
Abstract
The metabolic abnormalities of type 2 diabetes can be reversed reproducibly by bariatric surgery. By quantifying the major pathophysiological abnormalities in insulin secretion and insulin action after surgery, the sequence of events leading to restoration of normal metabolism can be defined. Liver fat levels fall within days and normal hepatic insulin sensitivity is restored. Simultaneously, plasma glucose levels return towards normal. Insulin sensitivity of muscle remains abnormal, at least over the weeks and months after bariatric surgery. The effect of the surgery is explicable solely in terms of energy restriction. By combining this information with prospective observation of the changes immediately preceding the onset of type 2 diabetes, a clear picture emerges. Insulin resistance in muscle, caused by inherited and environmental factors, facilitates the development of fatty liver during positive energy balance. Once established, the increased insulin secretion required to maintain plasma glucose levels will further increase liver fat deposition. Fatty liver causes resistance to insulin suppression of hepatic glucose output as well as raised plasma triacylglycerol. Exposure of beta cells to increased levels of fatty acids, derived from circulating and locally deposited triacylglycerol, suppresses glucose-mediated insulin secretion. This is reversible initially, but eventually becomes permanent. The essential time sequence of the pathogenesis of type 2 diabetes is now evident. Muscle insulin resistance determines the rate at which fatty liver progresses, and ectopic fat deposition in liver and islet underlies the related dynamic defects of hepatic insulin resistance and beta cell dysfunction. These defects are capable of dramatic reversal under hypoenergetic feeding conditions, completely in early diabetes and to a worthwhile extent in more established disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Puzzles are often most easily solved backwards. Isaac Newton exemplified this by using his newly invented and secret tool of calculus to establish the answer to unsolved mathematical problems, then presenting the proof in conventional mathematical terms having worked backwards to the question [1]. The same technique can be applied to unravelling the pathogenesis of disease, but there is a snag—a cure for the disease has to be discovered so that the pathogenesis may be observed, step by step, in reverse. However, it is now possible to achieve a cure in groups of people with type 2 diabetes. What happens during the period of return to normal insulin sensitivity and normal beta cell function?
Bariatric surgery and type 2 diabetes
The acid test for any new treatment is a prospective, randomised comparison with conventional treatment. Dixon and colleagues randomised patients with a BMI of 30–40 kg/m2 and type 2 diabetes duration of <2 years to a conventional treatment group or a surgically treated group [2]. The conventional group was treated using intensive lifestyle modification programmes, dietary advice, oral hypoglycaemic agents and insulin, and was followed up every 6 weeks. The surgically treated group underwent laparoscopic placement of an adjustable gastric band in addition to receiving the conventional treatment. The primary endpoint was remission of diabetes, defined as an HbA1c level of <6.2% and a fasting plasma glucose level of <7.0 mmol/l, off all treatment. At 2 years, remission was achieved in 13% of the conventional treatment group and 73% of the surgical group. Fasting plasma glucose fell from the baseline level of 8.8 mmol/l to 7.7 mmol/l in the conventional treatment group and to 5.9 mmol/l in the surgically treated group (Fig. 1). This was associated with weight change from 106 kg at baseline to 105 kg and 85 kg in the two groups, respectively (Fig. 1). The importance of the study lies in the demonstration, in a prospective, randomised setting, that normalisation of plasma glucose can be achieved in groups of individuals. Diabetologists have long known that the tiny proportion of iron-willed people who can substantially decrease their weight and maintain this, can exhibit a return to normal metabolism. Dixon and colleagues have demonstrated a more widely applicable phenomenon.

