
Abstract
Aims/hypothesis
Interruption of IL-2 signalling is an attractive therapeutic target in autoimmune disorders. In this study we evaluated the effect of a fusion protein composed of IL-2 and caspase-3 (IL2–cas) on NOD mice, as compared with disease induction by cyclophosphamide.
Methods
IL2–cas was assessed in NOD mice at various ages and in conjunction with cyclophosphamide administration. The effect of IL2–cas on diabetogenic cells was evaluated in adoptive transfer experiments and in cell suspension in vitro.
Results
IL2–cas induced apoptosis in T cells expressing the α chain of the IL-2 receptor (cluster of differentiation [CD]25) in vitro, with superior survival of T cells expressing CD4 and forkhead box P3 (FOXP3). The fusion protein decreased mixed lymphocyte reactivity, and pretreatment with IL2–cas decreased the efficacy of adoptive transfer of diabetes into NOD severe combined immunodeficiency mice. Administration of one dose of IL2–cas decreased the incidence of diabetes in NOD mice, showing a superior beneficial effect when administered at young age, and effectively blocked induction of hyperglycaemia by cyclophosphamide, reducing the severity of islet inflammation. Administration of IL2–cas caused an acute increase in CD25-FOXP3+ T cells in the lymph nodes, pancreas and thymus in NOD mice, with similar effects in wild-type mice. Administration of IL2–cas after onset of hyperglycaemia resulted in superior survival.
Conclusions/interpretation
Targeted elimination of cells expressing the IL-2 receptor by this fusion protein disrupts the autoimmune pathogenesis in prediabetic and diabetic NOD mice, despite depletion of CD25+ regulatory T cells. Furthermore, this particular fusion protein is permissive to the development of FOXP3+ T cells that might contribute to protracted protection from the progression of insulitis and overt hyperglycaemia.
Similar content being viewed by others

Introduction
Multiple immune deficits in NOD mice result in deregulation of homeostasis and eruption of destructive autoimmune insulitis [1–3]. Progressive development of pathogenic clones and islet infiltration starting at young age are evidence of continuous insufficiency of the suppressor pathways in controlling the reaction against self-antigens. Numerous therapeutic strategies have been implemented, with variable success, that aim to both reduce the activity of diabetogenic cells and enhance the suppressive efficacy of regulatory T cells (Treg). Generalised immunosuppression fails to correct the autoimmune process and is associated with adverse effects that are detrimental to the treatment of insulitis. Administration of one dose of cyclophosphamide (Cy) has been long recognised to induce diabetes in NOD mice. While causing transient lymphodepletion [4, 5], this agent is particularly toxic to naturally occurring Treg that express cluster of differentiation (CD)25 and Forkhead box P3 (FOXP3), causing both their depletion and functional inactivation in the pancreas and its draining lymph nodes [4]. Subsequent administration of agents that stimulate immuno-haematopoietic reconstitution partially reverse the toxic effects of Cy by increasing the pool of suppressor cells [6], as does adoptive transfer of Treg [4]. More selective therapy consists of administration of anti-CD3 antibodies, which were efficient in reversal of the destructive insulitis even after onset of overt hyperglycaemia [7]. However, peri-islet inflammatory cuffing persists in the non-diabetic mice, possibly because these antibodies inhibit, but do not eliminate, the pathogenic cells [8].
IL-2 is an attractive target of immunotherapy as an important constituent of immune activation [9, 10]. Subsets of naturally occurring Treg that emerge from the thymus as a distinct cell type [11, 12] are not only characterised by expression of high levels of the high-affinity α chain of the IL-2 receptor (CD25) [13], but also depend on IL-2 for their expansion and function [14, 15]. The implications and pleiotropic functions of IL-2 in autoimmune disorders have been extensively debated. The crucial role of IL-2 in suppressor cells has been demonstrated by eruption of anti-self-reactivity following IL-2 neutralisation in wild-type mice [16], and reduced disease severity in NOD mice following IL-2 supplementation [17]. Likewise, administration of multiple low-doses of a complex composed of IL-2 and anti-IL-2 antibody prevented insulitis in NOD mice by increasing the numbers of Treg in the pancreas [17]. These data depict a scenario where IL-2 deficiency within the islets is detrimental to the function of Treg and favours evolution of the autoimmune reaction, whereas administration of large doses of IL-2 precipitates the disease through expansion of effector cells [18].
A more specific approach to antagonise the pathogenic effects of IL-2 is targeting of cells expressing the IL-2 receptor. For example, an IL-2/Fc fusion protein partially blocked the development of diabetes in NOD mice by reducing T cells expressing the IL-2 receptor in the pancreas [19]. Other chimeric molecules have been designed by conjugation of IL-2 to toxic moieties that kill cells after internalisation through the IL-2/IL-2 receptor complex. A fusion protein composed of IL-2 and diphtheria toxin showed protective effects against development of diabetes in NOD mice and humans [20, 21], as well as in models of inflammatory colitis [22]. However, we recently showed that this fusion protein causes severe lymphopenia at the therapeutic doses, which might have detrimental consequences in the context of autoimmunity [23]. Homeostatic expansion of effector cells outpaces the recovery of Treg under conditions of lymphopenia [24], creating a transient period of instability with predominant autoimmune effector activity [9, 10]. In variance, a fusion protein composed of IL-2 and activated caspase-3 (IL2–cas) provided the same defence against inflammatory colitis without causing lymphopenia [23, 25]. In this study we evaluated the effect of IL2–cas in NOD mice, where targeted deletion of cells expressing the IL-2 receptor has two possible outcomes. One possibility is acute onset of destructive insulitis provoked by selective depletion of CD25+ Treg. Alternatively, targeted depletion of IL-2-receptor-positive effector cells can ameliorate the course of disease. We found that one dose of IL2–cas decreases the incidence of diabetes in an age-dependent manner, and antagonises the toxic effects of cyclophosphamide. Despite eradication of CD25+ Treg, protection afforded by this fusion protein was associated with increased FOXP3 levels.
