHM) were done by a single experienced investigator.
Clamp
After an overnight fast of 12 h, participants were admitted to the Clinical Research Unit at 07:30 hours. An indwelling cannula was inserted into an antecubital vein for infusion of stable-isotope tracers, glucose and insulin. To obtain arterialised venous blood samples, a retrograde cannula was inserted in a contralateral wrist vein and maintained in a thermoregulated box at 50°C. To keep the sampling line patent 0.9% NaCl was infused. [6,6-2H2]Glucose, [1,1,2,3,3-2H5]glycerol and L-[1-13C]valine were used as tracers (>99% enriched; Cambridge Isotopes, Andover, MA, USA) to study glucose kinetics, lipolysis and valine turnover respectively. At t = 0 h (08:00 hours), blood samples were drawn for determination of background enrichments. Then, a primed continuous infusion of isotopes was started: [6,6-2H2]glucose (prime: 11 μmol/kg; continuous: 0.11 μmol kg−1 min−1), [1,1,2,3,3-2H5]glycerol (prime: 1.6 μmol/kg; continuous: 0.11 μmol kg-1 min-1), and L-[1-13C]valine (prime: 13.7 μmol/kg; continuous: 0.153 μmol kg−1 min−1) and continued until the end of the clamp. After a 2 h equilibrium period (14 h of fasting), three blood samples were drawn for determination of basal glucose concentrations, isotope enrichment, and levels of glucoregulatory hormones and NEFA. Thereafter, a two-step hyperinsulinaemic–euglycaemic clamp was started: step 1 included an infusion of insulin at a rate of 20 mU m−2 min−1 (Actrapid 100 U/ml; Novo Nordisk, Alphen aan den Rijn, the Netherlands) to assess hepatic insulin sensitivity. Glucose 20% was started to maintain a plasma glucose concentration of 5 mmol/l. [6,6-2H2]Glucose was added to the glucose solution to achieve glucose enrichments of 1% in order to approximate the values for enrichment reached in plasma and thereby minimising changes in isotopic enrichment due to changes in the infusion rate of exogenous glucose. Plasma glucose concentrations were measured every 5 min at bedside. After 2 h of insulin infusion, five blood samples were drawn at 5 min intervals for the measurement of glucose concentrations and isotopic enrichments. Another blood sample was drawn for measurement of glucoregulatory hormones and NEFA. Hereafter, insulin infusion was increased to a rate of 60 mU m−2 min−1 (step 2) to assess peripheral insulin sensitivity. After 2 h of insulin infusion, blood sampling for glucose, isotope enrichments, glucoregulatory hormones and NEFA was repeated (ESM Fig. 1b).
Indirect calorimetry
Oxygen consumption (\( {\mathop V\limits^ \cdot }{\text{O}}_{2} \)) and carbon dioxide production (\( {\mathop V\limits^ \cdot }{\text{CO}}_{2} \)) were measured continuously during the final 20 min of both the basal state and during step 2 of the hyperinsulinaemic–euglycaemic clamp by indirect calorimetry using a ventilated hood system (Vmax model 2900; Sensormedics, Anaheim, CA, USA). The \( {\mathop V\limits^ \cdot }{\text{O}}_{2} \) and \( {\mathop V\limits^ \cdot }{\text{CO}}_{2} \) measurements during the last 10 min were used for further calculations.
Study medication
Prednisolone tablets were purchased from Pfizer (Sollentuna, Sweden) and matching placebo tablets were obtained from Xendo Drug Development (Groningen, the Netherlands). The tablets were encapsulated in order to allow the treatment to be blinded, as described previously [19]. Study medication was taken at 08:00 hours during the 2 week treatment except on days 13 and 14, when it was ingested at 06:00 hours. Patients kept a diary in which the exact time of medication intake during the study was registered.