Effect of intensive conventional therapy (blue lines) or gastric banding (red lines) on body weight (a), fasting plasma glucose (b), plasma triacylglycerol (c) and requirement for insulin or oral hypoglycaemic agents (d). In the gastric banding group, fasting plasma glucose and triacylglycerol returned to levels within the normal range. Data represent means (n = 30 for each group) and are replotted from reference [2] with permission
However, gastric banding is the least dramatic of possible surgical procedures to achieve weight loss. It involves the placement of an adjustable cuff around the gastro-oesophageal junction, and may be done via a laparoscope. At the other end of the scale, bilio-pancreatic diversion consists of distal gastric resection with closure of duodenal stump, trans-section of the small intestine, proximal anastomosis to stomach and anastomosis of the remaining ileum (carrying bilio-pancreatic secretions) to the distal ileum. Simple gastric bypass involves ileo-gastric anastomosis so that food bypasses the duodenum and upper jejunum. An earlier systematic review indicated resolution of diabetes in 99% after bilio-pancreatic diversion, 84% for gastric bypass and 48% for gastric banding [3]. Gastric bypass surgery, with its attendant risks, has been known for 20 years to bring about major weight loss and improve glucose tolerance in morbidly obese individuals. In the original series by Pories et al., 86 of 88 obese people with diabetes reverted to normal glucose tolerance within 4 months, and this remained normal over 6 years of follow-up [4].
Time-course of reversal of pathogenic factors
Recently, several detailed studies of metabolic changes following gastric bypass surgery have thrown further light upon reversal of type 2 diabetes. A prospective study of ten obese patients with type 2 diabetes undergoing bilio-pancreatic diversion, with re-study after 2 years, demonstrated weight change from 136 to 89 kg [5]. This was accompanied by a fall in the fasting plasma glucose level from 7.7 to 5.1 mmol/l and a decrease in the HbA1c level from 8.2% to 4.1%. The improvement in insulin sensitivity measured during a euglycaemic–hyperinsulinaemic clamp was not surprising in the context of the weight loss (the glucose disposal rate per kg of fat-free mass rose from 26.4 to 55.2 μmol min−1; it was 68.7 μmol min−1 in a non-diabetic post-bypass group). However, the return of beta cell sensitivity for insulin secretion in the type 2 diabetic patients (from 44 to 76 pmol min−1 m−2 [mmol/l]−1) to levels above those seen in the non-diabetic post-bypass control group (65 pmol min−1 m−2 [mmol/l]−1) was striking. The term ‘inexorable decline’ is traditionally applied to beta cell function in established type 2 diabetes. The recent data clearly demonstrate that this is not necessarily the case, at least in the first few years after diagnosis. What could underlie this reversal of the twin pathogenic factors of type 2 diabetes?
Insight into the time-course of change following a gastric bypass has been gained from detailed studies conducted over the first 4 weeks after surgery [6]. A group of morbidly obese people with a diabetes duration of between 1 and 3 years were studied. Preoperatively, the fasting blood glucose level was 7.8 mmol/l and, as would be expected, both beta cell sensitivity to glucose and whole body insulin sensitivity were low. After pancreatobiliary diversion, the group were re-studied at 1 and 4 weeks. Parenteral feeding provided 7.5 MJ/day (1,800 kcal/day) for the first six postoperative days, but this would represent a sudden decrease in energy consumption. As body weight was 152 kg preoperatively, a daily intake of about 13.4 MJ/day (3,200 kcal/day) would be required for weight maintenance [7]. By 7 days postoperatively, fat mass had decreased by 4.4 kg and the fasting blood glucose level had fallen to 5.9 mmol/l (Fig. 2). Insulin sensitivity, measured as the glucose infusion rate during euglycaemic–hyperinsulinaemic clamps, had doubled. Correspondingly, the fasting plasma insulin level had fallen from 136 to 108 pmol/l. By 4 weeks postoperatively, the fasting plasma glucose level had fallen further (to 4.8 mmol/l), with no further change in insulin sensitivity. The observed change in insulin sensitivity requires careful consideration.

Change in fasting plasma glucose (a), fasting plasma insulin (b) and insulin sensitivity (c) over the 4 weeks after bilio-pancreatic diversion in morbidly obese individuals with type 2 diabetes. The changes in plasma glucose and insulin sensitivity during the first week are striking. The yellow areas represent normal ranges. FFM, fat-free mass. Data represent mean ± SE (n = 10) and are replotted from reference [6] with permission
The euglycaemic–hyperinsulinaemic clamp is usually assumed to reflect muscle insulin sensitivity [8]. This is because elevations in plasma insulin levels rapidly bring about complete suppression of glucose production by the liver. Under these circumstances, the rate at which glucose is infused directly reflects the rate at which muscle tissue is stimulated to take up glucose. However, it has been known for some time that glucose production by the liver is not completely suppressed during the clamp in obese individuals [9]. Thus, this method underestimates glucose uptake by muscle, because the continuing production of glucose by the liver is ignored. The relevance of this for understanding pathogenesis has been elegantly demonstrated by Petersen and colleagues [10]. Figure 3 shows that the marked increase in glucose requirements during euglycaemic hyperinsulinaemia is almost entirely explained by greater suppression of liver glucose production, and that the total uptake of glucose by muscle is essentially unchanged. This figure also illustrates that the change in liver response to insulin represents a remarkable normalisation of insulin suppression of glucose production by the liver. This observation focuses attention upon the early changes in the liver during the onset of energy restriction.