Methods
Animal model and treatments
NOD, NOD severe combined immunodeficiency mice (NOD SCID) and C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and inbred in a barrier facility. All experimental procedures have been approved by the Institutional Animal Care Committee. Glucose was monitored at weekly intervals at constant hours by tail bleeding using a standard glucometer (Roche Diagnostics, Manheim, Germany). Hyperglycaemia was considered at blood glucose levels above 11 mmol/l in two consecutive measurements at 2 day intervals. The glucose tolerance test was performed by intraperitoneal administration of 2 g glucose and measurement of blood glucose at 30, 60 and 120 min. IL2–cas is composed of full-frame IL-2 conjugated head to tail to full-frame caspase-3 (without the leading procaspase segment) [23, 25]. The sequence was inserted into a PET-28 plasmid under control of the T7 promoter and protein retrieved from the inclusion bodies of Escherichia coli was purified under denatured conditions and refolded to a biologically active molecule. The fusion protein and cyclophosphamide (Sigma, St Louis, MO, USA) was administered intravenously or intraperitoneally in 200 μl PBS.
Cell preparation and analysis
Spleens, mesenteric/pancreatic lymph nodes, thymus and pancreas were gently minced on a 40 µm nylon mesh in HBSS to prepare single-cell suspensions. The CD4+ and CD4+CD25− T cells were isolated using the CD4+CD25+ Regulatory T Cell Isolation Kit, according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch-Gladbach, Germany). The yield of isolation was evaluated using fluorochrome-labelled primary antibodies: CD4 (clone RM 4-5), CD8 (clone 53-6.7), CD25 (clone PC61.5). Antibodies were purchased from eBioscience (San Diego, CA, USA) and BD Pharmingen (San Diego, CA, USA). FOXP3 was determined following permeabilisation and intracellular staining with a phycoerythrin-labelled antibody (FOXP3 staining buffer set NRRF-30, eBioscience). Measurements were performed with a Vantage SE flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Apoptosis and death were determined using Annexin-V (IQ products, Groningen, the Netherlands) and 7-aminoactinomycin-D (7-AAD, Sigma), respectively. Positive staining was determined on a log scale, normalised with control cells stained with isotype control antibodies. Proliferation was determined in lymphocytes labelled with 10 µmol/l 5-(and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular Probes, Carlsbad, CA, USA); CFSE dilution was quantified with the ModFit software.
In vitro apoptosis
Cells were incubated at a concentration of 2 × 106/ml in DMEM supplemented with 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate, 13.6 µmol/l folic acid, 270 µmol/l l-asparagine, 548 µmol/l l-arginine, 10 mmol/l HEPES, 50 µmol/l 2β-mercaptoethanol, 100 mg/ml streptomycin, 100 U/ml penicillin and 5% (vol./vol.) heat-inactivated fetal bovine serum (mixed lymphocyte reaction [MLR] medium). All the ingredients were purchased from Beit Haemek (Kibbutz Beit Haemek, Israel) and Sigma. Cells were incubated at 37°C in a humidified 5% CO2 atmosphere for 24 or 72 h with 1.2–3.6 μg/ml IL2–cas.
In vitro proliferation and mixed lymphocyte reaction
NOD responder cells (H2Kg7) labelled with CFSE were co-incubated in MLR medium with an equal number of stimulators irradiated at 30 Gy (Rad Source 2000; Rad Source, Alpharetta, CA, USA) from syngeneic (H2Kg7) or allogeneic mice (H2Kb), with and without 1 µg/ml IL2–cas. Proliferation of CD4+CD25− T cells was measured during 3 day incubation in MLR medium with 5 μmol/l concanavalin-A (ConA). All MLR assays were performed in triplicate and the data presented are representative of a minimum of three animals per group.
Histology
Mice killed by CO2 asphyxiation underwent whole-body desanguination by infusion of 25 ml PBS containing heparin into the abdominal aorta, fixed in ice-cold PBS containing 1.5% (vol./vol.) fresh paraformaldehyde (for 2 h at 0–4°C) and immersed in 30% (wt/vol.) sucrose overnight. Tissue embedded in OCT (Sakura Finetek, Torrance, CA, USA) was frozen in isopentane suspended in liquid nitrogen, sectioned (3–6 μm) with a Cryotome (Termo Shandon, Cheshire, UK) and counterstained with haematoxylin/eosin. Sections were visualised with an Axioplan 2 microscope (C. Zeiss, Brighton, MI, USA).
Statistical analysis
Data are presented as means ± standard deviations for each experimental protocol. The results in each experimental group were evaluated for reproducibility by linear regression of duplicate measurements. Differences between the experimental protocols were estimated with a post hoc Scheffe t test and a p value of less than 0.05 was considered significant.
Results
Influence of IL2–cas on T cells
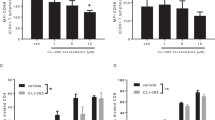
A fusion protein that internalises active caspase-3 is expected to induce apoptosis in lymphocytes that express the IL-2 receptor. Incubation of splenocytes from prediabetic (8 weeks) and diabetic NOD mice with escalating concentrations of the fusion protein for 24 h resulted in dose-dependent increase in apoptosis (Fig. 1a). Splenocytes of diabetic mice showed a lower (non-significant) but consistently reduced sensitivity to apoptosis induced by this fusion protein as compared with prediabetic mice. Preliminary studies showed that the optimal time to attain steady-state levels of apoptosis induced by IL2–cas is 3 days, suggesting that internalisation of the toxic moiety requires time to activate the apoptotic machinery in vitro. To determine specific targeting of the IL-2 receptor, CD4+ T cells isolated from the mesenteric and pancreatic lymph nodes of female NOD mice (aged 8 weeks) were incubated with 1 μg/ml IL2–cas for 72 h (Fig. 1b). The fusion protein induced apoptosis in CD25+ T cells (p < 0.05 vs medium) as expected, but was also toxic to CD25− T cells (p < 0.05 vs medium, Fig. 1c). Possibly, the toxic moiety was internalised by the IL-2/IL-2 receptor complex at a lower frequency of CD25 than the threshold of detection by flow cytometry, or the fusion protein was internalised by other components of the IL-2 receptor. Analysis of apoptosis in reference to FOXP3 expression revealed reduced sensitivity of CD4+FOXP3+ T cells to apoptosis as compared with CD4+FOXP3− T cells (p < 0.001) in medium, with no apparent significant effect of IL2–cas (Fig. 1d). Thus, the fusion protein preferentially targets CD25+ T cells, but different subsets of CD4+ T cells display variable sensitivities to caspase-induced apoptosis.