Glucose, lipid, and valine measurements
Plasma glucose concentrations were measured with the glucose oxidase method using a Biosen C-line Plus glucose analyser (EKF Diagnostics, Barleben/Magdeburg, Germany). Plasma NEFA concentrations were measured with an enzymatic colorimetric method (NEFA-C test kit; Wako Chemicals, Neuss, Germany) with an intra-assay variation of 1%, inter-assay variation of 4–15% and a detection limit of 0.02 mmol/l. [6,6-2H2]Glucose, [1,1,2,3,3,-2H5]glycerol and L-[1-13C]valine enrichment were measured with gas chromatography–mass spectrometry as described previously [20–22]. Briefly, temperature-programmed gas chromatography with split injection (Model 6890; Agilent Technologies, Palo Alto, USA) was coupled to a mass selective detector (model 5973 Agilent Technologies) in the electron impact ionisation mode for glucose and valine analysis and in the positive chemical ionisation mode for glycerol. Exact gas chromatography and mass spectometry variables are given in Ackermans et al., Ackermans et al. and Geukers et al. [20–22].
Glucoregulatory hormones
Insulin was determined on an Immulite 2000 system (Diagnostic Products, Los Angeles, CA, USA) with a chemiluminescent immunometric assay, intra-assay variation of 3–6%, an inter-assay variation of 4–6% and a detection limit of 15 pmol/l. Glucagon was determined with the Linco 125I radioimmunoassay (St Charles, MO, USA) with an intra-assay variation of 9–10%, an inter-assay variation of 5–7% and a detection limit of 15 ng/l.
Calculations
Endogenous glucose production (EGP) and the peripheral uptake of glucose (rate of disappearance, R d) were calculated using modified versions of the Steele equations for the non-steady state and were expressed as μmol kg−1 min−1 as described previously [23, 24]. Lipolysis (glycerol turnover) and proteolysis (valine turnover) were computed using formulae for steady state kinetics adapted for stable isotopes [20, 22] and were expressed as μmol kg−1 min−1. The equations are provided in ESM Fig. 3. The abbreviated Weir equation was used to calculate the 24 h energy expenditure. Glucose oxidation and fatty acid oxidation rates were derived from oxygen consumption and carbon oxide production as reported previously [25]. Glucose oxidation during insulin infusion was additionally expressed as percentage of glucose R d.
Statistics
Data are presented as mean values ± SD, or as median (interquartile range) in case of skewed distribution. Absolute changes from baseline (on treatment value − pre-treatment value) were compared between the groups using the Kruskal–Wallis test. Non-parametric analysis was chosen because of the relatively small number of participants and the uneven group sizes. Only in the case of a significant finding were prednisolone 7.5 mg and prednisolone 30 mg compared against placebo by post hoc testing, using the Mann–Whitney U test. To correct for multiple testing, Bonferroni correction was applied by multiplying the obtained p value from the Mann–Whitney U test by the numbers of comparisons (i.e. two) that were carried out. All statistical analyses were run on SPSS for Windows version 15.0 (SPSS, Chicago, IL, USA). A p < 0.05 was considered statistically significant.
Results
Anthropometric characteristics
The participant characteristics at inclusion are presented in Table 1; no differences were observed among the three treatment groups at baseline. BMI, lean body mass (LBM) and total fat mass were not altered by prednisolone treatment irrespective of the dose (ESM Table 1). Liver fat content averaged 1.3 ± 0.9% before treatment in all participants and was not changed by either study medication (ESM Table 1). VAT and SAT areas averaged 74 ± 19 and 136 ± 38 cm2, respectively, at baseline in all participants, and were not altered by treatment with either prednisolone dose (ESM Table 1).
Resting energy expenditure
Resting energy expenditure (REE) in the basal state was 100 ± 11 kJ kg−1 day−1 prior to treatment and was not changed by either prednisolone dose (Table 2). Insulin infusion increased REE before treatment to 106 ± 9 kJ kg−1 day−1 (p < 0.01), an effect that was not changed by prednisolone treatment (Table 2).
