
Abstract
Aims/hypothesis
In previous studies we have shown a significant involvement of the growth hormone (GH)–IGF axis in animal models of type 1 diabetes mellitus, but the role of this endocrine system in type 2 diabetes mellitus is less well characterised. We therefore examined the endocrine and renal GH–IGF axis changes in db/db mice, a model of type 2 diabetes mellitus and nephropathy.
Materials and methods
Obese and lean animals were followed, beginning at hyperglycaemia onset, for 4 weeks. Albuminuria and creatinine clearance, as well as kidney and glomerular morphology were assessed. Tissue protein levels were determined by western blotting and mRNA levels by RT-PCR.
Results
Serum GH and IGF1 levels immediately prior to killing were decreased and liver mRNA levels of insulin-like growth factor binding protein 1 (Igfbp1) were increased in obese animals. Kidney weight was increased in obese animals, associated with hyperfiltration, albuminuria and glomerular hypertrophy. Administration of a somatostatin analogue (PTR-313) did not improve any of these parameters of diabetic renal involvement. Renal Igf1 mRNA was decreased and renal Igfbp1 mRNA and protein were significantly increased in obese animals. Renal insulin-driven levels of phosphorylated forkhead box O1 (FOXO1) were decreased in obese animals.
Conclusions/interpretation
Diabetic db/db mice show significant renal changes (and IGFBP1 renal accumulation), similar to the findings in models of type 1 diabetes mellitus. A decreased signalling through the insulin receptor and decreased FOXO1 phosphorylation may allow Igfbp1 gene transcription. These renal changes are associated with low circulating IGF1 and GH levels and unchanged hepatic growth hormone receptor expression, unlike the condition in type 1 diabetes mellitus. This suggests that further GH inhibition to modulate renal complications in type 2 diabetes mellitus is not indicated.
Similar content being viewed by others

Introduction
Diabetic nephropathy (DN) is a common renal complication of both type 1 and 2 diabetes and the most common single cause of end-stage renal disease in the Western world. In addition, type 2 diabetic patients fare poorly on dialysis and have an excess mortality. While the pathogenesis of DN in type 1 diabetic patients has been better characterised, its pathogenesis in type 2 diabetic patients is less well understood [1]. Type 2 diabetes is characterised by insulin resistance and accumulation of fat in parenchymal organs. Many of the pathogenic theories on causes of DN in type 2 diabetes relate to chronic hyperglycaemia and its associated toxic effects on target organs [2]. In addition, insulin resistance is an early and pivotal change [3]. Changes in the other endocrine regulatory pathways related to glucose homeostasis, such as glucagon, cortisol, epinephrine and growth hormone (GH), are less well defined.
Nephropathy in type 1 diabetic patients has been better characterised over the past 10–15 years [4], including the role played by different growth factors, such as GH and IGF1. We have previously described an increase, similar to that in human disease, in circulating GH levels in the nonobese diabetic mouse model of type 1 diabetes mellitus. This was associated with decreased liver growth hormone receptor (Ghr, previously known as Ghr/bp) mRNA and decreased GH binding to liver membranes [5], suggesting a state of GH resistance. In addition, we have shown that renal cortical Ghr (i.e. Ghr/bp) mRNA levels and GHR (i.e GHR/BP) immunoreactivity are elevated in streptozotocin-induced type 1 diabetic rats at a late stage (1 month after manifestation diabetes) [6], in spite of an apparent opposite endocrine effect (decrease in liver GHR). In another study we showed that renal hypertrophy is accompanied by an increase in extractable IGF1 protein levels in the non-obese diabetic mouse up to 1 month after the onset of diabetes mellitus [7], suggesting a role for IGF1 in the development of DN. This increase in renal IGF1 protein was accompanied by an actual decrease in kidney Igf1 mRNA levels. Renal insulin-like growth factor binding protein 1 (IGFBP1) levels were elevated [8], hinting at a local trapping of circulating IGF1 by IGFBP1. Subsequently, we have shown that a GHR antagonist had salutary effects on DN markers, in association with a concomitant inhibition of renal IGF1 protein accumulation, but without affecting either somatic growth or circulating GH and IGF1 levels [9]. In contrast, administration of GH to streptozotocin-induced diabetic rats exacerbated DN in association with a marked increase in renal IGFBP1 message [10], adding another aspect to the pathogenic role of GH in DN. Thus, inhibition of GH action may have therapeutic implications for type 1 diabetic patients with DN.
