
Abstract
Aims/hypothesis
The role of dietary carbohydrate in the pathogenesis of type 2 diabetes is still a subject of controversial debate. Here we analysed the effects of diets with and without carbohydrate on obesity, insulin resistance and development of beta cell failure in the obese, diabetes-prone New Zealand Obese (NZO) mouse.
Materials and methods
NZO mice were kept on a standard diet (4% [w/w] fat, 51% carbohydrate, 19% protein), a high-fat diet (15, 47 and 17%, respectively) and a carbohydrate-free diet in which carbohydrate was exchanged for fat (68 and 20%, respectively). Body composition and blood glucose were measured over a period of 22 weeks. Glucose tolerance tests and euglycaemic-hyperinsulinaemic clamps were performed to analyse insulin sensitivity. Islet morphology was assessed by immunohistochemistry.
Results
Mice on carbohydrate-containing standard or high-fat diets developed severe diabetes (blood glucose >16.6 mmol/l, glucosuria) due to selective destruction of pancreatic beta cells associated with severe loss of immunoreactivity of insulin, glucose transporter 2 (GLUT2) and musculoaponeurotic fibrosarcoma oncogene homologue A (MafA). In contrast, mice on the carbohydrate-free diet remained normoglycaemic and exhibited hyperplastic islets in spite of a morbid obesity associated with severe insulin resistance and a massive accumulation of macrophages in adipose tissue.
Conclusions/interpretation
These data indicate that the combination of obesity, insulin resistance and the inflammatory response of adipose tissue are insufficient to cause beta cell destruction in the absence of dietary carbohydrate.
Similar content being viewed by others

Introduction
Type 2 diabetes is the consequence of a progressive failure of insulin-secreting beta cells, typically associated with obesity and insulin resistance. Two different scenarios have been proposed as the mechanism of this pathophysiological process. First, it has been suggested that beta cell damage is due to oxidative stress exerted by elevated glucose levels [1–3]. Alternatively, it has been proposed that obesity-induced ectopic fat accumulation in the pancreas causes apoptosis of beta cells [4, 5]. These mechanisms are not mutually exclusive and may well function in combination [6]. Little is known as to potential interactions, but understanding the in vivo contribution of either ‘glucotoxicity’ or ‘lipotoxicity’ to beta cell failure is essential in order to develop dietary and pharmacological interventions and ultimately prevent and cure diabetes.
In animal models, failure of beta cells can be accelerated or delayed by dietary modifications. In the db/db mouse strain, the prevalence of diabetes can be markedly reduced by the substitution of dietary protein for carbohydrate [7], suggesting that postprandial hyperglycaemia may play a role in the pathogenesis of islet cell failure. We have previously demonstrated that dietary fat content, in combination with the presence of diabetogenic alleles, markedly increases the prevalence of diabetes in parental New Zealand Obese mice (NZO) as well as in an outcross population of NZO with the lean SJL strain [8]. The NZO mouse is an optimal model for the study of obesity-associated diabetes (‘diabesity’). It presents morbid obesity associated with hyperphagia and reduced energy expenditure [9], rapidly developing a form of diabetes that is characterised by marked hyperglycaemia and hypoinsulinaemia associated with beta cell destruction [10–13]. Exposure to a ‘cafeteria-type’ high-fat diet (HFD) accelerated that process, apparently supporting the concept of lipotoxicity. However, the possibility that the diabetogenic effect of the HFD resulted from reduced insulin sensitivity and elevated postprandial blood glucose levels, which exerted a deleterious ‘glucotoxic’ effect on the beta cells, could not be ruled out entirely.
Thus, we systematically studied the effects of diets with and without carbohydrate on adiposity, postprandial blood glucose excursions and development of beta cell failure in the obese, diabetes-prone NZO mouse. Our data demonstrate that dietary carbohydrate plays an essential role in beta cell destruction and the manifestation of type 2 diabetes. Under a carbohydrate-free diet (CFD), even extreme obesity with a body fat mass of 60%, massive insulin resistance and a pronounced inflammatory response in adipose tissue are not sufficient to produce diabetes.
Materials and methods
Experimental animals
Male NZO from our own colony (NZO/HlBom: R. Kluge; German Institute of Human Nutrition, Nuthetal, Germany) and C57BL/6J mice (Charles River, Sulzfeld, Germany) were used throughout. Mice were housed singly at a temperature of 22°C with a 12 h light–dark cycle (lights on at 06.00 h) in type II macrolon cages with wooden gnawing sticks in order to avoid excessive growth of teeth. The animals were kept in accordance with the NIH guidelines for the care and use of laboratory animals, and all experiments were approved by the Ethics Committee of the State Ministry of Agriculture, Nutrition and Forestry (State of Brandenburg, Germany).