IL2–cas induces apoptosis in vitro. a Apoptosis of splenocytes from prediabetic NOD female mice aged 8 weeks and from new-onset diabetic mice exposed to escalating concentrations of IL2–cas for 24 h (n = 4); NOD prediabetic mice, grey bars; NOD diabetic mice, white bars. b Representative measurements of apoptosis and death using annexin-V (labelled with FITC) and 7-AAD, respectively, in the CD25− and CD25+ subsets of isolated CD4+ T cells. APC, allophycocyanin; PE, phycoerythrin. c–d CD4+ T cells isolated from the mesenteric lymph nodes of prediabetic NOD female mice aged 8 weeks were incubated with 1 μg/ml IL2–cas for 72 h (n = 4; dark grey bars) to determine apoptosis in reference to CD25 expression (c) and FOXP3 expression (d). Incubation in medium served as the control (light grey bars); *p < 0.05; ***p < 0.001
IL2–cas suppresses effector cells
To determine whether specific targeting of CD25+ T cells by IL2–cas increases or decreases immune reactivity, two functional assays were performed. First, activity of the fusion protein was evaluated in an in vivo MLR assay, where NOD mice (H2Kg7) aged 8 weeks were immunised with 5 × 106 splenocytes from C57BL/6 mice (H2Kb). NOD splenocytes were harvested after 2 days, labelled with CFSE and co-incubated with radiated syngeneic (H2Kg7) or allogeneic (H2Kd) stimulators. IL2–cas suppressed proliferation in response to both syngeneic and allogeneic stimuli (p < 0.05, Fig. 2a), suggesting that the fusion protein decreases reactivity rather than augments responsiveness by selective depletion of Treg. In a second experiment, the effect of IL2–cas on diabetogenic cells was evaluated in an adoptive transfer model into immunocompromised mice. Splenocytes of NOD mice with new-onset diabetes were incubated for 72 h with and without 1 μg/ml IL2–cas and 2 × 107 viable cells were adoptively transferred into NOD SCID mice. While all recipients of splenocytes incubated in medium developed diabetes, only three of the eight recipients of IL2–cas-pre-treated splenocytes developed the disease (Fig. 2b), demonstrating effective depletion of pathogenic cells. Similar depletion of diabetogenic cells has been achieved by pre-incubation with Fas ligand before adoptive transfer into immunocompromised mice [26].

IL2–cas reduces immune reactivity. a NOD mice (H2Kg7) aged 8 weeks (n = 5) were immunised with 5 × 106 splenocytes of C57BL/6 mice (H2Kb). After 2 days the spleens were harvested, and cells were labelled with CFSE and co-incubated with syngeneic (H2Kg7) or allogeneic (H2Kd) stimulators inactivated by irradiation (30 Gy). Proliferation was determined from CFSE dilution and quantified using the ModFit software. IL2–cas, black circles; medium, white circles; *p < 0.05. b Splenocytes of new-onset diabetic NOD mice were incubated with (n = 8, thick line) or without (n = 9, thinner line) 1 μg/ml IL2–cas for 72 h. Viable cells (2 × 107) were adoptively transferred into NOD SCID mice and hyperglycaemia was monitored in peripheral blood
Impact of cyclophosphamide and IL2–cas on diabetes in NOD mice
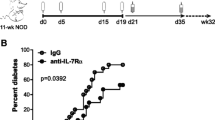
In view of the evidence that IL2–cas induces apoptosis in CD25+ T cells and decreases effector activity, the fusion protein was evaluated in NOD mice in vivo. On the one hand, an agent that mediates targeted killing of CD25+ T cells is expected to precipitate early onset of diabetes similar to cyclophosphamide [4–6] and anti-CD25 antibodies [27, 28]. On the other hand, decreased activity of reactive cells suggests that the fusion protein might ameliorate the course of insulitis as observed with diphtheria toxin in NOD mice [20] and IL2–cas in inflammatory colitis [22, 23, 25]. Our female NOD colony was monitored over an extended period of time to determine onset of diabetes at age 14 weeks, with an overall incidence of 83% diabetic mice at the age of 30 weeks (n = 312). Cyclophosphamide was chosen as a control agent that precipitates the onset of hyperglycaemia in NOD female mice in an age-dependent manner: administration of one dose of 200 μg/g Cy at ages 5.5, 8.5 and 11.5 weeks caused acute onset of hyperglycaemia within 2 weeks in 47%, 70% and 78% of the mice, respectively (Fig. 3a). In contrast, administration of 1 μg/g IL2–cas at ages 5.5, 8.5 and 11.5 weeks did not delay significantly the onset of disease relative to the female colony, but decreased its incidence to 26%, 38% and 50%, respectively (Fig. 3b). These data were consistent with reduced activity of effector cells in the MLR assay and partial depletion of diabetogenic cells as seen in the adoptive transfer experiment (Fig. 2). A common feature of Cy and IL2–cas was increased incidence of diabetes when immunomodulation was performed at an older age (Table 1), when the disturbance in Treg/effector T cell (Teff) balance is generally more pronounced. A significant difference was the acute onset of hyperglycaemia within 2 weeks after infusion of Cy, with a mean onset time of 12.8 ± 3.6 days (n = 62). In variance, recipients of IL2–cas developed diabetes close to the onset in the female colony, with mean onset time = 103 ± 24 days from its administration at the age of 5.5 weeks (n = 19) and 56 ± 6 days when given at the age of 11.5 weeks (n = 10). This delay in disease onset suggests that IL2–cas is more efficient in reducing the load of pathogenic cells at a younger age.