Glucose metabolism
Prednisolone treatment dose dependently and significantly increased FPG, despite fasting hyperinsulinaemia in the prednisolone 30 mg group (Table 3). Fasting EGP was only increased by prednisolone 30 mg (Fig. 1a), whereas suppression of EGP by insulin was impaired by both prednisolone doses (Fig. 1b). Peripheral glucose disposal was significantly decreased by prednisolone 30 mg, but not by prednisolone 7.5 mg (Fig. 1c). In the total study population, at baseline, glucose oxidation in the basal state was 5.8 (3.1–8.2) μmol kg−1 min−1, which was significantly increased during treatment with prednisolone 30 mg, but not with prednisolone 7.5 mg (Table 2). Pre-treatment, glucose oxidation increased to 20.0 (18.3–23.7) μmol kg−1 min−1 (9 ± 5% of R d) during hyperinsulinaemia (p < 0.001 compared with the basal state). Prednisolone treatment did not affect the absolute levels of glucose oxidation during hyperinsulinaemia; however, prednisolone 30 mg increased glucose oxidation relative to R d (Table 2).

The effects of prednisolone treatment on glucose metabolism. a Prednisolone treatment increased basal EGP. This was driven by prednisolone 30 mg once daily. b EGP during insulin infusion was dose dependently increased by prednisolone treatment. c The rate of glucose disappearance was decreased by prednisolone 30 mg, but not by prednisolone 7.5 mg once daily. Data represent means ± SEM. Black bars, before treatment; white bars, day 14 of treatment. Between-group changes from baseline were tested by Kruskal–Wallis (indicated by top line). Post hoc tests were done by Mann–Whitney U with Bonferroni correction for multiple testing (indicated by line with brackets). **p < 0.01; ***p < 0.001. PLB, placebo
Lipid metabolism
Lipolysis in the fasted state tended to be decreased by prednisolone treatment (p = 0.062), which was driven by the prednisolone 30 mg arm (p = 0.09) (Fig. 2a). Insulin-mediated suppression of lipolysis was markedly and dose dependently impaired by prednisolone during hyperinsulinaemic conditions (Fig. 2b, c). Similarly to changes in lipolysis, basal NEFA levels were decreased prednisolone treatment, which seemed to be driven by prednisolone 30 mg (Table 2). During step 1 of insulin infusion (20 mU m−2 min−1), insulin-mediated suppression of plasma NEFA levels was dose dependently decreased by prednisolone treatment (ESM Table 2). During the second step of insulin infusion (60 mU m−2 min−1), NEFA levels were reduced to detection level in the placebo group and prednisolone 7.5 mg group, but remained detectable in participants in the prednisolone 30 mg arm (ESM Table 2). Fasting triacylglycerol (TG) levels were increased by prednisolone 30 mg only (Table 3). In the total study population, pre-treatment, fasting fatty acid oxidation rate was 1.5 (1.3–1.8) μmol kg−1 min−1 and decreased during insulin infusion to 0.4 (0.2–0.7) μmol kg−1 min−1 (p < 0.001). Basal fatty acid oxidation was reduced by prednisolone 30 mg treatment, but fatty acid oxidation during insulin infusion was not affected by any of the treatment arms (Table 2).

The effects of prednisolone treatment on glycerol appearance (R a). a Prednisolone treatment tended to decrease basal lipolysis, which seemed caused by the prednisolone 30 mg once daily dose. b During insulin infusion targeted at 200 pmol/l, prednisolone treatment dose dependently increased glycerol turnover. This reached significance for prednisolone 30 mg, but remained a trend for prednisolone 7.5 mg once daily. c At higher insulin (600 pmol/l) during step 2 of the clamp, a similar pattern was observed, with a dose-dependent increase of whole-body lipolysis with prednisolone treatment. Data represent means ± SEM. Black bars, before treatment; white bars, day 14 of treatment. Between-group changes from baseline were tested by Kruskal–Wallis (indicated by top line). Post hoc tests were done by Mann–Whitney U with Bonferroni correction for multiple testing (indicated by line with brackets). *p < 0.05; **p < 0.01; † p = 0.062; ‡ p = 0.09. PLB, placebo
Protein metabolism
Prednisolone treatment did not affect proteolysis in the fasted state (Fig. 3a); however, prednisolone treatment increased proteolysis under hyperinsulinaemic conditions (Fig. 3b, c).