Diets
Standard diets (StD) and HFD were purchased from Altromin (Lage, Germany) in powdered form and were mixed with water (2:1 or 1:2) in order to generate similar textures in all diets. The StD (Art. no. 1324) contained (w/w) 4% fat, 50.5% carbohydrate and 19% protein, with 13, 63 and 24% of total digestible energy (11.8 kJ/g) from fat, carbohydrate and protein, respectively. The HFD (Art. no. C1057) contained 15% fat, 47% carbohydrate and 17% protein, with 34, 48 and 17% of total digestible energy (16.2 kJ/g) from fat, carbohydrate and protein. The CFD was prepared by mixing (w/w) 20% casein (Bayrische Milchindustrie eG, Landshut, Germany), 33.5% sunflower shortening, 33.5% palm fat (Ostthüringer Nahrungswerk Gera, Gera, Germany), 5% cellulose (J. Rettenmaier und Söhne, Ellwangen-Holzmühle, Germany), 0.5% safflower oil, 0.5% linseed oil (Kunella Feinkost, Cottbus, Germany), 5% mineral mixture and 2% vitamin mixture (Altromin, Lage, Germany). The diet contained 88 and 12% of total digestible energy (25.5 kJ/g) from fat and protein, respectively.
Analysis of body composition
Body composition (fat and lean mass) was measured by nuclear magnetic resonance (Bruker Minispec; Echo Medical Systems, Houston, TX, USA). Conscious mice were placed in an applied static magnetic field for 0.9 min as described previously [9, 14] and duplicate measurements of fat mass and lean mass were recorded for each time point and used for the data analysis.
Blood glucose
Blood glucose was measured weekly at 13.00 h in blood samples collected from the tail vein, using a glucose meter (MediSense Precision Q-I-D; Abbott, Wiesbaden, Germany).
Plasma insulin
Insulin levels were determined by ELISA with antiserum raised against rat insulin (kit no. INSKR020; CrystalChem, Chicago, IL, USA) and with mouse insulin (catalogue no. INSSM021; CrystalChem) as the standard.
Immunohistochemistry of pancreatic islets
Pancreatic tissue excised immediately after exsanguination of three to six mice from each diet group at week 10 or 22 was fixed in 4% formaldehyde for 24 h and embedded in paraffin according to standard procedures. Longitudinal serial sections (4 μm, at sampling intervals of 300 μm) were prepared. The following antibodies were used for immunostaining: sheep anti-insulin antiserum (1:6,000; Vector Laboratories, Burlingame, CA, USA), rabbit anti-glucagon antibody (1:2,000; Dako Diagnostika, Hamburg, Germany), rabbit antiserum against the C-terminus of rat glucose transporter 2 (GLUT2) (1:3,000) [15], affinity-purified rabbit anti-musculoaponeurotic fibrosarcoma oncogene homologue A (MafA) antiserum (1:500, BL1069; Bethyl Laboratories, Montgomery, TX, USA). Secondary antibodies were donkey peroxidase-linked anti-rabbit or sheep IgG-antiserum (1:1,000; Dianova, Hamburg, Germany). For detection of GLUT2 and MafA [16], biotin-conjugated donkey anti-rabbit IgG (Dianova) was used and was detected by incubation with streptavidin-biotin-horseradish peroxidase complex and diaminobenzidine (Dako Diagnostika) as substrate according to the manufacturer’s specifications. Specificity of staining was tested with pre-immune (GLUT2) or non-immune serum (MafA). Co-staining experiments indicated that anti-MafA antiserum [16] stained nuclei of insulin-positive cells. Microscopic investigation and photo documentation were performed with the combined light and fluorescence microscope ECLIPSE E-100 (Nikon, Düsseldorf, Germany) in combination with a video camera (CCD_1300CB; Vosskühler, Osnabrück, Germany) and the LUCIA analysis system (Nikon).
Quantitative analysis of islet morphology
Longitudinal sections of pancreas stained with anti-insulin, anti-glucagon and anti-GLUT2 antiserum were analysed with Lucia G image analysis software (Laboratory Imaging, Prague, Czechia) in a blinded procedure. In all islets of the sections (25–40), we determined the area of immunoreactive cells, the total islet area and the total section area. Beta cell area was determined as the area of all insulin-positive cells and calculated as percentage of the total islet area divided by 100. Islet area was calculated as mean of total islet area and as percentage of total section area.