Impact of cyclophosphamide and IL2–cas on diabetes onset in NOD mice. a Administration of 200 μg/g cyclophosphamide (arrows) at ages 5.5 (n = 15), 8.5 (n = 20) and 11.5 weeks (n = 27) precipitated acute onset of diabetes in female NOD mice. The incidence of spontaneous diabetes in the female NOD colony (n = 312) is shown as control. b Administration of 1 μg/g IL2–caspase (arrows) at 5.5 (n = 19), 8.5 (n = 16) and 11.5 weeks (n = 10) resulted in delayed onset and reduced incidence of diabetes. The incidence of spontaneous diabetes in the female NOD colony is shown. c Female NOD mice were injected with 200 μg/g cyclophosphamide and 1 μg/g IL2–cas after 3 days, at ages 5.5 weeks (n = 15), 8.5 weeks (n = 9) and 11.5 weeks (n = 19). For a–c: onset following administration at 5.5 weeks, dotted line; at 8.5 weeks, dot/dashed line; at 11.5 weeks, solid line; control, dashed line. d Disease severity was evaluated by histology in pancreatic sections of non-diabetic mice at the experimental endpoint of 28 weeks. Islets were scored according to 0 = no inflammation (lower image, white bar in graph) and 1 = inflammation (upper image, black bar in graph) in cryosections of naive NOD female mice (n = 63, islets from five mice), and administration at 8 weeks of IL2–cas (n = 38, islets from four mice) and sequential Cy and IL2–cas (n = 47, islets from three mice). Demonstrative images were acquired at ×20 magnification
IL2–cas alleviates Cy-induced diabetes
Although both Cy [4] and IL2–cas are particularly toxic to CD25+ T cells, the dissociated time courses of disease development suggested differential effects on immune cells. In a subsequent experiment IL2–cas was given 3 days after administration of Cy, the time point of maximal lymphodepletion [4, 5, 29]. Administration of 1–4 doses of 1 μg/g IL2–cas after 200 μg/g Cy to prediabetic female NOD mice decreased the incidence of hyperglycaemia in all age groups (Fig. 3c). Consistent with the age-dependent variations observed with each agent alone, diabetes was more efficiently prevented when both agents were administered at younger ages (Table 1). Interestingly, mice developed diabetes early after joint Cy/IL2–cas administration similar to disease onset after Cy, suggesting suboptimal IL2–cas dosage or timing of injection. Non-diabetic mice showed normal responses to a glucose tolerance test at the experimental endpoint of 28 weeks, questioning whether the severity of inflammatory insulitis has been modulated. Histological inspection of the islets revealed reduced severity of inflammatory insulitis in diabetes-free mice after treatment with IL2–cas with and without Cy, as compared with islets of age-matched non-diabetic naive NOD mice (Fig. 3d). Considering that in naive NOD female mice approximately 65% of the islets are inflamed at age 11 weeks [30], administration of IL2–cas was evidently associated with partial resolution of the inflammatory state, in addition to the competent insulin output in response to demand as determined by the glucose tolerance test.
IL2–cas modulates the course of disease after onset of hyperglycaemia
There are few cases where therapy is effective in arresting autoimmune diabetes in NOD mice after onset of hyperglycaemia [8]. To evaluate the capacity of the fusion protein to arrest disease progression, new-onset diabetic NOD female mice were given two to four doses of 1 μg/g IL2–cas at daily intervals. Initiation of therapy within 1 week after onset of hyperglycaemia resulted in superior survival as compared with untreated NOD mice (Fig. 4a). To determine how IL2–cas therapy impacted the activity of diabetogenic cells, 2 × 107 splenocytes harvested at 6 weeks after onset of hyperglycaemia were adoptively transferred into NOD SCID mice. While splenocytes from untreated female mice transferred the disease to 100% of the immunocompromised recipients within 8 weeks, splenocytes from diabetic mice treated with IL2–cas transferred the disease in 40% of the recipients (Fig. 4b). The decrease in activity of diabetogenic cells was consistent with superior survival, providing evidence that therapy improved glucose homeostasis even after the onset of overt diabetes.

IL2–cas ameliorates disease course after onset of overt hyperglycaemia. a NOD mice were given two to four daily doses of 1 μg/g IL2–cas after two consecutive measurements of blood glucose levels exceeding 11 mmol/l. Survival is presented for the treated group (n = 6, thick line) compared with untreated controls (n = 22, thinner line). b Splenocytes from diabetic mice were adoptively transferred into NOD SCID mice to determine the relative diabetogenic potential at 6 weeks after IL2–cas administration (n = 8, thinner line) as compared with untreated diabetic mice (n = 7, thick line)
Immunological consequences of immunomodulation
To determine the acute-phase consequences of administration of the two agents, lymphoid organs were evaluated at the nadir of lymphopenia caused by Cy in NOD mice [4, 5, 29]. Female NOD mice aged 8 weeks received one intraperitoneal dose of 200 μg/g Cy or 4 μg/g IL2–cas and after 3 days the spleens, mesenteric/pancreatic lymph nodes and thymus were compared with age- and sex-matched naive NOD mice (Fig. 5a). Neither of the agents caused significant changes in the CD4+ T cell subset (not shown); however, both agents decreased significantly (p < 0.001) the fraction of CD4+CD25+ cells (Fig. 5b). Consistently, the CD4+CD25+FOXP3+ T cell subset was depleted in all lymphoid organs by both agents (p < 0.001, Fig. 5c). However, these interventions differed in their effects on FOXP3+ cells: while Cy decreased fractional expression of FOXP3, a surprising increase in FOXP3+ cells was caused by IL2–cas in the lymph nodes, thymus (Fig. 5d) and pancreas (Fig. 5f). Notably, increased fractional expression of FOXP3 in the lymph modes and pancreas early after IL2–cas administration was associated with reduced the incidence of hyperglycaemia.

