
Abstract
Aims/hypothesis
Diabetic retinopathy is a progressive neurodegenerative disease, but the underlying mechanism is still obscure. Here, we focused on oxidative stress in the retina, and analysed its influence on retinal neurodegeneration, using an antioxidant, lutein.
Methods
C57BL/6 mice with streptozotocin-induced diabetes were constantly fed either a lutein-supplemented diet or a control diet from the onset of diabetes, and their metabolic data were recorded. In 1-month-diabetic mice, reactive oxygen species (ROS) in the retina were measured using dihydroethidium and visual function was evaluated by electroretinograms. Levels of activated extracellular signal-regulated kinase (ERK), synaptophysin and brain-derived neurotrophic factor (BDNF) were also measured by immunoblotting in the retina of 1-month-diabetic mice. In the retinal sections of 4-month-diabetic mice, histological changes, cleaved caspase-3 and TUNEL staining were analysed.
Results
Lutein did not affect the metabolic status of the diabetic mice, but it prevented ROS generation in the retina and the visual impairment induced by diabetes. ERK activation, the subsequent synaptophysin reduction, and the BDNF depletion in the diabetic retina were all prevented by lutein. Later, in 4-month-diabetic mice, a decrease in the thickness of the inner plexiform and nuclear layers, and ganglion cell number, together with increase in cleaved caspase-3- and TUNEL-positive cells, were avoided in the retina of lutein-fed mice.
Conclusions/interpretation
The results indicated that local oxidative stress that has a neurodegenerative influence in the diabetic retina is prevented by constant intake of a lutein-supplemented diet. The antioxidant, lutein may be a potential therapeutic approach to protect visual function in diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic retinopathy is considered a neurodegenerative disease in which visual dysfunction is initiated in early diabetes [1]. As recent studies reveal, many of the diabetic complications are associated with oxidative stress [2–4] as well as inflammation [4, 5]. However, the underlying mechanism in diabetic retinal degeneration remains to be elucidated. Moreover, a definitive therapy for its prevention is not available at this time.
Several intracellular signalling pathways downstream of inflammation are associated with oxidative stress [4–7]. One such pathway, angiotensin II type 1 receptor (AT1R) signalling, is pathogenic in the development of diabetic complications [3, 8]. In fact, the streptozotocin (STZ)-induced mouse model of diabetes has a decrease in responses of the oscillatory potentials (OPs) in electroretinograms (ERGs) through retinal AT1R signalling, as we have previously reported [8]. Another report showed that an angiotensin II converting enzyme inhibitor prevented the OP changes, supporting the idea that angiotensin II signal is important in diabetic retinopathy [9]. OPs reflect the functioning of the inner retina [10], and are already abnormal in early diabetes, in both human patients and experimental animals [8, 11–13]. This is at least in part because of the decrease in the level of synaptophysin caused by AT1R signalling in the retina [8]. Synaptophysin is a synaptic membrane protein that is abundant in the inner plexiform layer (IPL), where AT1R is also produced [14], and plays a critical role in OPs. In neurons, AT1R signalling activates extracellular signal-regulated kinase (ERK) to induce excessive degradation of synaptophysin, through the ubiquitin–proteasome system [8]. Therefore, AT1R signalling is one of the key modulators of diabetic retinopathy. However, whether or not these diabetic neurodegenerative changes can be prevented by suppressing reactive oxygen species (ROS) in the retina remains to be elucidated. On the other hand, retinal ganglion cells [15–18] and a subset of amacrine cells in the inner nuclear layer (INL) [19] are lost to apoptosis in diabetes, as shown by caspase-3 activation and TUNEL staining, and can be attenuated by administration of the soluble factor, brain-derived neurotrophic factor (BDNF) [19]. However, the relationship between BDNF and oxidative stress in diabetes is still obscure. Thus, evaluating the contribution of ROS in diabetic retinopathy may help establish a new therapy.