The effect of prednisolone treatment on valine appearance (R a). a Valine turnover in the basal state was not affected by prednisolone treatment. b Overall, valine turnover was increased during step 1 of the hyperinsulinaemic–euglycaemic clamp by prednisolone treatment. This seemed driven by prednisolone 30 mg in post hoc analysis, but failed to reach statistical significance. c A similar pattern was observed during step 2 of the clamp, where overall prednisolone treatment increased valine turnover; however, in post hoc testing, no significance was reached for either dose compared with placebo. Data represent means ± SEM. Black bars, before treatment; white bars, day 14 of treatment. Between-group changes from baseline were tested by Kruskal–Wallis (indicated by top line). Post hoc tests were done by Mann–Whitney U with Bonferroni correction for multiple testing (indicated by line with brackets). *p < 0.05; † p = 0.06. PLB, placebo
Glucoregulatory hormones
Prednisolone 7.5 mg did not change fasting plasma insulin levels or glucagon levels. Prednisolone 30 mg, in contrast, induced both fasting hyperinsulinaemia and hyperglucagonaemia (Table 3). Before treatment, insulin levels in the entire study population during the clamp were 171 (153–183) pmol/l (step 1) and 533 (485–564) pmol/l (step 2). These levels remained unchanged during the on-treatment clamps, although there was a tendency towards increased insulin levels in the placebo group (ESM Table 2). Glucagon levels remained increased during step 1, but not step 2, of the hyperinsulinaemic clamp during prednisolone 30 mg treatment (ESM Table 2).
Safety and tolerability
One participant in the prednisolone 30 mg group complained of gastric discomfort which was mild of nature and did not require any intervention. Otherwise, no treatment-related side effects were reported in either group.
Discussion
Low-dose GC treatment is chronically prescribed to a great number of patients, but to date it is uncertain whether low-dose GC treatment induces adverse metabolic effects, as is evident for higher GC doses. The present study is the first to demonstrate that low-dose GC therapy, that is, just above the normal daily cortisol replacement dose for adrenal insufficiency, significantly impairs the effects of insulin on glucose and lipid metabolism, but not on whole-body protein metabolism. Using a relatively long exposure time, i.e. 2 weeks, we report that treatment with prednisolone 7.5 mg impaired the ability of insulin to suppress EGP, whole-body lipolysis and plasma NEFA levels. These effects were present after only 2 weeks of treatment and occurred in the absence of changes in body weight, LBM, liver fat content and body fat distribution. Our data, demonstrating clear low-dose prednisolone-induced hepatic and adipose tissue insulin resistance, provide supportive mechanisms for observations made in large retrospective case–control studies that reported increased risk to develop diabetes [15] and increased need to initiate blood-glucose lowering therapy [14] during or following low-dose GC therapy.
In prospective studies, the reported incidence of diabetes at lower GC doses has been more modest, although there was considerable variation depending on the patient group exposed [16]. The numbers reported in these clinical trials may underestimate the actual incidence of diabetes. Although not well specified in the different studies, most studies relied on fasting glucose measurements to monitor changes in glucose tolerance. Our data demonstrate a very mild increase in fasting glucose and unchanged fasting insulin levels, but show evident changes induced by prednisolone 7.5 mg in metabolic fluxes under insulin-stimulated conditions. This observation is in line with previous studies reporting large postprandial glucose excursions with only modest changes in fasting glucose levels [19, 26]. These data imply that, in order to properly assess the effects of GCs on glucose metabolism, measurements under stimulated conditions should be performed, such as an OGTT or a glucose day curve.