Short-term immune consequences of cyclophosphamide and IL2–cas administration. Female NOD mice (8 weeks old) were given one (intraperitoneal) dose of 200 μg/g Cy or 4 μg/g IL2–cas and the lymphoid organs and pancreases were harvested after 3 days (n = 5). All data represent percentage distribution in the CD4+ T cell subset, and are compared with age-matched naive female NOD mice (n = 6). a Representative measurements of fractional expression of CD25 and FOXP3 in the CD4+ T cells subset in mice given IL2–cas (upper panels) and cyclophosphamide (lower panels). APC, allophycocyanin; PE, phycoerythrin. b Fractional expression of CD25. c Proportion of CD25+FOXP3+ T cells. d Fractional expression of FOXP3. e Distribution of CD25 and FOXP3 in CD4+ T cells infiltrating the pancreas (n = 4). For b–e: NOD, white bars; IL2–cas, black bars; Cy, grey bars. f Naive C57BL/6 mice were administered the same doses of Cy and IL2–cas and lymphoid organs were evaluated for CD25 and FOXP3 expression in CD4+ T cells after 3 days (n = 5); C57BL/6 mice, light grey bars; Cy, mid grey bars; IL2–cas, dark grey bars. *p < 0.05; **p < 0.01; ***p < 0.001; † p < 0.005
Elevated fractional expression of FOXP3 in the lymph nodes following administration of IL2–cas (0.9 μg/g, intravenous) has also been observed in a model of toxic colitis in BALB/c mice [23]. To determine whether the acute effects caused by the fusion protein and Cy are specific to NOD mice, wild-type C57BL/6 mice were infused (intraperitoneally) with 4 μg/g IL2–cas and 200 μg/g Cy. Both IL2–cas and Cy depleted CD25+ T cells in all lymphoid organs (not shown). However, while Cy reduced the CD4+FOXP3+ subset, IL2–cas preserved fractional expression of FOXP3 in all lymphoid organs (Fig. 5f). The higher incidence of CD4+FOXP3+ T cells in lymph nodes of NOD mice as compared with C57BL/6 mice may be attributable to lymphocyte activation caused by the autoimmune reaction. Considering that autoimmune inflammatory disorders, such as gastritis and diabetes, are triggered by IL-2 neutralisation in wild-type mice [16], C57BL/6 mice were monitored for 10 weeks after administration of two doses of 4 μg/g IL2–cas at an interval of 2 days (n = 5). These mice showed no signs of enteritis and hyperglycaemia, and this was confirmed by histological examination at the experimental endpoint.
Influence of IL2–cas on CD25− T cells
Immune homeostasis is based on dynamic changes in phenotypes and function of T cells, as demonstrated by reconstitution of all T cell lineages in NOD SCID mice after adoptive transfer of either CD25− or CD25+ T cells (A. Kaminitz, unpublished data). Increased fractional expression of FOXP3 in NOD mice after targeted depletion of CD25+ T cells may be caused either by relative insensitivity of CD4+FOXP3+ T cells to apoptosis, faster proliferation rates or induced expression of FOXP3 [4, 5, 18, 24]. To dissociate between these possibilities, we next determined whether IL2–cas induces FOXP3 expression and/or stimulates proliferation of various CD4+ T cell subsets in vitro. CD4+CD25− T cells were isolated from the mesenteric/pancreatic lymph nodes of female NOD mice aged 8 weeks (94 ± 3% CD4+CD25−) and incubated for 3 days. In the presence of IL2-cas, 12 ± 0.8% converted to express CD25 in liquid culture (p < 0.001 vs 6 ± 1.4% of cells in the medium). Most of these cells expressed high levels of CD25 (Fig. 6a), suggesting that upregulation was not caused by selective evasion from apoptosis of cells expressing low levels of the IL-2 receptor. Cells that expressed CD25 in culture (without stimulation) did not co-express FOXP3, which was detected in a small fraction of cells (2.5 ± 0.6%) that were mostly negative for the IL-2 receptor (Fig. 6b). The dissociated expression of CD25 and FOXP3 indicates that induction of the IL-2 receptor was independent of FOXP3 activation, and upregulation of this transcription factor is not frequent in CD25− T cells in vitro.

Influence of IL2–cas on CD4+CD25- cells in vitro. CD4+CD25- T cells (94 ± 3% purity) isolated from the mesenteric/pancreatic lymph nodes of female NOD mice aged 8 weeks (n = 3) were incubated with 1 μg/ml IL2–cas for 72 h and in medium (control). a Proportion with CD25 expression as compared with the fresh sample: fresh, grey bars; IL2–cas, black bars; control, white bars. b Representative measurements of CD25low and CD25high expression in the gated CD4+ T cells subset, and FOXP3 expression in reference to cells that expressed the IL-2 receptor. APC, allophycocyanin; PE, phycoerythrin. c Demonstrative measurements of proliferation from CFSE dilution using the ModFit software for simulation. IL2–cas, lower panels; control, upper panels. d Proliferation in response to 5 μmol/l ConA with reference to CD25 expression (n = 3): IL2–cas, black bars; control, white bars. ***p < 0.001
Subsequently, CD25− T cells were submitted to mitogenic stimulation with ConA (Fig. 6c). IL2–cas increased the cycling rates of cells that upregulated CD25 expression, in particular among those cells showing high levels of CD25 expression (the CD25high subset), demonstrating again that some cells expressing the IL-2 receptor were insensitive to apoptosis induced by the fusion protein (Fig. 6d). The faster proliferation induced by IL2–cas indicates that upregulation of CD25 was overestimated, such as the values for CD25 expression originated from expansion of a smaller fraction of cells that became CD25-positive. As delineated above, FOXP3 was detected in a small fraction of those cells that expressed the IL-2 receptor (Fig. 6b). Furthermore, faster proliferation rates in both CD25low and CD25high subsets suggest that internalisation of the fusion protein promoted cell cycling. Taken together, these data suggest that increased FOXP3 expression observed in IL2-cas-treated NOD mice originated primarily from the relative resistance of these cells to apoptosis, and not from induced expression of this transcription factor or fast proliferation of these cells.