Here, we focus on lutein, a xanthophyll carotenoid and an antioxidant, which is spread throughout the retina. Lutein is not synthesised in vivo and needs to be obtained through the diet, and is then delivered to the retina. It corresponds to the macular pigment in the retina with its optical isomer zeaxanthin. Long-term oral intake of lutein is reported to elevate serum lutein levels [20, 21], which correlate with the macular pigment density [20, 22], indicating that lutein constantly taken from the diet accumulates in the retina. Our previous data confirmed that lutein administration increases lutein levels in the choroid and retinal pigment epithelial cells in the eye, and suppresses inflammatory signalling in a model of laser-induced choroidal neovascularisation [7]. We previously reported in an endotoxin-induced uveitis model that lutein administration suppresses ROS and inflammatory signalling in the retina, and prevents the visual dysfunction [6] caused by rhodopsin degradation [23]. Therefore, growing evidence shows the role of lutein as a suppressor of ROS induced by inflammation [6, 7, 24, 25]. However, the effect of this antioxidant, lutein, in diabetic neurodegeneration is not fully understood.
In this study, we analysed whether constant lutein intake suppresses the neurodegenerative changes in the retina of STZ-induced diabetic mice. We first checked body weight or blood glucose level changes and the level of ROS generated in the retina of diabetic mice, with or without constant intake of lutein-supplemented diet. Then, visual function was measured, and biochemical and histological changes in the retina were analysed. We showed the neurodegenerative influence of oxidative stress in the diabetic retina that provides us with a possible mechanism involved in the pathogenesis of diabetic retinopathy.
Methods
Animals
All animal experiments described in this study were conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Diabetes was induced in 8-week-old C57BL/6 mice (Clea Japan, Tokyo, Japan) by daily i.p. injections of STZ (Sigma-Aldrich, St Louis, MO, USA) at a dose of 60 mg/kg body weight for 3 days. The blood glucose concentration was measured from a tail vein using a Medisafe Mini GR-102 (Terumo, Tokyo, Japan). The development of diabetes was defined as blood glucose >13.9 mmol/l (250 mg/dl) 7 days after the first injection of STZ. From then on, diabetic mice were constantly fed a 0.1% (wt/wt) lutein diet prepared by adding lutein (provided by Wakasa Seikatsu Co., Kyoto, Japan) to the mouse chow (purchased from Clea Japan), or the same chow with no additional lutein, until the time of analysis. These mice, and age-matched non-diabetic control mice, were then analysed 1 or 4 months after diabetes induction. Non-diabetic mice fed a lutein-supplemented diet were analysed, but there were no differences compared with the non-diabetic mice fed a control diet. Therefore, we show as controls the data from non-diabetic mice fed a control diet.
Measurement of ROS
Eyes were enucleated and immediately frozen in OCT compound (Sakura Finetek, Torrance, CA, USA). Unfixed cryosections (10 μm) were incubated with dihydroethidium (DHE; Invitrogen-Molecular Probes, Eugene, OR, USA) (5 μmol/l) for 20 min at 37°C, as previously described [6]. DHE specifically reacts with intracellular superoxide anion and is converted to the red fluorescent compound ethidium in nuclei. The sections were examined using a microscope equipped with a digital camera (Carl Zeiss, Jena, Germany) in the same exposure conditions, and the intensity of the staining in the INL was measured in the posterior retina at four points, two on either side of the optic nerve that were 200 and 500 μm apart, using the Image J program (version 1.37; National Institutes of Health, Bethesda, MD, USA). All the procedures in each sample, from preparing animals to taking photographs, were performed at the same time in parallel.
ERG analyses
Animals were dark-adapted for 12 h and prepared for the ERG procedure under dim red illumination. The mice were anaesthetised with pentobarbital sodium at a dose of 70 mg/kg body weight and placed on a heating pad that maintained their body temperature at 35–36°C throughout the experiment. The pupils were dilated with a solution of 0.5% (wt/vol.) tropicamide and 0.5% (wt/vol.) phenylephrine (Mydrin-P; Santen, Osaka, Japan). The ground electrode was a subcutaneous needle in the tail, and the reference electrode was placed subcutaneously between the eyes. The active contact lens electrodes (Mayo, Inazawa, Japan) were placed on the cornea. The recordings were performed with a PowerLab system 2/25 (AD Instruments, Bella Vista, NSW, Australia). The responses were differentially amplified with a gain of 1,000 using alternating current-coupled bioamplifier ML132 (AD Instruments) and filtered through a bandpass filter ranging from 0.3 to 500 Hz to yield a- and b-waves. The OPs were simultaneously recorded using a high-pass filter set to 100 Hz, so that an overall bandpass ranging from 100 to 500 Hz was achieved. Light pulses of 800 cd-s/m2 were delivered via a Ganzfeld System SG-2002 (LKC Technologies; Gaithersburg, MD, USA). With this system, OP1 is not separated from the a-wave response; thus we analysed amplitudes and implicit times of OP2, 3 and 4. The amplitude of total OPs was calculated by adding those of OP2, 3, and 4.