By including an additional treatment arm, we were able to study the dose-dependent effects of prednisolone treatment. Prednisolone 30 mg, an induction dosage often used in the clinic for short-term treatment, impaired the effects of insulin on EGP, glucose disposal and lipolysis to a greater extent than prednisolone 7.5 mg. In addition, prednisolone 30 mg increased proteolysis under hyperinsulinaemic conditions, increased glucagon levels and augmented fasting TG levels. Surprisingly, prednisolone 30 mg increased both basal glucose oxidation (absolute levels) and glucose oxidation during the clamp (expressed as % of R d). Increased basal glucose oxidation has, to our knowledge, not been reported previously following GC treatment. We hypothesise that prednisolone treatment induced metabolic inflexibility. This phenomenon refers to the inability to adjust fuel oxidation to substrate availability. As such, impaired fat oxidation in the fasted state was extensively shown in obese insulin-resistant and type 2 diabetic individuals [27]. In addition, following high-dose prednisolone treatment, FPG levels were, albeit it marginally, increased, which could have contributed to augmented glucose oxidation in the basal state. The finding that GC treatment increased glucose oxidation under hyperinsulinaemic conditions has been reported previously in non-diabetic individuals who were exposed to dexamethasone treatment [28]. GCs are well known to impair the insulin-stimulated glycogen synthesis pathway [29] and, therefore, increased glucose oxidation was proposed to serve as a mechanism to compensate for reduced glycogen synthesis in the presence of higher glucose levels within the skeletal muscle [28]. Our data similarly indicate preferential glucose oxidation compared with glycogen synthesis pathways, as absolute glucose oxidation rates were not altered despite lower glucose uptake. Glucose oxidation as a percentage of R d was thus increased. Finally, prednisolone 30 mg tended to decrease basal lipolysis and NEFA levels compared with placebo. In acute studies, cortisol infusion was shown to increase lipolysis in the fasted state. In these studies, hyperinsulinaemia was prevented by performing a pancreatic clamp with insulin infusion at basal levels [30, 31]. In studies addressing the effects of chronic GC treatment, typically no changes were observed in whole-body lipolysis in the fasted state [32], and fasting insulin levels were increased [33, 34]. In our study we found a tendency towards decreased lipolysis. As lipolysis is more sensitive to small changes in insulin concentration than EGP [35, 36], and plasma NEFA levels are closely correlated with changes in lipolysis, we hypothesise that decreased basal lipolysis and NEFA levels in individuals receiving prednisolone 30 mg may be because of the observed prednisolone-induced 80% increase in fasting plasma insulin levels. The negative correlation between the treatment-induced change in fasting plasma insulin and fasting NEFA levels (r = −0.564; p = 0.001) supports our hypothesis. Another hypothesis is that prednisolone 30 mg decreased the sympathetic tone to adipose tissue, resulting in lower rates of basal lipolysis [37].
The mechanisms underlying prednisolone-induced insulin resistance on the various metabolic fluxes have not been elucidated. GCs were shown to interfere with insulin signalling in vitro and in vivo in rodents in liver [38], adipose tissue [39] and skeletal muscle [40]. On the other hand, GCs may also directly activate genes involved in the regulation of various metabolic fluxes. As such, GCs were shown to induce phosphoenolpyruvate carboxykinase, the rate-controlling enzyme in hepatic glucose production [41]. In addition, hormone-sensitive lipase, a major regulator of adipose tissue lipolysis was demonstrated to be regulated by GCs [42]. Moreover, GCs directly activated muscle ring finger-1 and atrogin-1, proteins involved in skeletal muscle proteolysis [43]. Finally, GCs impaired glucose transport into skeletal muscle independent of changes in insulin signalling [6].
In the present study we exposed healthy volunteers to different doses of prednisolone, allowing us to specifically measure the metabolic effects of prednisolone. In clinical practice, prednisolone is used to treat patients with chronic inflammatory diseases. As systemic inflammation also impairs insulin sensitivity [44], the effects of GCs on various fluxes, given their combined anti-inflammatory and metabolic effects, could be more complex in this population.
One potential, yet inevitable, limitation of our experimental design when addressing the effects of real-life oral prednisolone treatment, as compared with previously reported, more artificial cortisol infusion studies, is that our clamp tests were performed under non-steady-state prednisolone levels. As prednisolone has a t 1/2 of 4–6 h, the final part of the clamp may have been performed under lower prednisolone levels. However, plasma prednisolone levels may be a poor marker of its dynamics, as many actions of prednisolone are genomic of nature, which take a number of hours to become effected. In addition, our participants were treated for 2 weeks, in contrast to the acute studies that have been mostly conducted up to now and, therefore, differences caused by the acute effects of prednisolone treatment may be very limited.