Physiological changes during 8% weight loss in obese individuals with type 2 diabetes compared with matched controls. The improvement in insulin sensitivity during the euglycaemic–hyperinsulinaemic clamp (a) would conventionally be interpreted as an improvement in the insulin sensitivity of muscle. However, determination of muscle glucose uptake during the clamps demonstrates no significant change (b), in sharp contrast to the simultaneous normalisation of the suppression of hepatic glucose production (c). The early improvement in glucose homeostasis during weight loss is associated with changes in the hepatic rather than peripheral tissue response to insulin. HGP, hepatic glucose production; LBM, lean body mass; T2D, type 2 diabetes. Data represent mean ± SE (n = 8) and are replotted from reference [10] with permission
The changes in liver volume and liver fat content during a period of very low energy intake have been examined [11]. The study was undertaken to define the rate of change of liver volume. This is important as a reduction in liver size before bariatric surgery makes the procedure easier to perform and considerably decreases the complication rate. Over a 12 week period, during which participants ingested 1.88–2.85 MJ/day (450–680 kcal/day), liver volume decreased by 30%. However, 80% of the change occurred during the first 2 weeks. During weight loss there is a tight relationship between liver volume and liver fat content [12], and it appeared that intra-hepatic fat was mobilised more rapidly than visceral or subcutaneous fat. This suggestion that liver fat stores are called upon first at the onset of negative energy balance has been examined directly by two studies using magnetic resonance methodology.
Petersen and colleagues observed that a loss of 8% of body weight over 7 weeks was associated with an 81% decrease in hepatic fat [10]. It is clear that liver fat is mobilised within days of onset of hypoenergetic feeding. Hollingsworth and colleagues observed the changes that accompanied weight loss of 3 kg over the first 10 days of a low-energy diet [13]. Liver fat fell significantly from 9.4 ± 1.8% at baseline to 8.2 ± 1.1% at 3 days and 7.0 ± 1.3 at 10 days (normal liver fat content <5%), demonstrating that very early substantial mobilisation of this fat depot occurs during dietary restriction.
Overall, this early and major decrease in liver fat occurs at the same time as normalisation of hepatic insulin sensitivity and a fall in fasting blood glucose level. This is entirely as would be expected from knowledge of the relationship between the extent of hepatic steatosis and suppression of hepatic glucose production by insulin [14, 15]. The reason for the rapid return of beta cell sensitivity to glucose is less certain. Even in normal individuals, insulin secretion can be blunted by constant hyperglycaemia [16], and the removal of ‘glucotoxicity’ could be a partial explanation. In addition, it is known that fat accumulation in and around the pancreatic islets is directly associated with impaired beta cell function in humans [17]. In obese rodents, the onset of hyperglycaemia is preceded by a rapid increase in pancreatic fat [18], and the mechanism of impairment of insulin secretion during chronic exposure of islet beta cells to fatty acids has been shown to be marked suppression of pyruvate cycling, which normally occurs during hyperglycaemia [19]. It is possible that such ectopic fat around the islets could be mobilised with the same dynamics as intra-hepatic fat, and that the early phase of fasting relieves the beta cells of the inhibitory burden of locally released fatty acids and diacylglycerol.
Fois gras in man and beast
Obesity is strongly related to non-alcoholic fatty liver disease [20]. This is true in early life as well as in adulthood. An autopsy study has demonstrated a dose relationship between the degree to which children are overweight and the presence of fatty liver [21]. In adolescence, 30% of obese individuals have fatty liver [22] and the extent of liver fat accumulation is inversely proportional to habitual daily physical activity, both in type 2 diabetic and non-diabetic individuals [23]. But which comes first? Are individuals with fatty liver more prone to insulin resistance and weight gain, or do lifestyle factors initiate and underlie fatty liver?