Immunoblot analyses
Isolated retinas were placed into lysis buffer (10 mmol/l TRIS–HCl [pH 7.6], 100 mmol/l NaCl, 1 mmol/l EDTA, 1% [wt/vol.] Triton X-100 and protease inhibitors). Each sample was separated by SDS-PAGE and electroblotted onto a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). After being blocked in 4% (wt/vol.) skimmed milk, the membrane was incubated at 4°C overnight with rabbit anti-phosphorylated ERK (1:1,000; Cell Signaling Technology, Beverly, MA, USA), mouse anti-synaptophysin (1:500; Sigma-Aldrich), rabbit anti-BDNF (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse anti-α-tubulin (1:2,000; Sigma-Aldrich) antibodies. The membrane was then incubated with a horseradish peroxidase-conjugated antibody against mouse immunoglobulins or with a biotinylated secondary antibody against rabbit immunoglobulins followed by avidin–biotin horseradish peroxidase complex (Vectastain Elite ABC Kit; Vector, Burlingame, CA, USA). The signals were visualised by chemiluminescence (ECL Blotting Analysis System; Amersham, Arlington Heights, IL, USA), measured by Image J software, and normalised to α-tubulin.
Structural analyses and immunohistochemistry
Retinas were fixed with 4% (wt/vol.) paraformaldehyde and prepared for paraffin sectioning. All sections (6 μm) were cut tangentially through the pupil and optic nerve, passing through the temporal ora serrata, the optic nerve and the nasal ora serrata. These paraffin sections cut at the centre of the eye were stained with haematoxylin and eosin. The thicknesses of the IPL, INL and outer nuclear layer (ONL) were measured in the posterior retina at four points, two on either side of the optic nerve that were 200 and 500 μm apart, using the Image J program. Thicknesses of IPL and INL were normalised to that of ONL at each point, to further standardise for bias introduced by skewed sectioning angle and to more precisely compare the length between the different sections. Previous reports show that ONL thickness [15] is not changed in the diabetic retina, although inner retinal thickness and, consequently, total retinal thickness are reduced by diabetes [4, 15]. The neuronal cell number in the ganglion cell layer (GCL) was quantified by counting the nuclei. For each sample, three sections were analysed and averaged.
To detect cleaved caspase-3, unstained sections were subjected to heat-induced antigen retrieval in a microwave oven for 5 min in 10 mmol/l citrate buffer, and the endogenous peroxidase was abolished by incubating the sections in 3% (wt/vol.) hydrogen peroxide in methanol for 10 min. Sections were incubated in blocking solution (10% normal bovine serum in PBS) and then incubated with a rabbit anti-cleaved caspase-3 antibody (1:100; Cell Signaling Technology), followed by a biotinylated secondary antibody and avidin–biotin horseradish peroxidase complexes (Vectastain Elite ABC Kit). The reaction product was developed by incubation for 10 min in Tyramide Signal Amplification Solution (Perkin Elmer Life Sciences, Boston, MA, USA), and the nuclei were counterstained with the nuclear dye bisbenzimide, diluted 1:1,000 from a 10 mg/ml stock solution (Hoechst 33258; Sigma-Aldrich) to check the retinal cell layers. Apoptotic cells were also detected in the sections using a TUNEL kit (Chemicon-Millipore, Billeria, MA, USA). Cleaved caspase-3- or TUNEL-positive cells in the GCL were counted as described above.
All the sections were examined using a microscope equipped with a digital camera (Carl Zeiss).
Statistical analysis
Data are expressed as means±SD. Statistical significance was tested with a one-way ANOVA with Tukey’s post hoc test, and differences were considered statistically significant at p < 0.05.
Results
Systemic effects of constant lutein intake in diabetic mice
Mice with STZ-induced diabetes, fed either control or lutein-supplemented diet, showed a significant decrease in body weight and a significant increase in blood glucose, compared with age-matched non-diabetic controls (Table 1). Treatment with a constant lutein diet for 1 or 4 months did not significantly change these metabolic variables in the diabetic mice.