The purpose of this study was to examine the endocrine GH-IGF changes in type 2 diabetes mellitus, as well as the early kidney changes in this disease, using a representative animal model, the db/db mouse.
Materials and methods
Animal experimentation
We purchased 4-week-old female db/db mice from Harlan Laboratories (Bicester, UK). Animal breeding complied with the National Institutes for Health Guide for the Care and Use of Laboratory Animals. The local institutional review committee approved the study protocol. Mice were still normoglycaemic at that age and became diabetic at age 6 weeks, when the experiment was started. Animals were housed in standard laboratory cages and had free access to normal mouse chow and unlimited tap water. The onset of diabetes mellitus was determined by the appearance and persistence of glycosuria, checked twice a week with chemstrips (Ketostix; Bayer, Newbury, UK). When the urine glucose test was positive, tail capillary glucose blood samples were examined with a glucometer (Elite; Bayer Diagnostics, Puteaux, France). Diabetes mellitus was diagnosed when blood glucose levels were above the normal values for these mice (above 9.9 mmol/l) on two consecutive days. The second day of persistent hyperglycaemia was counted as day 1 of diabetes mellitus. Lean heterozygous and nondiabetic db/N mice were used as controls. All animals were killed 4 weeks after the onset of diabetes mellitus, as previously described [11]. Body weight was measured twice weekly. Water intake was recorded every other day. Urine was collected for 24 h, using metabolic cages, on the day prior to killing. A standardised method of GH stimulation and secretion was applied [12], in which mice were anaesthetised with intraperitoneal pentobarbital (50 mg/kg) and blood was drawn from the retrobulbar plexus at least 5 min after injection. The serum was separated and frozen at −20°C for later measurements of glucose, IGF1 and GH. Immediately after killing, the right kidney and the liver were carefully removed and immediately frozen in liquid nitrogen and then stored at −70°C. A coronal 2-mm slice from the midportion of the left kidney was separated and fixed in a 4% paraformaldehyde solution for histomorphological assessment.
A subset of experiments was performed using a somatostatin analogue (PTR-313: Peptor, Rehovot, Israel) previously described by us as having protective effects on the kidney in a model of type 1 diabetes mellitus [11]. The agent was provided daily over 4 weeks by s.c. injections (1 mg kg−1 day−1) to diabetic animals, with control animals receiving the equivalent amount of saline. Animals were killed as previously described.
Immunoassays for serum GH
Serum GH was measured by RIA, as previously described [13], using a specific polyclonal antibody. The sensitivity of the method was 0.04 ng murine GH per tube; the CVs for tested sera at 13.9 and 201.1 μg/l were 13.6 and 5.9%, respectively. All sera were run in the same assay.
Immunoassays for serum IGF1
Serum IGF1 was measured as previously described [13], by the functional separation method, in which excess IGF2 blocks the interference of IGF binding proteins. The test sensitivity was 0.02 ng IGF1 per tube; the intra-assay CVs for sera with IGF1 levels ranging between 80, 212 and 412 ng/ml were 8.9, 2.9 and 9.6%, respectively. All sera were run in the same assay.
Urine albumin excretion
The urine samples, which were collected prior to killing, were stored at −20°C until assayed. A modified ELISA determined the urinary albumin concentration in the urine samples as previously described [13]. Intra- and inter-assay CVs were less than 5 and 10%, respectively. In the present study, all urine samples were run in one assay, in duplicates.