Discussion
This study demonstrates that targeted killing of cells expressing the high affinity IL-2 receptor provides partial protection from diabetes in NOD mice, antagonises destructive insulitis triggered by cyclophosphamide and improves survival after onset of hyperglycaemia. The fusion protein induces apoptosis in CD25+ T cells in vitro and in vivo, with a lesser but significant effect on CD25− T cells. The immunomodulatory consequences of IL2–cas include elevated fractional expression of FOXP3 in the lymph nodes and pancreases of diabetes-free mice, which is favourable to resolution of inflammation in some islets.
Despite the functional significance of CD25 and IL-2 signalling in naturally occurring Treg [11–16], the data presented here document deletion of diabetogenic cells by the fusion protein. Targeting cells that express the IL-2 receptor decreased the responsiveness to alloantigens in vitro, impaired adoptive transfer of the disease into NOD SCID mice and antagonised cyclophosphamide-induced destructive insulitis. Taken together, these data imply that the Treg/Teff ratio was tilted to the advantage of suppressor cells. Considering that CD4+CD25+FOXP3+ Treg were virtually eradicated in the lymphatics of NOD mice, reduced autoimmune reactivity evidently occurred through removal of the pathogenic cells. This is the likely explanation for the beneficial effect of the fusion protein after onset of overt diabetes, where arrest of complete islet destruction improved survival without exogenous administration of insulin. Treg were ineffective and therefore dispensable at these late stages of destructive insulitis. IL-2 targeted therapy can affect immune homeostasis in NOD mice by modulation of several activities of effector and regulatory cells. First, the fusion protein increased apoptosis in cells defined by flow cytometry as CD25−, suggesting its internalisation at very low levels of the high-affinity α chain. Second, Treg subsets display significant variations in sensitivity to apoptosis, which is differentially and continuously modulated by the inflammatory environment [31, 32]. Third, expression of the IL-2 receptor is a general feature of the physiological process of lymphocyte activation, thus the CD25+ subset is likely to include a certain number of effector cells [33]. Fourth, CD25 is variably downregulated and is shed from Treg without affecting their suppressive capacity [34].
The most significant difference in the contrasting effects of Cy and IL2–cas on the immune system of NOD mice was the expression of FOXP3, considered to be the characteristic marker of Treg [16, 34] that best defines a suppressive phenotype in the pancreas [18]. Administration of Cy to NOD and wild-type mice resulted in a sharp decline in FOXP3+ T cells, corroborating prior reports [4, 5]. The reason for selective depletion of Treg by Cy is not entirely clear: it may be caused by faster proliferation rates of Treg in vivo rendering them more susceptible to apoptosis [5, 35], or faster recovery of naive and pathogenic cells as compared with Treg through homeostatic expansion under lymphopenic conditions [24, 36]. The ensuing transient period of immune disequilibrium predisposes to immune reactivity, which is inefficiently suppressed by slower and delayed recovery of the Treg pool [9]. According to this scenario, the incomplete abrogation of diabetes when IL2–cas was administered after Cy might be caused by insufficient dosage of the fusion protein, or late timing after homeostatic expansion secondary to Cy-induced lymphopenia has already commenced. These possibilities are consistent with early onset of the disease in these mice, suggesting that the toxic effect of Cy was not sufficiently counterbalanced. A similar drastic decrease in FOXP3+ T cells has been induced by anti-CD25 antibodies, which either inhibited [37] or depleted Treg [27], and has been proposed to underlie the predisposition to diabetes following IL-2 neutralisation [16].
In contrast, expansion of CD25−FOXP3+ cells in the lymph nodes and pancreases of NOD mice caused by IL2–cas was associated with lower insulitis score and decreased incidence of hyperglycaemia. As CD25 is expressed in 50–60% of the CD4+FOXP3+ T cells, targeted killing of CD25+ lymphocytes was accompanied by expansion of the CD25−FOXP3+ subset in lymphoid organs. Consistent with superior survival of FOXP3+ T cells in vitro, CD25−FOXP3+ cells were markedly elevated early after administration of IL2–cas to NOD and wild-type mice. The positive findings in this study indicate that the basis for enrichment in the CD25−FOXP3+ subset is lower sensitivity of these cells to apoptosis induced by IL2–cas, similar to observations in models of experimental colitis [22, 23]. The mechanism of superior survival of this subset remains to be determined. We could not demonstrate upregulation of FOXP3 in CD25− cells in vitro, though this does not preclude that such expression occurs under inflammatory conditions in the pancreas and regional lymph nodes. Our findings are consistent with the observation that anti-CD25 antibodies do not mediate FOXP3 expression in CD4+CD25− T cells [37]. However, the data presented here vary from the absence of compensatory expansion of CD25−FOXP3+ T cells in response to anti-CD25 antibodies in a model of experimental autoimmune encephalomyelitis in wild-type mice [38]. Since anti-CD25 antibodies do not always deplete CD25+ T cells [27], but rather inhibit their function [37], the cytokine environment might be less favourable for expansion of the CD25−FOXP3+ T cells [39]. Reactive changes in CD25, CD62L and FOXP3 as phenotypic markers of putative Treg have been previously observed with immunomodulatory agents such as Cy, anti-IL-2, anti-CD25 and anti-CD3, and have been generally associated with positive outcome of immunomodulation in NOD mice [4, 5, 16, 28]. A similar reactive increase in FOXP3 expression has recently been associated with beneficial effects of Treg activation achieved by several immunomodulatory modalities, including IL-2/IL-2 antibody complexes, anti-thymocyte globulin, complete Freund's adjuvant, vasoactive intestinal peptide and rapamycin [18, 40–44]. Considering the very different mechanisms of action of these agents, it can be proposed that dominance of CD25−FOXP3+ T cells is mediated by superior survival and faster expansion.
An interesting observation was the CD25−-to-CD25+ conversion in a small subset of T cells in vitro, which was increased twofold by IL2–cas. Our incubation conditions were different from those used for expansion of adaptive CD25+ Treg, which are functionally competent and block the development of diabetes in NOD mice [45, 46]. CD25 expression in vitro did not involve FOXP3 as an intermediate transcription factor, and evidently this subset was insensitive to apoptosis induced by IL2–cas. The faster proliferation rates of CD25high cells in response to mitogenic stimuli suggest that the receptor was involved in expansion of this subset. It is tempting to speculate that internalisation of two factors involved in physiological process of lymphocyte activation—IL-2 and caspase-3—is responsible for the stimulation of cells that upregulated CD25 expression [47, 48]. The fact that IL2–cas did not aggravate insulitis indicates that the fusion protein does not expand diabetogenic cells.