Glucose tolerance test
NZO mice were fed an StD, HFD or CFD for 3 weeks and were fasted for 16 h prior to the experiment. Each animal received a single intraperitoneal injection of glucose (1.35 mg/g body weight). Samples for determination of blood glucose concentrations were obtained at 0, 15, 30, 60, 120 and 240 min from the tip of the tail.
Euglycaemic-hyperinsulinaemic clamp procedures
Animals were fed an StD, HFD or CFD for 2 weeks. Euglycaemic-hyperinsulinaemic clamps were performed as previously described [17] in 9-week old, conscious, catheterised mice that had fasted for 16 h. In vivo experiments lasted for 240 min and consisted of a 120-min basal period, directly followed by a 120-min euglycaemic-hyperinsulinaemic clamp. During the basal period a prime-continuous [3] glucose infusion (370 kBq bolus, 3.7 kBq/min) was started and continued to determine rates of whole-body glucose turnover. After the 120-min basal infusion period, a prime-continuous insulin infusion (10 mU kg−1 min−1) was started at time 0 min, raising insulin levels to within a physiological range. Steady-state conditions for plasma glucose concentration and specific activity were achieved within 70 min and a single 2-deoxy-d-[1-14C]glucose injection was administered at time 75 min. To determine plasma [3] glucose, 3H2O and 2-deoxy-d-[1-14C]glucose concentrations, blood samples were collected at times 80, 85, 90, 100, 110 and 120 min of the clamp and for measurement of basal [3-3H]glucose concentrations in the final 10 min of the basal period. Plasma glucose concentrations were measured with a Glukometer3000 (BST Bio Sensor Technologie, Berlin, Germany) via an enzymatic-amperometric reaction. A plasma sample for determination of basal insulin levels was obtained during the final 10 min of the basal period and for steady-state insulin levels at 120 min of the clamp.
Histology of adipose tissue
Adipose tissue was fixed in 4% buffered formaldehyde, rinsed in phosphate-buffered saline and dehydrated in a graded series of 50–100% ethanol followed by propylene oxide. Tissue was infiltrated with propylene/Epon-812 mixtures by gradually increasing the resin concentration. Sections of 0.5 μm were stained with Azur-II-Methylene Blue (Richardson stain). For quantification of macrophage infiltration in adipose tissue, tissue sections were stained with the macrophage-specific rat IgG2b monoclonal antibody (F480) against mouse macrophages (Caltag Laboratories, Burlingame, CA, USA), followed by incubation with mouse-adsorbed biotinylated rabbit anti-rat IgG (Vector Laboratories). Digital pictures were taken with a light microscope (Axioplan 2; Zeiss, Jena, Germany) using the measurement facilities of Imagic Access (Imagic, Glattburg, Switzerland). Multiple sections (separated by 70–80 μm each) obtained from gonadal and subcutaneous fat pads were analysed for adipocyte size and number, and for number of macrophages. For each treatment, ten slides were analysed, in a blinded study, by cell count of at least 50 fields (each representing approximately 500 adipocytes) per slide.
Statistical analysis
Differences in body weight, body fat and blood glucose under the different dietary regimens were tested with Dunnett’s pairwise multiple comparison t test, applying a general linear model for repeated measurement design. For un-paired samples, t tests were used to test differences in postprandial blood glucose and insulin levels, and in beta cell and islet area, after verifying normal distribution of data. All statistical analyses were performed with SPSS 11.5 (SPSS, Chicago, IL, USA), with differences being considered significant at p < 0.05.
Results
Carbohydrate-free, high-fat diet produces extreme obesity but protects NZO mice from developing severe hyperglycaemia
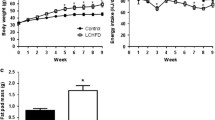
Figure 1 illustrates the development of body weight and body fat in male NZO mice under the different dietary regimens. Until 12 weeks of age, all groups progressively increased their body weight. Thereafter, only mice on the CFD continued to gain weight and accumulate fat (Fig. 1a,b), developing extreme obesity with an average body fat content of 60% at 22 weeks. In contrast, mice on StD and HFD remained constant or started to lose weight and body fat, apparently because of the development of severe glucosuria and hyperglycaemia (blood glucose >16.6 mmol/l; Fig. 1c).