Farmers are the supreme experts at the practical manipulation of nature. The production of fatty liver in ducks and geese has been honed over at least 2,500 years [24]. Currently, the birds are allowed increasing access to high-carbohydrate feed over a 12 week free-range period. During this phase, rapid weight gain occurs and the livers become fatty, enlarging to 50% of their final size. This is equivalent to severe fatty liver for man. In the final 12 days, gavage is used to administer a large, high-fat energy overload and exacerbate the hepatic fat excess. However, the first phase of fois gras production gives all the necessary clues to the genesis of fatty liver in man. Prolonged intake of energy in excess of requirement will bring about intra-hepatic triacylglycerol deposition.
The effect of modifying plasma insulin levels on fatty liver
As the predominant anabolic hormone of metabolism, insulin plays a major role in promoting the synthesis and storage of fatty acid/triacylglycerol. Under conditions of constant hyperinsulinaemia and a modest elevation of the plasma glucose level, fatty acid synthesis from glucose is accelerated [25]. The extent of portal hyperinsulinaemia will determine how much glucose is converted into fatty acid in the liver. This process will be significant when the 24 h energy intake exceeds expenditure over the same period [26, 27]. In groups of obese individuals, those with a higher plasma insulin level have markedly increased rates of de novo lipogenesis [26]. Newly synthesised triacylglycerol in the liver can be either oxidised, exported as VLDL or stored as hepatic triacylglycerol. As transport of fatty acid into mitochondria for oxidation is inhibited by malonyl-CoA, which is produced during lipogenesis [28], newly synthesised triacylglycerol is preferentially directed towards storage or export, thus increasing hepatic fat content and/or increasing the concentration of plasma VLDL triacylglycerol. From this biochemical perspective, increased insulin secretion in the presence of positive energy balance can explain the initiation of fatty liver. The process will be hastened in the presence of peripheral insulin resistance and hence higher plasma insulin levels.
The implication that the fatty liver in type 2 diabetes is not secondary to hyperglycaemia but is determined by portal hyperinsulinaemia can neatly be tested by examining the extent of fatty liver in type 1 diabetes. In this condition, hyperglycaemia coexists with portal insulin concentrations that are approximately 2.5-fold lower than normal, as there is no portal/peripheral gradient. Liver fat content in a group of 19 individuals with type 1 diabetes was reported to be one-third lower than that in matched controls [29]. This indicates that when the portal insulin concentration is low, liver fat content is low.
It is therefore important to examine the effect of decreasing insulin secretion. This may be achieved in four ways. First, hypoenergetic diets lead to lower insulin secretion rates, but in the context of negative energy balance it is not remarkable that liver fat content falls [30]. Second, increased physical activity increases muscle insulin sensitivity and decreases the plasma insulin level. The extent of habitual daily physical activity has been shown to be negatively correlated with the degree of hepatic steatosis, both in type 2 diabetic and non-diabetic groups [23], and this effect is additive to advice on weight loss [31]. Third, thiazolidinediones increase insulin sensitivity and decrease insulin secretion rates. These agents bring about a 54% decrease in liver fat content [32, 33]. Finally, use of subcutaneous insulin in type 2 diabetes brings about decreases in portal insulin delivery and liver fat [34].
Taken together, the biochemical and clinical trial information points to endogenously secreted insulin as the major promoter of hepatic steatosis. There is no reason to believe that hyperglycaemia per se promotes fat deposition in the liver, although its secondary effect will play a role in impaired glucose tolerance and early type 2 diabetes via increased insulin secretion rates and, hence, increased liver fat.