Immunohistochemistry
For immunohistochemistry studies, paraffin sections (4 μm) were deparaffinised in xylene, hydrated in gradual ethanol concentrations and reacted for 1 h at room temperature with a monoclonal antimouse IGF1 antibody (Upstate, Charlottesville, VA). This was followed by incubation with an appropriate biotinylated second antibody for 30 min and with biotin avidin-peroxidase complex for 30 min (Vectastain ABC kit; Vector, Burlingame, CA). The reaction was developed with 3,3-diaminobenzidine as a substrate. The intensity of the staining was evaluated under light microscopy in a semiquantitative way (+1 to +3) for the different glomerular areas.
Estimation of glomerular volume
The mean glomerular tuft volume (V G) was determined from the mean glomerular cross-sectional area (A G) by light microscopy, as previously described [14]. AG was determined as the average area of a total of 80 to 100 glomeruli (tuft omitting the proximal tubular tissue within the Bowman capsule); VG was calculated as: (VG = β/κ × [A G]3/2), where β = 1.38 is the shape coefficient for spheres (the idealised shape of glomeruli) and κ = 1.1 is a size distribution coefficient.
mRNA studies
Total RNA was prepared from frozen tissues by the standard methods. Evaluation of renal and liver Igf1 and Igfbp1 mRNAs was performed using the RT–PCR method, as previously described [10].
Igf1, Igfbp1 and β actin cDNA were amplified by PCR using specific primers. Igf1 sense: GGACCAGAGACCCTTTGCGGGG; Igf1 antisense: GGCTGCTTTTGTAGGCTTCAGTGG; Igfbp1 sense: TCCTCTGTCATCTCTGGGCTCTCA; Igfbp1 antisense: CGCCACGAGCACCTTGTTCA; β actin sense: GACGAGGCCCAGAGCAAGAG; β actin antisense: GGGCCGGACTCATCGTACTC. Every experiment was amplified with at least two different numbers of cycles to ensure that amplification was at the exponential phase of PCR. We found that 25–30 cycles for Igf1 and Igfbp1 and 20–25 cycles for β actin were in this range. Under these conditions we also found a linear dose response of the PCR product to increasing doses of cDNA. PCR products were quantified densitometrically using Fluorchem software (Alpha-Innotech, San Leandro, CA). To correct for differences in loading, we corrected densitometric values of Igf1 and Igfbp1 cDNAs with corresponding values of β-actin cDNA and calculated the Igf1:β-actin and Igfbp1:β-actin ratios.
Western immunoblot analysis
Western immunoblot analysis was performed from kidney and liver samples as previously described [10]. For the detection of kidney phosphorylated forkhead box O1 (FOXO1) (Ser256) and liver GHR, 200-μg portions of sample protein were loaded in each gel lane, subjected to 10% SDS polyacrylamide gel and electroblotted into nitrocellulose membranes. Blots were blocked for 1 h in TBS buffer containing 5% non-fat dehydrated milk, followed by overnight incubation with polyclonal antibody against IGFBP1 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal antibody against phospho-FOXO1 (Ser256) (Cell Signaling Technology, Beverly, MA) and polyclonal antibody against GHR (Santa Cruz) diluted in TBS containing 5% dried milk or diluted in TBS containing 5% BSA (phospho-FOXO1). Protein expression was quantitated densitometrically using Fluorchem software (Alpha-Innotech).
Other assays
During animal breeding blood glucose levels were determined with a glucometer (Bayer Diagnostics); the glucose oxidase method was used when the animals were killed. Urine creatinine values were assessed simultaneously to calculate creatinine clearance as an indicator of glomerular filtration rate.
Statistical analysis
The t test was used for the comparison of continuous variables. A p value of less than 0.05 was considered significant. Means are given as ± SEM.
Results
Growth and metabolic parameters
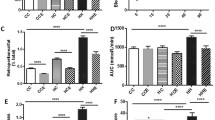
Body weight in obese animals when killed was significantly elevated (48 ± 3 g) in comparison with the lean controls (23 ± 2 g). Mean serum glucose levels in obese animals during the study were 25.3 ± 2.7 vs 6.4 ± 0.55 mmol/l in lean animals. No animals died in the course of the experiment. Serum GH levels at the time of killing were decreased in the obese group (16 ± 2 vs 26 ± 5 μg/l in lean animals; p < 0.05) (Fig. 1a). A similar decrease was seen for circulating IGF1 (237 ± 7 vs 433 ± 28 ng/ml in obese vs lean animals; p < 0.05) (Fig. 1b). Hepatic Ghr mRNA levels were unchanged in obese animals (90 ± 8% vs that of lean animals). Hepatic GHR protein content was analysed by western blot and found to be unchanged (Fig. 2). Hepatic Igfbp1 mRNA was increased in obese animals (243 ± 15% vs that of lean animals; p < 0.05) (Fig. 3).