Development of obesity and diabetes in NZO mice on the StD, HFD and CFD. Beginning at week 5, mice were kept on an StD (4% fat, 51% carbohydrate, 19% protein; n = 9; black symbols), HFD (15% fat, 47% carbohydrate, 17% protein; n = 8; open symbols) or CFD (68% fat, 20% protein; n = 8; grey symbols). Body weight (a) and total body fat mass (b) were monitored once weekly. c Time course of blood glucose levels in mice on the different diets. d Diabetes prevalence (hyperglycaemia >16.6 mmol/l) at week 12. Data represent means ± SE. p < 0.001 for difference between StD and CFD (a, b); p < 0.05 for difference between HFD and CFD (c)
To determine the time point of onset of diabetes in the different groups, blood glucose was measured once a week (Fig. 1c). On the CFD regimen, mice maintained average blood glucose levels below 10 mmol/l until the end of the observation period (22 weeks). In contrast, mice on the StD and HFD started to develop diabetes (hyperglycaemia with blood glucose levels >16.6 mmol/l) between weeks 9 and 12. At week 12, the prevalence of diabetes was 62.5% and 25% in mice on HFD and StD, respectively (Fig. 1d). At week 22, 75% of mice on StD and HFD were diabetic, with no diabetic mice in the CFD group. Thus, the increase in fat content from 4 to 15% (w/w) accelerated the onset of diabetes by approximately 3 weeks, whereas the restriction of carbohydrate fully prevented it.
The protective effect of the CFD, which contained vegetable fat only, appeared to be independent of the type of fat, since a CFD with lard as the fat component also prevented diabetes (blood glucose at week 22: CFD-vegetable fat, 6.09 ± 1.1 mmol/l; CFD-lard, 8.25 ± 2.8; HFD, 19.5 ± 9.7; means ± SD of six mice) (data not shown). Next we investigated whether the protective effect of the CFD could be reversed by adding carbohydrate to the diet at a later time point. NZO mice were fed the CFD for 7 weeks, at which time 30% of carbohydrate was subsequently added to the diet. Blood glucose levels increased from 9.4 ± 1.1 mmol/l to 15.2 ± 2.5, 25.6 ± 2.5 and 28.2 ± 0.8 mmol/l after 1, 2 and 3 weeks after addition of carbohydrate. The hyperglycaemia was accompanied by polyuria, polydipsia and massive weight loss (10.7 ± 1.3 g). Thus, addition of carbohydrate abolished the protective effect of CFD within 2–3 weeks.
Carbohydrate-free diet prevents destruction of beta cells in NZO mice
To provide direct evidence that CFD did indeed prevent destruction of pancreatic beta cells, we performed histopathology of the pancreas. Diabetes in mice on the carbohydrate-containing diets (StD, HFD) was associated with a marked reduction in islet cells immunoreactive for insulin (Fig. 2a,b), GLUT2 and the beta-cell-specific transcription factor MafA (Electronic Supplementary Material [ESM] Figs. 1 and 2). Simultaneously, the relative number of alpha cells, as detected with anti-glucagon antiserum, increased in both groups, indicating that hypoinsulinaemia in NZO was caused by selective beta cell damage with no reduction of alpha cells. At week 22, essentially no normal islets were visible in pancreatic sections of the diabetic mice on StD or HFD. In contrast, in CFD-fed mice all islets appeared normal with regard to immunoreactivity of insulin, glucagon, GLUT2 and MafA (Fig. 2c,d, ESM Fig. 1c).