Assembling the time-course to type 2 diabetes
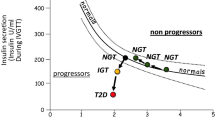
It has long been known that subnormal insulin sensitivity is the best predictor of the risk of developing type 2 diabetes. Recent studies have emphasised the primacy of extrahepatic insulin resistance [35]. In such individuals, raised plasma insulin levels compensate in terms of plasma glucose control, but set the scene for enhanced lipogenesis in the liver. The best available information suggests that excess fat deposition in the liver is uniformly present before the onset of classical type 2 diabetes [22, 23, 36–39]. Liver fat content is supra-normal in type 2 diabetes, with minimal crossover of range with normal individuals [23]. Such studies using magnetic resonance spectroscopy are able to quantify liver fat precisely, as are biopsy studies. A necessarily small biopsy series has shown all of a group of ten type 2 diabetic volunteers to have hepatic steatosis [40]. When ultrasound is used, only more severe degrees of steatosis are detected, and the prevalence of fatty liver will be underestimated. A large ultrasound-based study has shown 70% of people with type 2 diabetes to have fatty liver [39]. Nonetheless, even using ultrasound to classify individuals at baseline as either having or not having fatty liver, the prognostic power of predicting onset of type 2 diabetes within 8 years is impressive (Fig. 4) [36]. This study of 3,189 individuals with normal glucose tolerance at baseline showed a very low incidence of type 2 diabetes over 8 years if fatty liver had been excluded at baseline. Even when adjusted for age and BMI, the hazard ratio for diabetes was 5.5 (range 3.6–8.5).

Cumulative incidence of new-onset diabetes in men with previously normal glucose tolerance. At baseline, liver fat content was assessed by ultrasound, and the fatty liver group (red line) and non-fatty liver group (blue line) were prospectively followed. If fatty liver was not present at baseline, the risk of developing type 2 diabetes was very low, even though the method used for detection of fatty liver has low sensitivity and some individuals with fatty liver would have been included in the non-fatty liver group (log-rank test p < 0.001 for difference between the groups). Data are 6 monthly cumulative incidence rates for 3,135 and 9,744 person-years of follow-up for the fatty liver and non-fatty liver groups, respectively. Figure replotted from reference [36] with permission
In support of this, Suzuki demonstrated a temporal progression from weight gain to raised levels of liver enzymes, to hypertriglyceridaemia and, eventually, glucose intolerance [41]. It must be noted that the association of raised liver enzyme concentrations with fatty liver is strong between groups with and without type 2 diabetes but not absolute for diabetic individuals, who may exhibit an increase within the normal range. In a group of 84 children with biopsy-confirmed non-alcoholic fatty liver disease, most were insulin resistant and 12% already had abnormal glucose tolerance [42]. Certainly, weight loss can slow or halt the pathogenesis of type 2 diabetes. When a group of individuals with impaired glucose tolerance was followed over time, weight loss of only 1 kg decreased the incidence of diabetes by 20% [43].
The clearest prospective information of the time course to development of type 2 diabetes comes from the West of Scotland Coronary Prevention Study [38]. In a large group of men followed for 2 years, 9% developed diabetes during the period of observation. Two years before the onset of type 2 diabetes, this subgroup was characterised by having elevated levels of plasma triacylglycerol and alanine aminotransferase (Fig. 5). There was a steady rise in the level of this liver enzyme in the run-up to the time of diagnosis, suggesting that the burden on the liver cells was steadily increasing. In contrast, although fasting plasma glucose concentration was marginally above that of the group who did not develop diabetes, it remained fairly constant until the few months immediately prior to the diagnosis of diabetes, when a rapid rise occurred. There was not a gradual decompensation with a linear increase in fasting plasma glucose, as is often assumed. In established type 2 diabetes, hepatic glucose production in everyday life remains relatively unsuppressed [44] and postprandial plasma insulin levels are elevated by approximately 2.5-fold [45].

Changes observed during follow-up in the West of Scotland Coronary Prevention Study. Individuals who developed type 2 diabetes (red lines) during the study were compared with those who remained normoglycaemic (blue lines). It can be seen that they were characterised as having raised plasma glucose levels for at least 2 years (a). Over this period, plasma alanine aminotransferase (ALT) steadily rose (b). A relatively sudden increase in plasma glucose then occurred. Triacylglycerol levels were elevated but relatively constant from at least 2 years before diagnosis (c). Data replotted from reference [38] with permission
The difference in the time-course of change in pathogenic factors during the natural history of type 2 diabetes and during its reversal by bariatric surgery deserves comment (see text box: Steps in the pathogenesis of type 2 diabetes). Fatty liver becomes established years before the onset of type 2 diabetes. It appears necessary but not sufficient for its development. The subsequent period of years of over-exposure of the islets to an elevated plasma triacylglycerol level and a modest increase in the plasma glucose level is necessary to precipitate beta cell decompensation. Individuals are likely to exhibit different degrees of islet sensitivity in response to the adverse effects of these factors. However, in established diabetes, sudden mobilisation of the liver fat allows normal physiology to be reinstated within days, provided that long-term damage to the beta cells has not yet occurred. The published data suggest that 2–3 years of type 2 diabetes does not usually produce irreversible beta cell damage.