Diabetes-induced oxidative stress in the retina was prevented by lutein
To show the levels of ROS generated in the retina, we performed a DHE reaction that detects one of the ROS, superoxide anion, in the tissue. DHE staining was observed in all the layers of the retinas of 1-month-diabetic mice fed a control diet (Fig. 1a, b); however, it was suppressed throughout the retina in diabetic mice that had been fed only with a lutein diet from the onset.

Diabetes-induced oxidative stress in the retina was reduced by lutein. a DHE staining in the retinal section. Original magnification ×200; scale bar, 30 μm. b Fluorescence intensity in the INL relative to that of non-diabetic mice, measured by the Image J program. Note that ROS increased throughout the retina of mice diabetic for 1 month; however, this change was prevented by constant intake of lutein from the onset of diabetes. Non-diabetic mice, n=4; diabetic mice fed control diet, n=4; diabetic mice fed lutein diet, n=5. Values are means±SD. *p < 0.05, **p < 0.01
Therefore, diabetes-induced oxidative stress in the retina was efficiently reduced by constant lutein intake.
Diabetes-induced visual impairment was suppressed by lutein
To determine the effect of constant lutein intake on visual function in diabetes, we measured ERGs in 1-month-diabetic mice (Fig. 2a, b). The amplitude of the OPs (OP3 and total OPs), which reflects the functional status of the inner retina, was decreased in the diabetic mice. However, reduction in the OP amplitudes was successfully prevented by constant lutein intake from the onset of diabetes. No noteworthy changes in the implicit time of OPs and in the a- or b-waves were detected in either the non-diabetic mice and diabetic mice, and this was also the case after lutein treatment (data not shown).

Diabetes-induced visual dysfunction was suppressed by lutein. Representative wave responses from an individual mouse in each group to one flash (a). Constant lutein intake from the onset of diabetes significantly inhibited the reduction of OP3 and total OPs (ΣOPs; summation of OP2, 3, and 4) amplitude in 1-month-diabetic mice (b). Black columns, non-diabetic mice, n=5; white columns, diabetic mice fed control diet, n=6; grey columns, diabetic mice fed lutein diet, n=6. Values are means±SD. *p < 0.05
Therefore, constant intake of the antioxidant lutein protected the inner retina from the functional deficits usually induced by diabetes.
Diabetes-induced ERK activation and reduction in synaptophysin were suppressed by lutein intake
To elucidate lutein’s effect on ERK activation, we performed immunoblot analyses of phosphorylated (i.e. activated) ERK. The activation of ERK in the retina of 1-month-diabetic mice was significantly inhibited by lutein intake (Fig. 3a, d).

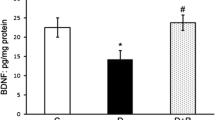
Diabetes-induced biochemical changes in the retina are prevented by lutein as shown by immunoblot analysis. a, d Diabetes-induced ERK activation detected by ERK phosphorylation (pERK) in the retina of 1-month-diabetic mice was prevented by constant intake of lutein. Non-diabetic mice, n=8; diabetic mice fed control diet, n=8; diabetic mice fed lutein diet, n=7. b, e A decrease in synaptophysin in the retina of 1-month-diabetic mice was prevented by constant intake of lutein. Non-diabetic mice, n=6; diabetic mice fed control diet, n=6; diabetic mice fed lutein diet, n=6. c, f The level of BDNF was also reduced in the retina of 1-month-diabetic mice, and was significantly rescued by constant intake of lutein. Non-diabetic, mice n=6; diabetic mice fed control diet, n=6; diabetic mice fed lutein diet, n=6. Bar graph values are relative to those of the non-diabetic controls. Values are means±SD. *p < 0.05, **p < 0.01
Next, we analysed the synaptic vesicle protein synaptophysin, which plays a critical role in the cellular source of OPs. Immunoblot analysis showed that the synaptophysin protein level was reduced 1 month after diabetes onset (Fig. 3b, e). However, constant lutein intake from the onset of diabetes preserved the synaptophysin level. In the retina of non-diabetic mice fed the lutein-supplemented diet, levels of activated ERK and synaptophysin were not changed (data not shown).
Thus, in the diabetic retina, ERK activation and subsequent synaptophysin reduction were inhibited by constant lutein intake.
Diabetes-induced reduction in BDNF was suppressed by lutein intake
Since diabetes decreases one of the major trophic factors for retinal neurons, BDNF, we also measured BDNF by immunoblot analysis in the 1-month-diabetic retina. Importantly, the decrease in BDNF caused by diabetes was attenuated by constant intake of the lutein-supplemented diet (Fig. 3c, f).