Loss of insulin-producing beta cells in NZO mice on the StD and HFD, but not on the CFD. Beta cells in pancreatic sections obtained at the indicated time points from StD-fed mice (a), HFD-fed mice (b) and CFD-fed mice (c) were visualised by immunohistochemistry with anti-insulin antiserum as described in Materials and methods. In parallel sections, alpha cells were stained with anti-glucagon antiserum. Sections were counterstained with haematoxylin. Scale bars depict 100 μm. d–f Quantification of beta cell and islet area in pancreatic sections of mice at 22 weeks as indicated. For comparison, data from 22-week old C57BL/6J mice (B6) on a standard diet were determined. *p < 0.05; ***p < 0.001
A quantitative analysis of the area of cells immunoreactive for insulin in pancreatic sections indicated a 75 to 90% loss of functioning beta cells in mice on the StD and HFD regimens, as compared with NZO mice on CFD or with normal C57BL/6J mice (Fig. 2d). Similar results were obtained when the area of cells immunoreactive for GLUT2 was determined (area of GLUT2-positive cells, as % of total islet area: C57BL/6J 78.9 ± 6.2; NZO-StD 18.4 ± 2.7; NZO-HFD 13.9 ± 2.8; NZO-CFD 68.4 ± 3.8). The remaining cells that were not immunoreactive for either insulin or glucagon probably represent dysfunctional, in part necrotic beta cells. Mean islet area as well as total islet area normalised per total area of the pancreatic sections was larger in CFD-fed than in StD-fed, HFD-fed and C57BL/6J mice, indicating beta cell hyperplasia in response to the CFD (Fig. 2e,f). Moreover, the relative number of glucagon-positive cells was higher in NZO mice on StD and HFD than in those on CFD (C57BL/6J 20.5 ± 2.8; NZO-StD 57.8 ± 6.3; NZO-HFD 38.0 ± 4.4; NZO-CFD 26.5 ± 2.3; values: % of total). Thus, the data support the conclusion that the CFD specifically prevents beta cell destruction, which is the cause of hyperglycaemia in NZO mice on StD and HFD.
High-fat and carbohydrate-free diets markedly aggravate insulin resistance
To exclude the possibility that the CFD had somehow ameliorated insulin resistance in NZO mice, despite its high adipogenic potency, we performed glucose tolerance tests in mice at an early time point (week 8) before the onset of diabetes. Compared with C57BL/6J, NZO mice were severely glucose intolerant, with the highest glucose levels in mice on the CFD (ESM Fig. 3a). In addition, fasting blood glucose was elevated in CFD mice (p < 0.05), presumably due to markedly increased hepatic glucose output (Fig. 3c). Total plasma insulin concentrations (AUC from 0–240 min) during the glucose load were higher in HFD and CFD than in StD-fed mice (ESM Fig. 3b). Thus, HFD and CFD appeared to markedly aggravate insulin resistance in NZO mice. This conclusion was confirmed by euglycaemic-hyperinsulinaemic clamp experiments (Fig. 3). Both the glucose infusion rates required to maintain euglycaemia, whole-body glucose utilisation and glycolysis, and the effect of insulin on hepatic glucose output were significantly lower in HFD- and CFD- than in StD-fed mice. Since 2-deoxyglucose uptake into gastrocnemius muscle was not reduced, the fat-containing diets appeared to predominantly reduce hepatic insulin sensitivity under the conditions of the experiment (hyperinsulinaemia). In conclusion, both glucose tolerance tests and glucose clamp experiments indicated that CFD prevented diabetes despite producing a similar degree of insulin resistance to that produced by the diabetogenic HFD.

Effect of HFD and CFD on insulin resistance in NZO mice. Euglycaemic (8 mmol/l), hyperinsulinaemic clamp experiments were performed in 9-week-old, weight-matched NZO mice as described in Materials and methods. Mice were fed the different diets for 2 weeks and were fasted overnight before the experiment. a Glucose infusion rates, b whole-body glucose utilisation, c basal hepatic glucose production (HGP), d suppression of HGP, e whole-body glycolysis and f 2-deoxyglucose uptake in gastrocnemius muscle. Data represent means ± SE of eight animals. *p < 0.05; ***p < 0.001; # p < 0.02; ## p < 0.005; ###p < 0.0001
Insulin resistance not only corresponds with total fat, but also in particular with the visceral fat stores [18]. To test whether the different diets had modified the distribution of fat stores, we measured the epididymal fat pads of mice treated with the different diets as a measure of visceral fat stores. Weights of epididymal fat pads normalised per total body weight were similar in mice on StD (3.9 ± 0.2%), HFD (4.2 ± 0.2%) or CFD (3.63 ± 0.5%). Thus, the different diets did not modify the extent of visceral obesity in NZO.
Increased fat accumulation under CFD is associated with early macrophage infiltration into adipose tissue
It has previously been suggested that circulating cytokines released from adipose tissue are responsible for insulin resistance and beta cell failure [19–21]. In mice, obesity is associated with macrophage infiltration into adipose tissue [22, 23]. Since our data imply that obesity as such is not sufficient to generate diabetes, it was important to determine whether macrophage infiltration (as a parameter of the inflammatory state of adipose tissue) was correlated with diabetes or with obesity under the different dietary regimens. Figure 4a illustrates the immunohistochemical detection of macrophages in adipose tissue. As indicated by a quantitative analysis of the tissue slices (Fig. 4b), the number of macrophages increased from approximately 75–400 per 100 μg of adipose tissue (corresponding with an increase from three to 20 macrophages per 100 adipocytes) during the development of obesity (between weeks 5 and 16) under all three dietary regimens. At week 10, significantly more macrophages were detected in mice on CFD than in those on StD or HFD. These data dissociate the inflammatory response to obesity from the development of diabetes and suggest that this response may be necessary but is not sufficient to produce beta cell failure.