We conclude that GC treatment, even at a low so-called maintenance dose that is prescribed to large numbers of patients for prolonged periods of time, impairs both glucose metabolism and lipolysis by impairing the metabolic actions of insulin on the liver and adipose tissue. The results of this study uncover the mechanisms by which GC doses just above cortisol replacement levels, particularly when used chronically, may impair glucose tolerance in susceptible patients. Physicians treating patients with low-dose GCs should be aware of the induction of metabolic disturbances and should not solely rely on fasting measurements. In addition, our study indicates that insulin-sensitising therapies could be considered when treating patients with GC therapy. As such, the thiazolidinedione troglitazone was shown to prevent GC-induced glucose intolerance in healthy humans [45]. Troglitazone, however, is no longer available for use in humans, and therefore a search for other options to alleviate GC-induced insulin resistance seems justified. In this regard, the current development of the so-called dissociated GC receptor agonists, which aim to separate the anti-inflammatory and dysmetabolic effects of GCs, may be very interesting [46].
Abbreviations
- DEXA:
-
Dual emission X-ray absorptiometry
- EGP:
-
Endogenous glucose production
- FPG:
-
Fasting plasma glucose
- GC:
-
Glucocorticoid
- LBM:
-
Lean body mass
- R d :
-
Rate of disappearance
- REE:
-
Resting energy expenditure
- SAT:
-
Subcutaneous adipose tissue
- VAT:
-
Visceral adipose tissue
References
Bijlsma JW, van der Goes MC, Hoes JN, Jacobs JW, Buttgereit F, Kirwan J (2010) Low-dose glucocorticoid therapy in rheumatoid arthritis: an obligatory therapy. Ann NY Acad Sci 1193:123–126
Hernandez-Rodriguez J, Cid MC, Lopez-Soto A, Espigol-Frigole G, Bosch X (2009) Treatment of polymyalgia rheumatica: a systematic review. Arch Intern Med 169:1839–1850
Salvarani C, Cantini F, Hunder GG (2008) Polymyalgia rheumatica and giant-cell arteritis. Lancet 372:234–245
Parker BJ, Bruce IN (2007) High dose methylprednisolone therapy for the treatment of severe systemic lupus erythematosus. Lupus 16:387–393
Lichtenstein GR, Abreu MT, Cohen R, Tremaine W (2006) American Gastroenterological Association Institute medical position statement on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology 130:935–939
van Raalte DH, Ouwens DM, Diamant M (2009) Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur. J Clin Invest 39:81–93
Buttgereit F, Da Silva JA, Boers M et al (2002) Standardised nomenclature for glucocorticoid dosages and glucocorticoid treatment regimens: current questions and tentative answers in rheumatology. Ann Rheum Dis 61:718–722
Wajngot A, Giacca A, Grill V, Vranic M, Efendic S (1992) The diabetogenic effects of glucocorticoids are more pronounced in low- than in high-insulin responders. Proc Natl Acad Sci USA 89:6035–6039
Larsson H, Ahren B (1999) Insulin resistant subjects lack islet adaptation to short-term dexamethasone-induced reduction in insulin sensitivity. Diabetologia 42:936–943
Gravholt CH, Dall R, Christiansen JS, Moller N, Schmitz O (2002) Preferential stimulation of abdominal subcutaneous lipolysis after prednisolone exposure in humans. Obes Res 10:774–781
Taskinen MR, Kuusi T, Yki-Jarvinen H, Nikkila EA (1988) Short-term effects of prednisone on serum lipids and high density lipoprotein subfractions in normolipidemic healthy men. J Clin Endocrinol Metab 67:291–299
Short KR, Bigelow ML, Nair KS (2009) Short-term prednisone use antagonizes insulin’s anabolic effect on muscle protein and glucose metabolism in young healthy people. Am J Physiol Endocrinol Metab 297:E1260–E1268
Fernandez-Rodriguez E, Stewart PM, Cooper MS (2009) The pituitary-adrenal axis and body composition. Pituitary 12:105–115
Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J (1994) Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 154:97–101
Gulliford MC, Charlton J, Latinovic R (2006) Risk of diabetes associated with prescribed glucocorticoids in a large population. Diabetes Care 29:2728–2729
Hoes JN, Jacobs JW, Verstappen SM, Bijlsma JW, van der Heijden GJ (2009) Adverse events of low- to medium-dose oral glucocorticoids in inflammatory diseases: a meta-analysis. Ann Rheum Dis 68:1833–1838
Tushuizen ME, Bunck MC, Pouwels PJ et al (2007) Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 30:2916–2921
Tushuizen ME, Pouwels PJ, Bontemps S et al (2010) Postprandial lipid and apolipoprotein responses following three consecutive meals associate with liver fat content in type 2 diabetes and the metabolic syndrome. Atherosclerosis 211:308–314