By assimilating the knowledge gained from studies of the reversal of type 2 diabetes into the prospective observation of the development of the condition a clear temporal pattern, the twin cycle hypothesis, can be described (Fig. 6). Energy imbalance allows the onset of fatty liver. Peripheral insulin resistance, either as a familial trait or as a result of lifestyle factors, will expedite this process because of hyperinsulinaemia. The increased liver fat brings about a degree of hepatic insulin resistance that involves failure to suppress fasting glucose production. The degree of hepatic steatosis will be exacerbated by the portal hyperinsulinaemia, and a raised plasma triacylglycerol level signals the presence of excess liver fat [46]. Exposure of the beta cells to excess fatty acids derived from circulating and locally deposited triacylglycerol suppresses glucose-mediated insulin secretion [17–19].

The twin vicious cycles of type 2 diabetes. During the chronic intake of more energy than is expended each day, fat will accumulate in the liver. As this process is promoted by insulin, individuals with a degree of insulin resistance (determined by family or lifestyle factors) will accumulate liver fat more readily than others. As the liver fat increases, the process of glucose production by the liver becomes less sensitive to suppression by insulin, plasma glucose tends to rise and basal insulin secretion rates rise. This forms vicious cycle A (in blue). The increased liver fat will increase secretion of triacylglycerol from the liver. All tissues will be exposed to more triacylglycerol than required, but the pancreatic islets are susceptible to local triacylglycerol accumulation. The red arrows represent proximal steps to the onset of type 2 diabetes. The post-meal response to glucose becomes blunted, and this is exacerbated by the modestly raised plasma glucose. Vicious cycle B (in red) causes impaired glucose regulation, and eventually the fatty acid and glucose inhibitory effects on the islets reach a threshold, leading to a relatively sudden onset of clinical diabetes. Other processes may contribute to beta cell dysfunction in addition to the fatty acid effect, contributing to the heterogeneity in clinical phenotype
The twin vicious cycles then grind onwards. Raised intrahepatic triacylglycerol leads to decreased suppression of hepatic glucose output, which increases basal insulin secretion and further increases intrahepatic triacylglycerol. Separately but sequentially, supra-normal plasma glucose and fatty acid delivery causes decreased postprandial beta cell function and decreased glucose-responsive insulin secretion, leading to further excursions in plasma glucose. At a threshold determined by individual factors, beta cell decompensation occurs and the plasma glucose level rises relatively rapidly, leading to biochemical or symptomatic presentation of type 2 diabetes. Over several years of diabetes, the cumulative effects of excessive glucose and fatty acid exposure bring about a degree of impairment of beta cell function that is irreversible. There is continued decline for as long as the causative factors of energy overload and raised liver fat persist. At some point, insulin therapy is required for reasonable control of plasma glucose.
Conclusions
The essential time sequence of the pathogenesis of type 2 diabetes is now evident. Muscle insulin resistance determines the rate at which fatty liver progresses, and ectopic fat deposition in the liver and islets underlies the related dynamic defects of hepatic insulin resistance and beta cell dysfunction. These defects are capable of dramatic reversal under hypoenergetic feeding conditions, completely in early diabetes and to a worthwhile extent in more established disease.
It is now clear that we are dealing with an ecological phenomenon, the aetiology of which demonstrably lies in positive energy balance. Nowhere is this more clearly illustrated than in the Pima Indians, a population in which the prevalence of type 2 diabetes has risen from 0% to 40% over half a century, in step with a change from subsistence farming to inactivity and surplus food [47]. When our species finds itself living under conditions of exercise dearth and energy excess, the mechanisms that evolved over millennia to allow populations to withstand famines become maladaptive.