Serum GH (a) and IGF1 (b) in the control lean (C) and obese diabetic (D) mice. Values are mean±SEM; n = 8 in each group. *p < 0.05

Western blot analysis (a) of liver GHR content, using 200-μg tissue lysates, in control lean (C ) and obese diabetic (D) mice. Total lysates were separated on SDS-PAGE, followed by immunoblot analysis, using anti-GHR antibody. The 130-kDa band corresponds to GHR. The blot shown is representative of five independent experiments. b Densitometry analysis (represented in arbitrary densitometric units) summarising the five independent experiments

Liver Igfbp1 mRNA levels. a Representative ethidium bromide stains of liver Igfbp1 mRNA, using RT-PCR. The two pairs of lanes depicted per experimental group each represent duplicates from two animals. b Densitometric analysis of five separate RT-PCR reactions (representing five different animals per group). The Igfbp1:β-actin ratio is represented in arbitrary densitometric units. *p < 0.05 vs C
Kidney weight and renal functional studies
Kidney weights were increased in the obese group (169 ± 3 vs 134 ± 3 mg in lean animals; p < 0.05) (Fig. 4a). Creatinine clearance was calculated from 24 h urine collection in all animals, using metabolic cages prior to sacrifice, and served as a marker of glomerular filtration rate (Fig. 4b). It was markedly elevated in the obese animals in comparison with controls (3.5 ± 0.3 vs 1.7 ± 0.4 μl min−1 g−1 body weight in lean animals, p < 0.05). Albuminuria was also measured from the 24 h urine collections (Fig. 4c). A similar trend to that described for creatinine clearance was observed for albuminuria, i.e. a marked increase in obese animals (0.76 ± 0.01 vs 0.1 ± 0.02 μg 24 h−1 g−1 bodyweight in lean animals, i.e. 760 ± 90% that of lean animals; p < 0.05). Glomerular volume increased significantly in the obese group (4.3 ± 0.4 × 105 vs 2.1 ± 0.1 × 105 μm3 in the lean group, i.e. 205 ± 20% that of lean animals; p < 0.05) (Fig. 4d).

Mean absolute kidney weight (KW) (a), creatinine clearance (b), urinary albumin excretion (c) and glomerular volume (d) at 4 weeks in the control lean (C) and obese diabetic (D) groups. BW bodyweight. Values are mean±SEM; n = 8 in each group. *p < 0.05 vs C
Renal IGF1 and IGFBP1
Kidney Igf1 mRNA levels were significantly decreased at 4 weeks after the onset of diabetes in the obese group, compared with the control groups (70 ± 7% that of lean animals; p < 0.05) (Fig. 5). Renal immunostainable IGF1 showed no clear differences between the experimental groups. Steady-state renal Igfbp1 mRNA levels were increased in the obese group (142 ± 5% that of lean animals; p < 0.05) (Fig. 6a,b). A similar pattern was observed for IGFBP1 protein content, as assessed by western blot analysis (241 ± 39% that of lean animals) (Fig. 6c,d). Renal phosphorylated FOXO1 levels were decreased in diabetic animals (52 ± 9% that of lean animals; p < 0.005) (Fig. 7).