Partial resolution of inflammation and protection from diabetes through elimination of diabetogenic cells achieved with IL2–cas offers an additional therapeutic option, with several advantages in the context of autoimmune insulitis. First, direct introduction of toxic moieties through the IL-2/IL-2 receptor complex overcomes the relative resistance of NOD lymphocytes to apoptosis (as compared with wild-type mice) [49, 50], and the decreased susceptibility of diabetogenic cells to Treg-mediated inhibition [33, 51, 52]. Inasmuch as B cell lymphoma protein 2 (BCL-2) is associated with the survival of islet-infiltrating T cells [18], IL2–cas was found to reduce the BCL-2/apoptosis regulator BAX ratio in a model of toxic colitis [25], suggesting that the fusion protein sensitises pathogenic lymphocytes to apoptosis. Thus, fusion proteins comprising IL-2 and toxic moieties are of therapeutic value irrespective of concomitant depletion of CD25+ Treg in NOD mice and models of inflammatory colitis [22, 23], which are associated with aberrant sensitivity of the pathogenic cells to apoptosis. In contrast, anti-CD3 and anti-CD25 antibodies primarily inhibit cell function and variably mediate their physical elimination [7, 8, 27, 37], which might be beneficial to preserve Treg but does not eliminate the diabetogenic potential. Second, decreased suppressive function [53, 54] and lower numbers of Treg [55, 56] are considered as mechanisms that contribute to the evolution of insulitis in NOD mice. Although such a decline in Treg has been not observed in other studies [51, 52], concurrent elimination of pathogenic cells has the distinct advantage of direct repression of the autoimmune process. Third, FOXP3+ T cells in the pancreas and draining lymphatics are less sensitive to apoptosis induced by IL2–cas, resulting in expansion of C25−FOXP3+ cells. Increased fractional expression of FOXP3 in the thymus, the origin of naturally occurring Treg [11, 12] might be beneficial to restoration and maintenance of self-tolerance through resetting of the regulatory/effector equilibrium.
Abbreviations
- 7-AAD:
-
7-Aminoactinomycin-D
- BCL-2:
-
B cell lymphoma protein 2
- CD:
-
Cluster of differentiation
- CD25:
-
α chain of the IL-2 receptor
- CFSE:
-
5-(and 6-)Carboxyfluorescein diacetate succinimidyl ester
- ConA:
-
Concanavalin-A
- Cy:
-
Cyclophosphamide
- FOXP3:
-
Forkhead box P3
- IL2–cas:
-
IL-2 and activated caspase-3
- MLR:
-
Mixed lymphocyte reaction
- SCID:
-
Severe combined immunodeficiency
- Teff:
-
Effector T (cells)
- Treg:
-
Regulatory T (cells)
References
Delovitch TL, Singh B (1997) The non-obese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 7:727–738
Kishimoto H, Sprent J (2001) A defect in central tolerance in NOD mice. Nat Immunol 2:1025–1031
Anderson MS, Bluestone JA (2004) The NOD mouse. A model of immune dysregulation. Annu Rev Immunol 20:447–485
Brode S, Raine T, Zaccone P, Cooke A (2006) Cyclophosphamide-induced type-1 diabetes in the NOD mouse is associated with a reduction of CD4+CD25+Foxp3+ regulatory T cells. J Immunol 177:6603–6612
Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H (2005) Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105:2862–2868
Hadaya K, Kared H, Masson A, Chatenoud L, Zavala F (2005) G-CSF treatment prevents cyclophosphamide acceleration of autoimmune diabetes in the NOD mouse. J Autoimmun 24:125–134
Chatenoud L, Primo J, Bach JF (1997) CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 158:2947–2954
Chatenoud L, Bluestone JA (2007) CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 7:622–632
Fehervari Z, Yamaguchi T, Sakaguchi S (2006) The dichotomous role of IL-2: tolerance vs immunity. Trends Immunol 27:109–111
Yarkoni S, Kaminitz A, Sagiv Y, Yaniv I, Askenasy N (2008) Involvement of IL-2 in homeostasis of regulatory T cells: the IL-2 cycle. BioEssays 30:875–888
Suri-Payer E, Amar AZ, Thornton AM, Shevach EM (1998) CD4+ CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol 160:1212–1218
Kuniyasu Y, Takahashi T, Itoh M, Shimizu J, Toda G, Sakaguchi S (2000) Naturally anergic and suppressive CD25+CD4+ T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int Immunol 12:1145–1155
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Malek TR, Bayer AL (2004) Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 4:665–674
Nelson BH (2004) IL-2, regulatory T cells, and tolerance. J Immunol 172:3983–3988
Setoguchi R, Hori S, Takahashi T, Sakaguchi S (2005) Homeostatic maintenance of natural Foxp3+CD25+CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 201:723–735
Serreze DV, Hamaguchi K, Leiter EH (1989) Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun 2:759–776
Tang Q, Adams JY, Penaranda C et al (2008) Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28:687–697
Zheng XX, Steele AW, Hancock WW et al (1999) IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J Immunol 163:4041–4048
Pacheco-Solva A, Bastos MG, Pankewycz O, Nichols J, Murphy JR, Strom TB, Rubin-Kelley VE (1992) IL-2 toxin treatment blocks diabetogenic autoimmunity in NOD mice. Eur J Immunol 22:697–702
Woodworth TG (1993) Early clinical studies of IL-2 fusion toxin in patients with severe rheumatoid arthritis and recently onset insulin-dependent diabetes mellitus. Clin Exp Rheumatol 8:S177–S180
Sagiv Y, Kaminitz A, Lorberboum-Galski H, Askenasy N, Yarkoni S (2009) IL-2 targeted therapy in inflammatory bowel disease. Ann NY Acad Sci 1173:791–797
Yarkoni S, Sagiv Y, Kaminitz A, Askenasy N (2009) Targeted therapy to IL-2 receptor using diphtheria toxin and caspase-3 fusion proteins modulates regulatory T cells and ameliorates inflammatory colitis. Eur J Immunol 39:2850–2864
King C, Ilic A, Koelsch K, Sarvetnick N (2004) Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 117:265–277