Diabetes-induced histological changes in the inner retina were suppressed by lutein
We next analysed the retinas of 4-month-diabetic mice which had been fed either the lutein or the control diet from the onset. The thickness of the retinal layers were measured in paraffin sections and each thickness was normalised to ONL thickness, as described in the “Methods”. The normalised thickness of the IPL, where neurites and the synapses of inner retinal cells are present, was significantly reduced in the retina of 4-month-diabetic mice; however, the change was clearly avoided in the retina of the lutein-fed diabetic mice (Fig. 4a). Moreover, the normalised INL thickness was reduced (Fig. 4a), suggesting that INL cells were decreased, and GCL cell numbers also declined (Fig. 4b) in the retinas of 4-month-diabetic mice that had been fed the control diet. However, the cell loss was significantly suppressed by long-term lutein intake (Fig. 4a, b).

Diabetes-induced histological changes were suppressed by lutein. Thickness of each retinal layer was measured in paraffin sections after haematoxylin and eosin staining. Original magnification ×400. a Relative thicknesses of IPL and INL, normalised to ONL measured at the same point, respectively, were reduced in the retina of 4-month-diabetic mice, but remained normal in the diabetic mice fed the lutein diet all the time from diabetes onset. b The neuronal cell number in the GCL in one cross-section of the mice diabetic for 4 months was decreased; however, this change was significantly suppressed by lutein. Black columns, non-diabetic mice, n=6; white columns, diabetic mice fed control diet, n=5; grey columns, diabetic mice fed lutein diet, n=5. Values are means±SD. *p < 0.05, **p < 0.01
Therefore, these data showed that the diabetes-induced reduction in inner retinal cells and their neurites was prevented by constant intake of lutein.
Diabetes-induced apoptosis was inhibited by lutein
We further analysed the levels of apoptosis markers in the retinas of 4-month-diabetic mice. Cleaved caspase-3, the activated form, was clearly upregulated in GCL cells and weakly in INL cells in the retina of diabetic mice (Fig. 5a, c, arrowheads in a). Moreover, TUNEL-positive cells were also clearly observed in the GCL (Fig. 5b, d, arrowheads in b). However, the number of caspase-3-activated or TUNEL-positive cells in the GCL were significantly reduced in the lutein-fed diabetic mice, indicating that the apoptosis induced in the diabetic retina was suppressed by constant intake of lutein.

Diabetes-induced apoptosis was inhibited by lutein. a Caspase-3 activation detected by immunohistochemistry of cleaved caspase-3. Caspase-3 activation was obvious in GCL and weak in INL cells of 4-month-diabetic retinas (arrowheads, cleaved caspase-3-positive cells); however, the activation was suppressed by constant intake of lutein. b TUNEL staining in the GCL of 4-month-diabetic retinas (arrowheads) was also suppressed by lutein treatment. Cleaved caspase-3-positive cells (c) and the TUNEL-positive cells (d) in GCL were counted. a, c Non-diabetic mice, n=4; diabetic mice fed control diet, n=4; diabetic mice fed lutein diet, n=4. b, d Non-diabetic mice, n=5; diabetic mice fed control diet, n=6; diabetic mice fed lutein diet, n=6. Value are means±SD. *p < 0.05, **p < 0.01. a Original magnification ×200, scale bar, 30 μm. b Original magnification ×400; scale bar, 40 μm
Discussion
We have demonstrated that diabetes-induced oxidative stress in the retina is reduced by a constant intake of lutein (Fig. 1) without changing body weight or blood glucose levels (Table 1). Importantly, impairment of visual function measured by OPs of the ERGs (Fig. 2), as well as ERK activation, the subsequent synaptophysin reduction and BDNF depletion after 1 month of diabetes, were all suppressed by lutein (Fig. 3). Later, at 4 months of diabetes, histological changes (Fig. 4) caused by apoptosis (Fig. 5) in the retina of diabetic mice were inhibited by feeding the lutein-supplemented diet all the time after onset of diabetes.
Superoxide anion can be generated in the mitochondria under high glucose [26]. However, lutein prevented oxidative stress in the retina of diabetic mice without body weight or blood glucose level changes (Table 1), suggesting that lutein may have scavenged ROS, at least in the retinal cells. This is consistent with the fact that lutein is delivered into the IPL, which consists of the neurites of inner retinal cells, as well as photoreceptor cells [27, 28].