Representative ethidium bromide stain of renal Igf1 mRNA (a), using RT-PCR. The two pairs of lanes depicted per experimental group each represent duplicates from two animals. Amplification of β-actin mRNA was done, using the same amount of initial total RNA. b Densitometric analysis of five separate RT-PCR reactions (representing five different animals per group). The renal Igf1:β-actin ratio is presented as arbitrary units. *p < 0.05 vs C

Representative ethidium bromide stain (a) of renal Igfbp1 mRNA from control (C) and obese diabetic (D) mice, using RT-PCR. The two pairs of lanes depicted per experimental group each represent duplicates from two animals. Amplification of β-actin mRNA was done using the same amount of initial total RNA. b Densitometric analysis of five separate RT-PCR reactions (representing five different animals per group). The renal Igfbp1:β-actin ratio is represented in arbitrary densitometric units. c Western blot analysis of kidney IGFBP1 content, using 200-μg tissue lysates, in control lean and obese diabetic mice. Total lysates were separated on SDS-PAGE, followed by immunoblot analysis, using anti-IGFBP1 antibody. The 38-kDa “doublet” band corresponds to IGFBP1. The blot shown is representative of five independent experiments. d Densitometry analysis summarising the five independent experiments. Data are expressed in arbitrary densitometric units. *p < 0.05 vs C

a Western blot analysis of kidney phosphorylated (p)-FOXO1 content, using 200-μg tissue lysates, in control lean (C) and obese diabetic (D) mice. Total lysates were separated on SDS-PAGE, followed by immunoblot analysis, using anti-p-FOXO1 antibody. The 75-kDa band corresponds to p-FOXO1. The blot shown is representative of five independent experiments. b Densitometry analysis summarising the five independent experiments. Data are expressed in arbitrary densitometric units. *p < 0.05 vs C
Intervention with the somatostatin analogue (PTR 313) decreased circulating IGF1 levels in control animals injected with the agent (337 ± 10 vs 434 ± 28 ng/ml in lean animals, p < 0.05), showing its basic ability to act as somatostatin in these animals too. However, no significant effect by this agent (measured by body weight, circulating GH, circulating IGF1, kidney weight, creatinine clearance and albuminuria) could be shown when it was provided to the diabetic animals.
Discussion
This study emphasises the major differences between type 1 and type 2 diabetes mellitus regarding both its GH–IGF endocrine and local–renal changes. An apparently similar combination of endocrine GH–IGF changes (low circulating IGF1 and GH levels) was previously described in type 2 diabetic humans [15]. The unchanged hepatic GHR levels in the model used in our study are also very different from their marked decrease in type 1 diabetes mellitus [4], again similar to indirect observations in humans (unchanged serum GHR levels, a surrogate marker of liver GHR production) [16]. The exact effects of type 2 diabetes mellitus on GHR signalling in the model used by us have not been fully elucidated yet. In vitro studies suggest that hepatic GH resistance may develop in response to chronic hyperinsulinaemia [17]. The result of these changes may be a decrease in circulating IGF1 and IGF1 action, which may by itself be a mechanism for decreased beta cell survival in long-term type 2 diabetes mellitus [18, 19]. Free serum IGF1 levels (which depend on levels of the significant IGF binding proteins) were not measured in our study. Increased IGFBP1 levels (mainly its phosphorylated and highly IGF1-bound fraction) may prevent the mitogenic actions of IGF1 on blood vessels and are inversely related to adverse outcomes of type 2 diabetes mellitus, such as hypertension or macrovascular disease [20]. In our experiment, given the increased levels of both renal and hepatic IGFBP1 and the decrease in total IGF1, free IGF1 was expected to be low.
The main endocrine difference between type 1 and type 2 models is the decrease in serum GH in obese db/db mice, whereas GH levels increase in type 1 diabetes mellitus [5]. There are significant similarities between the metabolic syndrome and untreated GH deficiency in adults, including abdominal/visceral obesity and insulin resistance. Abdominal/visceral distribution of adipose tissue is associated with endocrine disturbances, including increased activity of the hypothalamic–pituitary–adrenal axis and a blunted secretion of GH and sex steroids [21], partly explaining the low concentrations of circulating IGF1. GH replacement in men with abdominal obesity can diminish the negative metabolic consequences of visceral obesity [22], suggesting that low levels of this hormone are important for the metabolic aberrations in obese type 2 diabetic patients [23]. In our study we showed that the addition of a GH antagonist (somatostatin) did not affect any of the renal parameters (hypertrophy, hyperfiltration and albuminuria), contrary to previous reports on beneficiary effects of GH antagonists in type 1 diabetes mellitus [13], thus supporting the concept that no further suppression of GH is indicated in type 2 diabetes mellitus.