van Raalte DH, Nofrate V, Bunck MC et al (2010) Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur J Endocrinol 162:729–735
Ackermans MT, Ruiter AF, Endert E (1998) Determination of glycerol concentrations and glycerol isotopic enrichments in human plasma by gas chromatography/mass spectrometry. Anal Biochem 258:80–86
Ackermans MT, Pereira Arias AM, Bisschop PH, Endert E, Sauerwein HP, Romijn JA (2001) The quantification of gluconeogenesis in healthy men by (2)H2O and [2-(13)C]glycerol yields different results: rates of gluconeogenesis in healthy men measured with (2)H2O are higher than those measured with [2-(13)C]glycerol. J Clin Endocrinol Metab 86:2220–2226
Geukers VG, Oudshoorn JH, Taminiau JA et al (2005) Short-term protein intake and stimulation of protein synthesis in stunted children with cystic fibrosis. Am J Clin Nutr 81:605–610
Finegood DT, Bergman RN, Vranic M (1987) Estimation of endogenous glucose production during hyperinsulinemic–euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 36:914–924
Steele R (1959) Influences of glucose loading and of injected insulin on hepatic glucose output. Ann NY Acad Sci 82:420–430
Frayn KN (1983) Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 55:628–634
Clore JN, Thurby-Hay L (2009) Glucocorticoid-induced hyperglycemia. Endocr Pract 15:469–474
Corpeleijn E, Saris WH, Blaak EE (2009) Metabolic flexibility in the development of insulin resistance and type 2 diabetes: effects of lifestyle. Obes Rev 10:178–193
Henriksen JE, Alford F, Vaag A, Handberg A, Beck-Nielsen H (1999) Intracellular skeletal muscle glucose metabolism is differentially altered by dexamethasone treatment of normoglycemic relatives of type 2 diabetic patients. Metabolism 48:1128–1135
Ruzzin J, Wagman AS, Jensen J (2005) Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 48:2119–2130
Djurhuus CB, Gravholt CH, Nielsen S et al (2002) Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans. Am J Physiol Endocrinol Metab 283:E172–E177
Djurhuus CB, Gravholt CH, Nielsen S, Pedersen SB, Moller N, Schmitz O (2004) Additive effects of cortisol and growth hormone on regional and systemic lipolysis in humans. Am J Physiol Endocrinol Metab 286:E488–E494
Macfarlane DP, Forbes S, Walker BR (2008) Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol 197:189–204
Birkenhager JC, Timmermans HA, Lamberts SW (1976) Depressed plasma FFA turnover rate in Cushing’s syndrome. J Clin Endocrinol Metab 42:28–32
Miyoshi H, Shulman GI, Peters EJ, Wolfe MH, Elahi D, Wolfe RR (1988) Hormonal control of substrate cycling in humans. J Clin Invest 81:1545–1555
Bonadonna RC, Groop LC, Zych K, Shank M, DeFronzo RA (1990) Dose-dependent effect of insulin on plasma free fatty acid turnover and oxidation in humans. Am J Physiol 259:E736–E750
Gelding SV, Coldham N, Anyaoku V, Heslop K, Halliday D, Johnston DG (1993) Differential metabolic actions of biosynthetic insulin analogues in normal man assessed by stable isotopic tracers. Diabet Med 10:470–476
Nakada MT, Stadel JM, Poksay KS, Crooke ST (1987) Glucocorticoid regulation of beta-adrenergic receptors in 3T3-L1 preadipocytes. Mol Pharmacol 31:377–384
Rojas FA, Hirata AE, Saad MJ (2003) Regulation of insulin receptor substrate-2 tyrosine phosphorylation in animal models of insulin resistance. Endocrinology 21:115–122
Lundgren M, Buren J, Ruge T, Myrnas T, Eriksson JW (2004) Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J Clin Endocrinol Metab 89:2989–2997
Saad MJ, Folli F, Kahn JA, Kahn CR (1993) Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest 92:2065–2072
Vegiopoulos A, Herzig S (2007) Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol 275:43–61
Slavin BG, Ong JM, Kern PA (1994) Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res 35:1535–1541
Schakman O, Gilson H, Thissen JP (2008) Mechanisms of glucocorticoid-induced myopathy. J Endocrinol 197:1–10
Dessein PH, Joffe BI (2006) Insulin resistance and impaired beta cell function in rheumatoid arthritis. Arthritis Rheum 54:2765–2775
Willi SM, Kennedy A, Wallace P, Ganaway E, Rogers NL, Garvey WT (2002) Troglitazone antagonizes metabolic effects of glucocorticoids in humans: effects on glucose tolerance, insulin sensitivity, suppression of free fatty acids, and leptin. Diabetes 51:2895–2902
Rosen J, Miner JN (2005) The search for safer glucocorticoid receptor ligands. Endocr Rev 26:452–464
Acknowledgements
This paper was written within the framework of project T1-106 of the Dutch Top Institute Pharma.