Two important questions emerge: Does the species have the ability to adjust its environment and avoid the primary causes of type 2 diabetes? What distinguishes those individuals who appear resistant to the metabolic challenge, remaining normoglycaemic in the face of little exercise and much eating?
References
Hall RA, Hall MR (eds) (1962) Unpublished scientific papers of Isaac Newton. Cambridge University Press, Cambridge, pp 11–14
Dixon JB, O’Brien PE, Playfair J et al (2008) Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA 299:316–323
Buchwald H, Avidor Y, Braunwald E et al (2004) Bariatric surgery: a systematic review and meta-analysis. JAMA 292:1724–1737
Pories WJ, Caro JF, Flickinger EG, Meelheim HD, Swanson MS (1987) The control of diabetes mellitus (NIDDM) in the morbidly obese with the Greenville Gastric Bypass. Ann Surg 206:316–323
Camastra S, Manco M, Mari A et al (2007) Beta-cell function in severely obese type 2 diabetic patients: long-term effects of bariatric surgery. Diabetes Care 30:1002–1004
Guidone C, Manco M, Valera-Mora E et al (2006) Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 55:2025–2031
Gibney ER, Murgatroyd P, Wright A, Jebb S, Elia M (2003) Measurement of total energy expenditure in grossly obese women: comparison of the bicarbonate–urea method with whole-body calorimetry and free-living doubly labelled water. Int J Obes Relat Metab Disord 27:641–647
DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP (1981) The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterisation. Diabetes 30:1001–1007
Firth R, Bell P, Rizza R (1987) Insulin action in non-insulin dependent diabetes mellitus: the relationship between hepatic and extrahepatic insulin resistance and obesity. Metabolism 36:1091–1095
Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI (2005) Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54:603–608
Colles SL, Dixon JB, Marks P, Strauss BJ, O’Brien PE (2006) Preoperative weight loss with a very-low-energy diet: quantitation of changes in liver and abdominal fat by serial imaging. Am J Clin Nutr 84:304–311
Dixon JB, Bhathal PS, O’Brien PE (2001) Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 121:91–100
Hollingsworth KG, Abubacker MZ, Joubert I, Allison ME, Lomas DJ (2006) Low-carbohydrate diet induced reduction of hepatic lipid content observed with a rapid non-invasive MRI technique. Br J Radiol 79:712–715
Seppala-Lindroos A, Vehkavaara S, Hakkinen AM et al (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 87:3023–3028
Ryysy L, Hakkinen A-M, Goto T et al (2000) Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 49:749–758
Ferner RE, Ashworth L, Tronier B, Alberti KGMM (1986) Effects of short-term hyperglycaemia on insulin secretion in normal humans. Am J Physiol 250:E655–E661
Tushuizen ME, Bunck MC, Pouwels PJ et al (2007) Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 30:2916–2921
Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH (1994) β-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte–β-cell relationships. Proc Natl Acad Sci USA 91:10878–10882
Boucher A, Lu D, Burgess SC et al (2004) Biochemical mechanism of lipid-induced impairment of glucose-stimulated insulin secretion and reversal with a malate analogue. J Biol Chem 279:27263–27271
Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S (2005) Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 42:44–52
Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C (2006) Prevalence of fatty liver in children and adolescents. Pediatrics 118:1388–1393
Perseghin G, Bonfanti R, Magni S et al (2006) Insulin resistance and whole body energy homeostasis in obese adolescents with fatty liver disease. Am J Physiol Endocrinol Metab 291:E697–E703
Perseghin G, Lattuada G, De Cobelli F et al (2007) Habitual physical activity is associated with intrahepatic fat content in humans. Diabetes Care 30:683–688
Toussaint-Samat M (1994) History of food. Blackwell, Oxford, pp 424–433
Sidossis LS, Stuart CA, Shulman GI, Lopaschuk GD, Wolfe RR (1996) Glucose plus insulin regulate fat oxidation by controlling the rate of fatty acid entry into the mitochondria. J Clin Invest 98:2244–2250
Schwarz JM, Linfoot P, Dare D, Aghajanian K (2003) Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr 77:43–50