In accord with clinical and experimental data [8, 11, 13, 29], OPs in ERGs were changed in early diabetes. Pharmacological studies using tetrodotoxin and glycine suggested the cellular origin of OPs as neurons with synapse formation in the inner retina [30–32]. Lack of synaptophysin induces a decrease in synaptic vesicles, which disturbs neurotransmitter release and synaptic network activity [33]. Thus, the present data showing that OPs were preserved when synaptophysin levels were rescued by ROS reduction are realistic.
In addition, it should be also noted that synaptic activity promotes cell survival [34, 35] and vision protection in the following phase (discussed below), through increasing the levels of intracellular calcium ion in neurons, which are triggered by synaptic activity, i.e. the neuronal electric stimuli [34, 35].
Our results showed that ERK activation was suppressed and the synaptophysin level was preserved when the ROS level was decreased by lutein (Figs 1 and 3), indicating that ROS activated ERK to reduce synaptophysin levels in diabetes, as AT1R signalling did in our previous study [8]. In cardiac fibroblasts, angiotensin II increases the intracellular ROS and ERK activation through AT1R, to induce cardiac hypertrophy. The increase is cancelled by the NADH/NADPH oxidase inhibitor that reduces intracellular ROS, indicating that angiotensin II stimulates ROS production via AT1R and NADH/NADPH oxidase, to activate ERK [36]. Thus, ROS generation in the diabetic retina may have promoted ERK activation downstream of angiotensin II, and lutein inhibited this AT1R-mediated diabetic change in the retina.
We also found that reduction of BDNF was attenuated by lutein, indicating that this change was partly caused by excessive oxidative stress. Since BDNF is regulated by neuronal synaptic activity [37, 38], the positive effect of lutein on BDNF may have been through preservation of the synaptophysin level and the subsequent neuronal synaptic activity in the retina of diabetic mice.
As regards the roles of BDNF, it regulates neurotransmitter release and neuronal activity [38, 39]. Thus, BDNF reduction may also have been involved in visual impairment shown by OPs in ERGs. Another role of BDNF is to promote survival of inner retinal cells [19, 40]. Thus, this oxidative stress-mediated BDNF reduction may, to some extent, have contributed to the obvious histological changes subsequently appearing in the inner retina.
Histological changes in the inner retina were clearly observed in 4-month-diabetic mice (Figs 4 and 5). Consistent with previous reports showing caspase-3 activation and TUNEL staining [16, 18, 41, 42], cell loss in the STZ-induced diabetic retina of this study was also through apoptosis pathway. This long-term effect may be caused by multiple signals activated by diabetes. However, lutein’s ability to maintain synaptophysin levels and neuronal network activity (discussed above), together with the resulting maintenance of BDNF production, may be involved in promoting the survival of neurons in the inner retina, interrupting the positive feedback loop for progressive neurodegeneration. Interestingly, in another progressive neurodegenerative disease, Alzheimer’s disease, reductions in synaptophysin [43] and BDNF [44] are also observed, and oxidative stress is involved in the pathogenesis. However, whether or not lutein will have any effect on the progression of Alzheimer’s disease is still unknown.
Taken as a whole, the current evidence clearly indicates that the visual impairment occurring in early diabetes is regulated by oxidative stress, which was prevented by constant lutein intake.
The relationship between a lutein diet and eye disease has been mainly analysed in human age-related macular degeneration (AMD) and cataract [45]. AMD may partly be exacerbated by blue light (the blue-light hazard), and lutein may act as a blue-light filter to prevent subsequent photochemical injury to the retina that is mediated by oxidative stress. Several previous human studies have suggested, and a large-scale clinical study (the Age-Related Eye Disease Study 2) is now ongoing, to show the effect of lutein in reducing risk of progression of AMD, which is, at least in part, accelerated by light exposure [46]. But, the present animal study showed that lutein does have the effect, in vivo, of suppressing oxidative stress independently of blue-light exposure. This antioxidative effect is also reported in another field: increase in dietary intake of lutein is protective against the development of early atherosclerosis from the results of epidemiological human data, and in vitro and in vivo mouse model experiments [21]. Future clinical studies aimed at analysing the effect of a daily lutein administration in human diabetic retinopathy are anticipated. Other epidemiological data, showing that a higher plasma concentration of lutein/zeaxanthin and lycopene is associated with significantly lower odds of diabetic retinopathy [47], may also support this idea.