In spite of these endocrine dissimilarities between models of type 1 and type 2 diabetes, local renal changes in type 2 diabetes mellitus are similar to those found in type 1 diabetes mellitus (renal and glomerular hypertrophy, hyperfiltration, albuminuria) [24]. In both models changes can be seen after a similar short period of hyperglycaemia. The local renal IGF changes associated with the type 2 diabetic phenotype (decreased renal Igf1 mRNA and increased renal IGFBP1 accumulation) are still similar to those seen in models of type 1 diabetes.
The lack of a difference in immunostaining for renal IGF1 could be due to the low sensitivity of this semi-quantitative method. We have previously shown that renal IGFBP1 levels were also elevated in a model of nephropathic type 1 diabetes mellitus [7]. In that model, the change in renal (and serum) IGFBP1 was ascribed to decreased serum insulin, which is known to be a potential regulator of IGFBP1 gene expression [25]. However, we have now shown that renal IGFBP1 is also increased in our model of type 2 diabetes mellitus, where circulating GH is decreased and hyperinsulinaemia has been described [26]. This potential contradiction may be explained by recently discovered pathways of insulin resistance involving the downregulation of the transcription factor FOXO1 [27], a known insulin-controlled positive regulator of IGFBP1 transcription [28]. Insulin inhibits FOXO1 activity by phosphorylating it via v-akt murine thymoma viral oncogene homologue 1 (AKT1), thus preventing its entrance into the nucleus. FOXO1 inhibits hepatic gluconeogenesis, in addition to its involvement in the inhibition of PDX1 in pancreatic beta cells, potentially contributing to beta cell dysfunction [29]. In our study we showed a decrease in levels of phosphorylated FOXO1, thus enabling the unphosphorylated FOXO1 fraction to enter into the nucleus and activate target genes including Igfbp1. The correlation between IGFBP1 expression and the effects of insulin has also been shown in humans [19]. Therefore, a state of insulin resistance as seen in type 2 diabetes mellitus would be expected to increase IGFBP1 expression through the regulatory effects of FOXO1. Although original studies implied that IGFBP1 has an independent effect on cells via its binding to the integrin receptor [30], most studies using different models have shown that IGFBP1 has mainly inhibitory effects on IGF1 actions [31, 32]. Thus, IGFBP1 accumulation may actually inhibit the already decreased Igf1 expression seen in the model we used.
In summary, kidney changes in db/db mice appear in conjunction with similar local IGF1/IGFBP1 changes as seen in type 1 diabetes mellitus. However, circulating GH levels are initially decreased in these obese mice. The potential role of GH in the exacerbation of renal diseases and the potential role of GH inhibition in models of DN in type 1 diabetes have been previously discussed. However, given the basically low GH levels in these obese diabetic mice and in human type 2 diabetes mellitus, modulation of such IGF1/IGFBP1 renal changes by further GH inhibition does not seem to be indicated. Alternatively, the renal complications and local GH–IGF axis changes in type 2 diabetes mellitus may be affected to a greater degree by the local diabetic environment, which may lead to the upregulation of glucose transporters, such as solute carrier family 2 (facilitated glucose transporter), member 1 (previously known as GLUT1) [33], or the activation of the diacylglycerol–protein kinase C–extracellular signal-regulated kinase pathway by hyperglycaemia [34]. Possible interventions in this model could then include an improvement in renal insulin signalling [35, 36]. This is exemplified by the use of insulin sensitisers in humans, such as peroxisome proliferator-activated receptor gamma agonists, which have been beneficial in the treatment of renal injury in early diabetic nephropathy [37, 38]. The potential role of restoring GH levels [39] and the particular independent effects of renal IGFBP1 accumulation remain to be determined.