Schwarz JM, Neese RA, Turner S, Dare D, Hellerstein MK (1995) Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and whole-body fuel selection. J Clin Invest 96:2735–2743
McGarry JD, Mannaerts GP, Foster DW (1977) A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest 60:265–270
Perseghin G, Lattuada G, De Cobelli F et al (2005) Reduced intrahepatic fat content is associated with increased whole-body lipid oxidation in patients with type 1 diabetes. Diabetologia 48:2615–2621
Nobili V, Marcellini M, Marchesini G et al (2007) Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care 30:2638–2640
Sreenivasa Baba C, Alexander G, Kalyani B et al (2006) Effect of exercise and dietary modification on serum aminotransferase levels in patients with nonalcoholic steatohepatitis. J Gastroenterol Hepatol 21:191–198
Belfort R, Harrison SA, Brown K et al (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355:2297–2307
Ravikumar B, Gerrard J, Dalla Man C et al (2008) Pioglitazone decreases fasting and postprandial endogenous glucose production in proportion to decrease in hepatic triglyceride content. Diabetes doi:10.2337/db07-1828
Juurinen L, Tiikkainen M, Hakkinen AM, Hakkarainen A, Yki-Jarvinen H (2007) Effects of insulin therapy on liver fat content and hepatic insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab 292:E829–E835
Bock G, Chittilapilly EG, Basu R et al (2007) Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose. Role of increased rates of gluconeogenesis. Diabetes 56:1703–1711
Shibata M, Kihara Y, Taguchi M, Tashiro M, Otsuki M (2007) Nonalcoholic fatty liver disease is a risk factor for type 2 diabetes in middle-aged Japanese men. Diabetes Care 30:2940–2944
Cali AM, Zern TL, Taksali SE et al (2007) Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect proatherogenic state. Diabetes Care 30:3093–3098
Sattar N, McConnachie A, Ford I et al (2007) Serial metabolic measurements and conversion to type 2 diabetes in the West of Scotland Coronary Prevention Study: specific elevations in alanine aminotransferase and triglycerides suggest hepatic fat accumulation as a potential contributing factor. Diabetes 56:984–991
Targher G, Bertolini L, Padovani R et al (2007) Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 30:1212–1218
Turnbull AJ, Mitchison HC, Peaston RTP et al (1997) The prevalence of hereditary haemochromatosis in a diabetic population in north-east England. Quart J Med 90:271–275
Suzuki A, Angulo P, Lymp J et al (2005) Chronological development of elevated aminotransferases in a nonalcoholic population. Hepatology 41:64–71
Nobili V, Marcellini M, Devito R et al (2006) NAFLD in children: a prospective clinical-pathological study and effect of lifestyle advice. Hepatology 44:458–465
Rasmussen SS, Glumer C, Sandbaek A, Lauritzen T, Borch-Johnsen K (2008) Determinants of progression from impaired fasting glucose and impaired glucose tolerance to diabetes in a high-risk screened population: 3 year follow-up in the ADDITION study, Denmark. Diabetologia 51:249–257
Singhal P, Caumo A, Carey PE, Cobelli C, Taylor R (2002) Regulation of endogenous glucose production after a mixed meal in type 2 diabetes. Am J Physiol Endocrinol Metab 283:E275–E283
Carey PE, Halliday J, Snaar JEM, Morris PG, Taylor R (2003) Direct assessment of muscle glycogen storage after mixed meals in normal and type 2 diabetic subjects. Am J Physiol 284:E286–E294
Adiels M, Westerbacka J, Soro-Paavonen A et al (2007) Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 50:2356–2365
Bennett PH (1999) Type 2 diabetes among the Pima Indians of Arizona: an epidemic attributable to environmental change. Nutr Rev 57:S51–S54
Acknowledgements
This review is based on a lecture given by the author at the Diabetes UK Annual Professional Meeting, April 2008. The concepts were developed during research supported by Wellcome Programme Grant 073561.
Duality of interest
The author has received lecture or consultancy fees unconnected with this review from Merck Sharp and Dohme, Novartis, Eli Lilly, NovoNordisk, Takeda and Servier Laboratories.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Taylor, R. Pathogenesis of type 2 diabetes: tracing the reverse route from cure to cause. Diabetologia 51, 1781–1789 (2008). https://doi.org/10.1007/s00125-008-1116-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1116-7