In summary, we have shown that lutein prevents diabetes-induced visual dysfunction caused by damage to the inner neural retina that ultimately leads to retinal cell death. Lutein inhibited oxidative stress, thereby preserving the neuroprotective pathways in the early diabetic retina.
Abbreviations
- AT1R:
-
Angiotensin II type 1 receptor
- BDNF:
-
Brain-derived neurotrophic factor
- DHE:
-
Dihydroethidium
- ERG:
-
Electroretinogram
- ERK:
-
Extracellular signal-regulated kinase
- GCL:
-
Ganglion cell layer
- INL:
-
Inner nuclear layer
- IPL:
-
Inner plexiform layer
- ONL:
-
Outer nuclear layer
- OP:
-
Oscillatory potential
- ROS:
-
Reactive oxygen species
- STZ:
-
Streptozotocin
References
Barber AJ (2003) A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry 27:283–290
Dandona P, Thusu K, Cook S et al (1996) Oxidative damage to DNA in diabetes mellitus. Lancet 347:444–445
Lee HB, Yu MR, Yang Y, Jiang Z, Ha H (2003) Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 14:S241–S245
Zheng L, Du Y, Miller C et al (2007) Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 50:1987–1996
Rivero A, Mora C, Muros M, Garcia J, Herrera H, Navarro-Gonzalez JF (2009) Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci (Lond) 116:479–492
Sasaki M, Ozawa Y, Kurihara T et al (2009) Neuroprotective effect of an antioxidant, lutein, during retinal inflammation. Invest Ophthalmol Vis Sci 50:1433–1439
Izumi-Nagai K, Nagai N, Ohgami K et al (2007) Macular pigment lutein is antiinflammatory in preventing choroidal neovascularization. Arterioscler Thromb Vasc Biol 27:2555–2562
Kurihara T, Ozawa Y, Nagai N et al (2008) Angiotensin II type 1 receptor signaling contributes to synaptophysin degradation and neuronal dysfunction in the diabetic retina. Diabetes 57:2191–2198
Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ (2003) ACE inhibition salvages the visual loss caused by diabetes. Diabetologia 46:401–408
Dong CJ, Agey P, Hare WA (2004) Origins of the electroretinogram oscillatory potentials in the rabbit retina. Vis Neurosci 21:533–543
Kizawa J, Machida S, Kobayashi T, Gotoh Y, Kurosaka D (2006) Changes of oscillatory potentials and photopic negative response in patients with early diabetic retinopathy. Jpn J Ophthalmol 50:367–373
Hancock HA, Kraft TW (2004) Oscillatory potential analysis and ERGs of normal and diabetic rats. Invest Ophthalmol Vis Sci 45:1002–1008
Shirao Y, Kawasaki K (1998) Electrical responses from diabetic retina. Prog Retin Eye Res 17:59–76
Kurihara T, Ozawa Y, Shinoda K et al (2006) Neuroprotective effects of angiotensin II type 1 receptor (AT1R) blocker, telmisartan, via modulating AT1R and AT2R signaling in retinal inflammation. Invest Ophthalmol Vis Sci 47:5545–5552
Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW (1998) Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791
Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB (2004) Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci 45:3330–3336
Kern TS, Barber AJ (2008) Retinal ganglion cells in diabetes. J Physiol 586:4401–4408
Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB (2008) Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes 57:889–898
Seki M, Tanaka T, Nawa H et al (2004) Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes 53:2412–2419
Curran-Celentano J, Hammond BR Jr, Ciulla TA, Cooper DA, Pratt LM, Danis RB (2001) Relation between dietary intake, serum concentrations, and retinal concentrations of lutein and zeaxanthin in adults in a Midwest population. Am J Clin Nutr 74:796–802
Dwyer JH, Navab M, Dwyer KM et al (2001) Oxygenated carotenoid lutein and progression of early atherosclerosis: the Los Angeles Atherosclerosis Study. Circulation 103:2922–2927
Broekmans WM, Berendschot TT, Klopping-Ketelaars IA et al (2002) Macular pigment density in relation to serum and adipose tissue concentrations of lutein and serum concentrations of zeaxanthin. Am J Clin Nutr 76:595–603
Ozawa Y, Nakao K, Kurihara T et al (2008) Roles of STAT3/SOCS3 pathway in regulating the visual function and ubiquitin–proteasome-dependent degradation of rhodopsin during retinal inflammation. J Biol Chem 283:24561–24570