Abbreviations
- A G :
-
mean glomerular cross-sectional area
- DN:
-
diabetic nephropathy
- FOXO1:
-
forkhead box O1
- GH:
-
growth hormone
- GHR:
-
growth hormone receptor
- IGFBP1:
-
insulin-like growth factor binding protein 1
- V G :
-
mean glomerular tuft volume
References
Wolf G, Ritz E (2003) Diabetic nephropathy in type 2 diabetes prevention and patient management. J Am Soc Nephrol 14:1396–1405
Sheetz MJ, King GL (2002) Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 288:2579–2588
Petersen KF, Shulman GI (2006) Etiology of insulin resistance. Am J Med 119:S10–S16
Schrijvers BF, De Vriese AS, Flyvbjerg A (2004) From hyperglycemia to diabetic kidney disease: the role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev 25:971–1010
Landau D, Segev Y, Eshet R, Fyvbjerg A, Phillip M (2000) Growth hormone (GH)–IGF–I axis changes in hyperglycemic nonobese diabetic (NOD) mice. Int J Exp Diab Res 1:9–186
Landau D, Domene H, Flyvbjerg A et al (1998) Differential renal growth hormone (GH) receptor and GH binding protein expression in experimental diabetes mellitus. Growth Horm IGF Res 8:39–45
Segev Y, Landau D, Marbach M, Schadeh N, Flyvbjerg A, Phillip M (1997) Renal hypertrophy in hyperglycemic non-obese diabetic mice is associated with renal accumulation of insulin-like growth factor (IGF) I. J Am Soc Nephrol 8:436–444
Landau D, Chin E, Bondy C et al (1995) Expression of insulin-like growth factor binding proteins in the rat kidney: effects of long-term diabetes. Endocrinology 136:1835–1842
Segev Y, Landau D, Rasch R, Flyvbjerg A, Phillip M (1999) Growth hormone receptor antagonism prevents early renal changes in the non-obese diabetic (NOD) mouse. J Am Soc Nephrol 10:2374–2381
Landau D, Israel E, Rivkis I et al (2003) The effect of growth hormone on the development of diabetic kidney disease in rats. Nephrol Dial Transplant 18:694–702
Landau D, Segev Y, Afargan M et al (2001) A novel somatostatin analogue prevents the development of renal diabetic complications in the non-obese diabetic (NOD) mouse. Kidney Int 60:505–512
Takahashi K, Daughaday WH, Kipnis DM (1971) Regulation of immunoreactive growth hormone secretion in male rats. Endocrinology 88:909–917
Segev Y, Eshet R, Rivkis I et al (2004) Comparison between somatostatin analogues and ACE inhibitor in the NOD mouse model of diabetic kidney disease. Nephrol Dial Transplant 19:3021–3028
Pagtalunan ME, Rasch R, Rennke HG et al (1995) Morphometric analysis of effects of angiotensin II on glomerular structure in rats. Am J Physiol 268:F82–F88
Bang P, Brismar K, Rosenfeld RG et al (1994) Fasting affects serum insulin-like growth factors (IGFs) and IGF-binding proteins differently in patients with noninsulin-dependent diabetes mellitus versus healthy nonobese and obese subjects. J Clin Endocrinol Metab 78:960–967
Kratzsch J, Keliner K, Zilkens T et al (1996) Growth hormone-binding protein related immunoreactivity is regulated by the degree of insulinopenia in diabetes mellitus. Clin Endocrinol (Oxf) 44:673–678
Ji S, Guan R, Frank SJ, Messina JL (1999) Insulin inhibits growth hormone signaling via the growth hormone receptor/JAK2/STAT5B pathway. J Biol Chem 274:13434–13442
Uchida T, Nakamura T, Hashimoto N et al (2005) Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med 11:175–182
van Haeften TW, Twickler TB (2004) Insulin-like growth factors and pancreas beta cells. Eur J Clin Invest 34:249–255
Heald AH, Siddals KW, Fraser W et al (2002) Low circulating levels of insulin-like growth factor binding protein-1 (IGFBP-1) are closely associated with the presence of macrovascular disease and hypertension in type 2 diabetes. Diabetes 51:2629–2636