Shteingart S, Rapoport M, Grodzovski I, Sabag O, Lichtenstein M, Eavri R, Lorberboum-Galski H (2009) Therapeutic potency of IL2-caspase3 targeted treatment in a murine experimental model of inflammatory bowel disease (IBD). Gut 58:790–798
Franke DD, Yolcu ES, Alard P, Kosiewicz MM, Shirwan H (2007) A novel multimeric form of FasL modulates the ability of diabetogenic T cells to mediate type 1 diabetes in an adoptive transfer model. Mol Immunol 44:2884–2892
Ly D, Mi QS, Hussain S, Delovitch TL (2006) Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol 177:3695–3704
Mellanby RJ, Thomas D, Phillips JM, Cooke A (2007) Diabetes in non-obese diabetic mice is not associated with quantitative changes in CD4+ CD25+ Foxp3+ regulatory T cells. Immunology 12:115–128
Mahiou J, Walter U, Lepault F, Godeau F, Bach JF, Chatenoud L (2001) In vivo blockade of the Fas-Fas ligand pathway inhibits cyclophosphamide-induced diabetes in NOD mice. J Autoimmun 16:431–440
Kaminitz A, Mizrahi K, Yaniv I, Farkas DL, Stein J, Askenasy N (2009) Low levels of allogeneic but not syngeneic hematopoietic chimerism reverse autoimmune insulitis in prediabetic NOD mice. J Autoimmun 33:83–91
Pandiyan P, Lenardo MJ (2008) The control of CD4+CD25+Foxp3+ regulatory T cell survival. Biol Direct 3:6
Yolcu ES, Kaminitz A, Ash S, Sagiv Y, Askenasy N, Yarkoni S (2008) Apoptosis as a mechanism of T regulatory cell homeostasis and suppression. Immunol Cell Biol 86:650–658
Gregori S, Giarratana N, Smiroldo S, Adorini L (2003) Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol 171:4040–4047
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY (2005) Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity 22:329–341
Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon BL (2003) Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med 198:737–746
Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK (2005) Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med 202:1375–1386
Kohm AP, McMahon JS, Podojil JR et al (2006) Cutting edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol 176:3301–3305
Stephens LA, Gray D, Anderton SM (2005) CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci USA 102:17418–17423
Askenasy N, Kaminitz A, Yarkoni S (2008) Mechanisms of T regulatory cell function. Autoimmun Rev 7:370–375
Rosignoli F, Torroba M, Juarranz Y et al (2006) VIP and tolerance induction in autoimmunity. Ann NY Acad Sci 1070:525–530
Battaglia M, Stabilini A, Draghici E et al (2006) Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes 55:1571–1580
Simon G, Parker M, Ramiya V et al (2008) Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes 57:405–414
Monti P, Scirpoli M, Maffi P et al (2008) Rapamycin monotherapy in patients with type 1 diabetes modifies CD4+CD25+FOXP3+ regulatory T cells. Diabetes 57:2341–2347
Tian B, Hao J, Zhang Y et al (2009) Upregulating CD4+CD25+FOXP3+ regulatory T cells in pancreatic lymph nodes in diabetic NOD mice by adjuvant immunotherapy. Transplantation 87:198–206
Tang Q, Henriksen KJ, Bi M et al (2004) In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med 199:1455–1465
Weber SE, Harbertson J, Godebu E et al (2006) Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. J Immunol 176:4730–4739
Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW (1995) Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3:521–530
Alam A, Cohen LY, Aouad S, Sekaly RP (1999) Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J Exp Med 190:1879–1890
Yang W, Hussain S, Mi QS, Santamaria P, Delovitch TL (2004) Perturbed homeostasis of peripheral T cells elicits decreased susceptibility to anti-CD3-induced apoptosis in prediabetic nonobese diabetic mice. J Immunol 173:4407–4416
Decallonne B, van Etten E, Giulietti A, Casteels K, Overbergh L, Bouillon R, Mathieu C (2003) Defect in activation induced cell death in non-obese diabetic (NOD) T lymphocytes. J Autoimmun 20:219–226
You S, Belghith M, Cobbold S et al (2005) Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T cells. Diabetes 54:1415–1422
D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, Mathis D (2008) The defect in T cell regulation in NOD mice is an effect on the T cell effectors. Proc Natl Acad Sci USA 105:19857–19862
Gregg RK, Jain R, Schoenleber SJ et al (2004) A sudden decline in active membrane bound TGF-beta impairs both T regulatory cell function and protection against autoimmune diabetes. J Immunol 173:7308–7316
Tritt M, Sgouroudis E, d'Hennezel E, Albanese A, Piccirillo CA (2008) Functional waning of naturally occurring CD4+ regulatory T cells contributes to the onset of autoimmune diabetes. Diabetes 57:113–123
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12:431–440
Wu AJ, Hua H, Munson SH, McDevitt HO (2002) Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci USA 99:12287–12292
Acknowledgements
This work was funded by grants from the Frankel Trust for Experimental Bone Marrow Transplantation (N. Askenasy), JDRF innovative grant 5-2005-1102 (N. Askenasy), and support from Target-In. The excellent technical assistance of N. Binkowski, E. Zozovski and A. Zemliansky (Frankel Laboratory, Schneider Children’s Medical Center of Israel) is gratefully acknowledged.
Duality of interest
S. Yarkoni serves as the Chief Executive Officer of GASR Biotechnology and has significant equity in this company. Y. Sagiv is an employee of GASR Biotechnology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yarkoni, S., Kaminitz, A., Sagiv, Y. et al. Targeting of IL-2 receptor with a caspase fusion protein disrupts autoimmunity in prediabetic and diabetic NOD mice. Diabetologia 53, 356–368 (2010). https://doi.org/10.1007/s00125-009-1604-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1604-4