Sundelin SP, Nilsson SE (2001) Lipofuscin-formation in retinal pigment epithelial cells is reduced by antioxidants. Free Radic Biol Med 31:217–225
Jin XH, Ohgami K, Shiratori K et al (2006) Inhibitory effects of lutein on endotoxin-induced uveitis in Lewis rats. Invest Ophthalmol Vis Sci 47:2562–2568
Nishikawa T, Edelstein D, Du XL et al (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Snodderly DM, Auran JD, Delori FC (1984) The macular pigment. II. Spatial distribution in primate retinas. Invest Ophthalmol Vis Sci 25:674–685
Snodderly DM, Brown PK, Delori FC, Auran JD (1984) The macular pigment. I. Absorbance spectra, localization, and discrimination from other yellow pigments in primate retinas. Invest Ophthalmol Vis Sci 25:660–673
Berka JL, Stubbs AJ, Wang DZ et al (1995) Renin-containing Muller cells of the retina display endocrine features. Invest Ophthalmol Vis Sci 36:1450–1458
Wachtmeister L (1998) Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res 17:485–521
Heynen H, Wachtmeister L, van Norren D (1985) Origin of the oscillatory potentials in the primate retina. Vision Res 25:1365–1373
Yonemura D, Kawasaki K (1979) New approaches to ophthalmic electrodiagnosis by retinal oscillatory potential, drug-induced responses from retinal pigment epithelium and cone potential. Doc Ophthalmol 48:163–222
Spiwoks-Becker I, Vollrath L, Seeliger MW, Jaissle G, Eshkind LG, Leube RE (2001) Synaptic vesicle alterations in rod photoreceptors of synaptophysin-deficient mice. Neuroscience 107:127–142
Dudek H, Datta SR, Franke TF et al (1997) Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science 275:661–665
Ikegami K, Koike T (2000) Membrane depolarization-mediated survival of sympathetic neurons occurs through both phosphatidylinositol 3-kinase-and CaM kinase II-dependent pathways. Brain Res 866:218–226
Sano M, Fukuda K, Sato T et al (2001) ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 89:661–669
Kohara K, Kitamura A, Morishima M, Tsumoto T (2001) Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291:2419–2423
Binder DK, Scharfman HE (2004) Brain-derived neurotrophic factor. Growth Factors 22:123–131
Lohof AM, Ip NY, Poo MM (1993) Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 363:350–353
Johnson JE, Barde YA, Schwab M, Thoenen H (1986) Brain-derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J Neurosci 6:3031–3038
Barber AJ, Antonetti DA, Kern TS et al (2005) The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci 46:2210–2218
Gastinger MJ, Singh RS, Barber AJ (2006) Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci 47:3143–3150
Zhan SS, Beyreuther K, Schmitt HP (1993) Quantitative assessment of the synaptophysin immuno-reactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia 4:66–74
Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW (1991) BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7:695–702
Alves-Rodrigues A, Shao A (2004) The science behind lutein. Toxicol Lett 150:57–83
Ozawa Y, Ishida S, Tsubota K (2008) Age-related macular degeneration (AMD); from pathogenesis and approved therapies to proposed treatments for prevention. Anti-Aging Medicine 5:81–82
Brazionis L, Rowley K, Itsiopoulos C, O’Dea K (2009) Plasma carotenoids and diabetic retinopathy. Br J Nutr 101:270–277
Acknowledgements
We thank Y. Oike (Kumamoto University, Japan) for advice on preparing the manuscript, and I. Kawamori and H. Koizumi for technical assistance. This study was supported by a grant from Wakasa Seikatsu Co., and partly by a grant-in-aid from the Ministry of Education, Science and Culture of Japan to Y. Ozawa.
Duality of interest
M. Sasaki and Y. Ozawa received financial support from, and S. Kobayashi is an employee of, Wakasa Seikatsu Co., who provided the lutein. The remaining authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sasaki, M., Ozawa, Y., Kurihara, T. et al. Neurodegenerative influence of oxidative stress in the retina of a murine model of diabetes. Diabetologia 53, 971–979 (2010). https://doi.org/10.1007/s00125-009-1655-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1655-6