Macrophage infiltration into adipose tissue of NZO mice on StD, HFD and CFD. Mice were kept on the respective diets from week 5 to week 16. a Adipose tissue was fixed at the indicated time points and macrophages were stained with a macrophage-specific antiserum. The scale bar depicts 100 μm. b Macrophages and adipocytes were counted in 10 sections per animal. In separate experiments, the number of adipocytes per tissue weight was determined and macrophage counts were calculated per tissue mass. Open bars, 5 weeks; grey bars, 10 weeks; black bars, 16 weeks. Error bars represent SE of means of three animals. ***p < 0.001; # p = 0.009
Carbohydrate-free diet prevents postprandial blood glucose excursions
To determine whether carbohydrate restriction had eliminated postprandial blood glucose excursions, non-diabetic mice (week 8) were starved overnight, re-fed and 2-h postprandial blood glucose was measured (ESM Fig. 4a). In addition, food intake was monitored during the re-feeding period and was found to be higher in mice on CFD than on those on StD (ESM Fig. 4b). Both StD and HFD increased blood glucose concentrations, whereas in the absence of dietary carbohydrate (CFD) blood glucose levels dropped after feeding. Thus, the diabetogenic potencies of the diets appeared to correlate with the extent of postprandial glycaemia.
Discussion
The present data demonstrate that obesity-associated beta cell failure (diabesity) is promoted not only by dietary fat, but also by dietary carbohydrate in a mouse model of type 2 diabetes. Whereas a conventional HFD (15% fat) with 47% carbohydrate accelerated the development of diabetes, a CFD with high fat (68%) completely protected against beta cell failure. Conclusions based on these data are consistent with an earlier study reporting that substitution of carbohydrate for protein prevented diabetes in the db/db mouse [7]. Thus, an essential role of dietary carbohydrate for the development of diabetes has been demonstrated in two genetically diverse mouse models of diabesity under two different dietary regimens. In addition, the present data indicate that even under the conditions of extreme diet-induced obesity (body fat was 60% of total weight at week 22) with severe insulin resistance and inflammatory response of adipose tissue, dietary carbohydrate was essential for beta cell failure. Since postprandial blood glucose excursions were completely eliminated with the CFD, the data provide further proof that glucose toxicity plays a major role in beta cell failure of type 2 diabetes [1–3].
Diabetes in NZO under StD and HFD was associated with a dramatic decrease in immunoreactivity of insulin in pancreatic islets. This effect cannot only be due to degranulation of beta cells, since immunoreactivity of two other proteins specific for beta cells, GLUT2 and MafA, was decreased to a similar extent. Thus, diabetes in NZO reflects a profound destruction of beta cell function. Reduced expression of GLUT2 in beta cells of NZO mice and in other models of diabetes (Chinese hamster and Psammomys obesus) has been reported to precede the loss of insulin immunoreactivity [24–26]. More recently, loss of the beta-cell-specific transcription factor MafA [16, 27] due to oxidative stress has been suggested to be a crucial factor in beta cell failure. It remains to be shown that the development of diabetes in NZO follows a similar scenario with an early loss of MafA, and that a carbohydrate-restricted diet prevents the formation of reactive oxygen species.
Although our data indicate that dietary carbohydrate is essential for the development of diabetes in obese mice, they do not exclude a major role of lipids. With outcross populations, a threshold body weight (45 g at 12 weeks) is required for the development of diabetes [8, 12, 28, 29]. In the presence of dietary carbohydrate, an increase of dietary fat accelerated the development of diabetes (Fig. 2) [8]. Thus, obesity is absolutely required for diabetes to develop in NZO mice, although on its own it is apparently not sufficient. The present data are therefore consistent with a scenario in which the combination of fatty acids or triacylglycerol accumulating in the beta cells (‘lipotoxicity’) [5] with elevated glucose concentration (‘glucotoxicity’) [1, 2] confers beta cell toxicity.