Franco C, Bengtsson BA, Johannsson G (2001) Visceral obesity and the role of the somatotropic axis in the development of metabolic complications. Growth Horm IGF Res 11:S97–S102
Johannsson G, Marin P, Lonn L et al (1997) Growth hormone treatment of abdominally obese men reduces abdominal fat mass, improves glucose and lipoprotein metabolism, and reduces diastolic blood pressure. J Clin Endocrinol Metab 82:727–734
Johannsson G, Bengtsson BA (1999) Growth hormone and the metabolic syndrome. J Endocrinol Invest 22:41–46
Feliers D, Duraisamy S, Faulkner JL et al (2001) Activation of renal signaling pathways in db/db mice with type 2 diabetes. Kidney Int 60:495–504
Lee PD, Giudice LC, Conover CA et al (1997) Insulin-like growth factor binding protein-1: recent findings and new directions. Proc Soc Exp Biol Med 216:319–357
Orland MJ, Permutt MA (1987) Quantitative analysis of pancreatic proinsulin mRNA in genetically diabetic (db/db) mice. Diabetes 37:341–347
Schinner S, Scherbaum SR, Bornstein SR et al (2005) Molecular mechanisms of insulin resistance. Diabet Med 22:674–682
Tomizawa M, Kumar A, Perrot V et al (2000) Insulin inhibits the activation of transcription by a C-terminal fragment of the forkhead transcription factor FKHR. A mechanism for insulin inhibition of insulin-like growth factor-binding protein-1 transcription. J Biol Chem 275:7289–7295
Nakae J, Biggs WH 3rd, Kitamura T et al (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32:245–253
Galiano RD, Zhao LL, Clemmons DR et al (1996) Interaction between the insulin-like growth factor family and the integrin receptor family in tissue repair processes. Evidence in a rabbit ear dermal ulcer model. J Clin Invest 98:2462–2468
Sakai K, D’Ercole AJ, Murphy LJ et al (2001) Physiological differences in insulin-like growth factor binding protein-1(IGFBP-1) phosphorylation in IGFBP-1 transgenic mice. Diabetes 50:32–38
Gockerman A, Prevette T, Jones JI et al (1995) Insulin-like growth factor (IGF)-binding proteins inhibit the smooth muscle cell migration responses to IGF-I and IGF-II. Endocrinology 136:4168–4173
Heilig CW, Concepcion LA, Riser BL et al (1995) Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest 96:1802–1814
Haneda M, Koya D, Kikkawa R (2001) Cellular mechanisms in the development and progression of diabetic nephropathy: activation of the DAG–PKC–ERK pathway. Am J Kidney Dis 38:S178–S181
Catena C, Cavarape A, Novello M et al (2003) Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int 64:2163–2171
Coward RJ, Welsh GI, Yang J et al (2005) The human glomerular podocyte is a novel target for insulin action. Diabetes 54:3095–3102
Nakamura T, Ushiyama C, Suzuki S et al (2001) Effect of troglitazone on urinary albumin excretion and serum type IV collagen concentrations in Type 2 diabetic patients with microalbuminuria or macroalbuminuria. Diabet Med 18:308–313
Bakris G, Viberti G, Weston WM et al (2003) Rosiglitazone reduces urinary albumin excretion in type II diabetes. J Hum Hypertens 17:7–12
Nam SY, Kim KR, Cha BS et al (2001) Low-dose growth hormone treatment combined with diet restriction decreases insulin resistance by reducing visceral fat and increasing muscle mass in obese type 2 diabetic patients. Int J Obes Relat Metab Disord 25:1101–1107
Acknowledgements
This was study was supported by grants from the USA–Israel Binational Science Foundation (no. 2003055) and Peptor (Rehovot, Israel).
Duality of interest
The authors have no potential conflicts of interest related to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Segev, Y., Eshet, R., Yakir, O. et al. Systemic and renal growth hormone–IGF1 axis involvement in a mouse model of type 2 diabetes. Diabetologia 50, 1327–1334 (2007). https://doi.org/10.1007/s00125-007-0663-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0663-7