Duality of interest
D. H. van Raalte, M. Brands, N. J. van der Zijl, M. H. Muskiet, P. J. W. Pouwels, M. T. Ackermans and H. P. Sauerwein declare that there is no duality of interest associated with this manuscript. Through M. Diamant, the VU University Medical Centre received research grants from Amylin Pharmaceuticals, Eli Lilly, GlaxoSmithKline, Merck, Novartis, Novo Nordisk, sanofi-aventis and Takeda, consultancy fees from Eli Lilly, Merck, Novo Nordisk, Astra Zeneca/BMS and sanofi-aventis and speaker fees from Eli Lilly, Merck and Novo Nordisk. M. J. Serlie received speaker fees from MSD, Novo Nordisk, Sorgente and Fresenius.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. H. van Raalte and M. Brands contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Variables of body composition and body fat distribution before and during 2 week treatment with placebo, prednisolone 7.5 mg or prednisolone 30 mg (PDF 102 kb)
ESM Table 2
Metabolic variables during the two-step hyperinsulinaemic–euglycaemic clamp before and during 2 week treatment with placebo, prednisolone 7.5 mg or prednisolone 30 mg (PDF 68 kb)
ESM Fig. 1
Study design (a) and overview of the two-step hyperinsulinaemic–euglycaemic clamp protocol (b). M, start study medication; PLB, placebo; PRED, prednisolone; QD, once daily; S, screening; T1, MRI/DEXA scan; T2, clamp; V, follow-up visits (PDF 106 kb)
ESM Fig. 2
Patient flow diagram: 100% of the randomised participants completed the study (PDF 15 kb)
ESM Fig. 3
Kinetic equations for the steady state (a) and for the non-steady state (b). Ra = rate of appearance; TTRtracer = tracer/tracee ratio of the tracer infused (=1); Itracer = tracer infusion rate; TTRp = tracer/tracee ratio in plasma; TTRexo = tracer/tracee ratio of exogenous glucose infusion; Iexo = infusion rate of exogenous glucose; Rd = rate of disappearance; Ra(t) = average Ra between t = 1 and t = 2; TTRp(t) = average TTRp between t = 1 and t = 2; pV = distribution volume; Cunl(t) = average plasma concentration tracee between t = 1 and t = 2; dTTRp(t)/dt = change in plasma TTRp between t = 1 and t = 2; Rd(t) = average Rd between t = 1 and t = 2; dCunl(t)/dt = change in tracee concentration between t = 1 and t = 2; EP(t) = average endogenous production between t = 1 and t = 2; Iunl = tracee infusion rate (PDF 18 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Raalte, D.H., Brands, M., van der Zijl, N.J. et al. Low-dose glucocorticoid treatment affects multiple aspects of intermediary metabolism in healthy humans: a randomised controlled trial. Diabetologia 54, 2103–2112 (2011). https://doi.org/10.1007/s00125-011-2174-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2174-9