Considerable data have indicated that cytokines originating from adipose tissue play a major role in the pathogenesis of insulin resistance and beta cell failure in both human and mouse diabetes [21]. Obesity appears to represent an inflammatory state and circulating cytokines are generated by the adipose tissue or by immigrating macrophages [22, 23]. The present data, however, indicate that the inflammatory consequences of obesity, as manifested by massive macrophage infiltration of adipose tissue, are not sufficient to produce beta cell failure in the NZO mouse. Yet since a certain degree of obesity is required for the development of diabetes, the data do not exclude the possibility that inflammatory cytokines are involved in the pathogenesis of insulin resistance and diabetes.
An earlier study reported that pancreatic islets of diabetic NZO mice are infiltrated by leucocytes [13], suggesting a role of the immune system in beta cell damage in NZO. In the present study, we observed infiltration of islets at a later stage of beta cell destruction, when cells had become necrotic. As illustrated in Fig. 2a–c, loss of functioning beta cells occurred without any lymphocytic infiltration. Thus, we believe that infiltration might be a secondary phenomenon occurring at a later stage of beta cell damage. Although we cannot fully discount the possibility that leucocyte infiltration is a contributing factor to the pathogenesis of diabetes, we feel it is unlikely to be the primary cause of beta cell destruction. This conclusion is also supported by the present finding that diet is a major factor modifying the development of diabetes.
Several lines of evidence obtained in human studies suggest that the quality of dietary carbohydrate is a determinant of the risk of developing diabetes. In prospective cohort studies, a diet with a high glycaemic load was associated with an increased risk of type 2 diabetes [30]. Likewise, the consumption of soft drinks and white bread increases the diabetes risk [31, 32]. Furthermore, the glucosidase inhibitor acarbose reduces the risk of impaired glucose tolerance progressing to overt diabetes [33]. Thus, there is substantial evidence suggesting that postprandial glucose excursions play a crucial role in the development of human beta cell failure. However, national as well as international diabetes associations have so far refrained from recommending a diet with a low glycaemic load as a preventive measure. Importantly, the study presented here used a diet that is not palatable for humans, and our experimental data cannot be directly translated into nutritional recommendations. However, our data do underline the need to further investigate the role of carbohydrate in the development of human diabetes and to test preventive strategies accordingly.
Abbreviations
- CFD:
-
carbohydrate-free diet
- GLUT2:
-
glucose transporter 2
- NZO:
-
New Zealand obese mouse
- HFD:
-
high-fat diet
- StD:
-
standard diet
- MafA:
-
musculoaponeurotic fibrosarcoma oncogene homologue A
References
Leahy JL, Bonner-Weir S, Weir G (1992) Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 15:442–455
Yki-Jarvinen H (1992) Glucose toxicity. Endocr Rev 13:415–431
Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A (2001) Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 50(Suppl 1):S154–S159
Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH (1994) Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci USA 91:10878–10882
Unger RH (2003) Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 144:5159–5165
Poitout V, Robertson RP (2002) Secondary beta-cell failure in type 2 diabetes—a convergence of glucotoxicity and lipotoxicity. Endocrinology 143:339–342
Leiter EH, Coleman DL, Ingram DK, Reynolds MA (1983) Influence of dietary carbohydrate on the induction of diabetes in C57BL/KsJ-db/db diabetes mice. J Nutr 113:184–195
Plum L, Giesen K, Kluge R et al (2002) Characterization of the diabetes susceptibility locus Nidd/SJL in the New Zealand obese (NZO) mouse: islet cell destruction, interaction with the obesity QTL Nob1, and effect of dietary fat. Diabetologia 45:823–830
Jürgens HS, Schürmann A, Kluge R et al (2006) Hyperphagia, lower body temperature, and reduced running wheel activity precede development of morbid obesity in New Zealand obese mice. Physiol Genomics 13:234–241
Crofford OB, Davis CK (1965) Growth characteristics, glucose tolerance and insulin sensitivity of New Zealand obese mice. Metabolism 14:271–280
Ortlepp JR, Kluge R, Giesen K et al (2000) A metabolic syndrome of hypertension, hyperinsulinemia, and hypercholesterolemia in the New Zealand Obese (NZO) mouse. Eur J Clin Invest 30:195–202
Reifsnyder PC, Leiter EH (2002) Deconstructing and reconstructing obesity-induced diabetes (diabesity) in mice. Diabetes 51:825–832
Junger E, Herberg L, Jeruschke K, Leiter EH (2002) The diabetes-prone NZO/Hl strain. II. Pancreatic immunopathology. Lab Invest 82:843–853
Tinsley FC, Taicher GZ, Heiman ML (2004) Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res 12:150–160
Hellwig B, Brown FM, Schürmann A, Shanahan MF, Joost HG (1992) Localization of the binding domain of the inhibitory ligand forskolin in the glucose transporter GLUT4 by photolabeling, proteolytic cleavage and a site-specific antiserum. Biochim Biophys Acta 1111:178–184
Kitamura YI, Kitamura T, Kruse JP et al (2005) FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab 2:153–163
Ren JM, Marshall BA, Mueckler MM, McCaleb M, Amatruda JM, Shulman GI (1995) Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. J Clin Invest 95:429–432
Carey DG (1998) Abdominal obesity. Curr Opin Lipidol 9:35–40
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91
Greenberg AS, McDaniel ML (2002) Identifying the links between obesity, insulin resistance and beta-cell function: potential role of adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur J Clin Invest 32(Suppl 3):24–34
Rajala MW, Scherer PE (2003) The adipocyte: at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Chankiewitz E, Peschke D, Herberg L et al (2006) Did the gradual loss of GLUT2 cause a shift to diabetic disorders in the New Zealand obese mouse (NZO/Hl)? Exp Clin Endocrinol Diabetes 114:262–269
Jörns A, Tiedge M, Sickel E, Lenzen S (1996) Loss of GLUT2 glucose transporter expression in pancreatic beta cells from diabetic Chinese hamsters. Virchows Arch 428:177–185
Jörns A, Tiedge M, Ziv E, Shafrir E, Lenzen S (2002) Gradual loss of pancreatic beta-cell insulin, glucokinase and GLUT2 glucose transporter immunoreactivities during the time course of nutritionally induced type-2 diabetes in Psammomys obesus (sand rat). Virchows Arch 440:63–69
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280:11107–11113
Leiter EH, Reifsnyder PC, Flurkey K, Partke HJ, Junger E, Herberg L (1998) NIDDM genes in mice. Deleterious synergism by both parental genomes contributes to diabetic thresholds. Diabetes 47:1287–1295
Plum L, Kluge R, Giesen K, Altmüller J, Ortlepp JR, Joost HG (2000) Type-2-diabetes-like hyperglycaemia in a backcross model of New Zealand obese (NZO) and SJL mice: Characterization of a susceptibility locus on chromosome 4 and its relation with obesity. Diabetes 49:1590–1596
Hu FB, Manson JE, Stampfer MJ et al (2001) Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med 345:790–797
Schulze MB, Manson JE, Ludwig DS et al (2004) Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 292:927–934
Heidemann C, Hoffmann K, Spranger J et al (2005) A dietary pattern protective against type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition (EPIC)–Potsdam Study cohort. Diabetologia 48:1126–1134
Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M, STOP-NIDDM Trial Research Group (2002) Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet 359:2072–2077
Acknowledgements
The authors are grateful to S. Woods, University of Cincinnati, for helpful discussions and manuscript editing, to D. Accili (Dept. Medicine, Columbia University, New York) for a gift of the anti-FoxO1 antiserum and to C. Plaue, A. Teichmann, A. Seelig and E. Meyer for skilful technical assistance. This work was supported in part by the European Community’s FP6 EUGENE2 (LSHM-CT-2004-512013), the German Research Foundation (Jo 117/11-2) and the German Ministry of Education, Research and Technology (NGFN2 and 0313040A).
Duality of interest
None of the authors has a duality of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. S. Jurgens and S. Neschen contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Loss of GLUT2 and MafA expression in beta cells of NZO mice on StD (a) and HFD (b), but not on CFD (c). Beta cells in pancreatic sections obtained at the indicated time points were visualised by immunohistochemistry with anti-GLUT2 or anti-MafA antiserum as described in Materials and methods. Scale bars: 100 μm(GIF 296 kb)
ESM Fig. 2
Reduced expression and subcellular redistribution of GLUT2 and MafA in beta-cells of NZO mice on the StD and HFD in comparison with beta cells of NZO mice kept on the CFD. Beta cells in pancreatic sections obtained at week 10 were visualised by immuno-histochemistry with anti-GLUT2 or anti-MafA antiserum as described in Materials and methods. Scale bars: 50 μm(GIF 402 kb)
Rights and permissions
About this article
Cite this article
Jürgens, H.S., Neschen, S., Ortmann, S. et al. Development of diabetes in obese, insulin-resistant mice: essential role of dietary carbohydrate in beta cell destruction. Diabetologia 50, 1481–1489 (2007). https://doi.org/10.1007/s00125-007-0662-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0662-8