
Abstract
Aims/hypothesis
Plasminogen activator inhibitor-1 (PAI-1, also known as serpin peptidase inhibitor, clade E [nexin, plasminogen activator inhibitor type 1], member 1 [SERPINE1]) plays a pathogenetic role in renal fibrosis. It is upregulated in experimental and human diabetic nephropathy. These studies assessed the effect of PAI-1 deficiency and overproduction on renal disease in experimental diabetes.
Materials and methods
Diabetes was induced by injection of streptozotocin in 6-week-old PAI-1-deficient mice, transgenic mice overexpressing Pai-1 and control mice. Animals were killed after 24 weeks of diabetes or after observation alone.
Results
Pai-1 mRNA was upregulated in kidneys from genetically normal mice with diabetes and in non-diabetic Pai-1 transgenic mice. PAI-1 was not further increased in kidneys from Pai-1 transgenic mice with diabetes. Diabetes-associated albuminuria and glomerular injury, as well as renal α-smooth muscle actin production, were ameliorated in diabetic PAI-1-deficient mice, an amelioration associated with attenuated increases in renal matrix metallopeptidase-2 expression and activity. Diabetic Pai-1 transgenic mice did not develop increased albuminuria or glomerular injury, but the tubulointerstitial area was modestly enhanced. In addition to the findings in diabetic mice, abnormalities also developed in 30-week-old PAI-1-deficient and Pai-1 transgenic mice without diabetes. PAI-1 deficiency resulted in increased tubulointerstitial area, TGFB1 protein and α-smooth muscle actin. Non-diabetic 30-week-old Pai-1 transgenic mice developed similar renal abnormalities and increased matrix metallopeptidase-2 activity, together with a modest increase in serum glucose and HbA1c.
Conclusions/interpretation
These results demonstrate that endogenous PAI-1 deficiency protects mice from glomerular injury in longer term diabetes and that endogenous PAI-1 maintains normal renal interstitial structure in ageing not associated with diabetes.
Similar content being viewed by others

Introduction
Plasminogen activator inhibitor-1 (PAI-1, also known as serpin peptidase inhibitor, clade E [nexin, plasminogen activator inhibitor type 1], member 1 [SERPINE1]) is a serine protease inhibitor that inhibits plasminogen activation. It regulates fibrinolysis and plasmin-mediated matrix metallopeptidase (MMP) activation, leading to inhibition of extracellular matrix (ECM) degradation and potentially to activation of fibrotic processes [1, 2]. PAI-1 is potentially profibrogenic and therefore a therapeutic target in fibrotic renal diseases. While PAI-1 expression is minimal in normal human kidneys [3, 4], it is induced in kidney disease associated with fibrosis, including diabetic nephropathy [4, 5]. PAI-1 has been implicated in the progression of disease within a number of different tissues and organs including the kidney [6–13].
Diabetic nephropathy is a leading cause of end-stage renal disease, with significant morbidity and mortality [14]. It is characterised by thickening of glomerular and tubular basement membranes and accumulation of ECM components in the mesangium and tubulointerstitium, leading to glomerular and tubulointerstitial fibrosis. Stimuli relevant to diabetic nephropathy induce PAI-1, including glucose [15], as well as various other relevant stimuli including advanced glycation end-products [16], reactive oxygen species [17], angiotensin II [18] and TGFB1 [19]. A role for PAI-1 in type 2 diabetes and the metabolic syndrome has been suggested by studies in experimental models. PAI-1-deficient db/db mice developed less adiposity, hyperglycaemia and hyperinsulinaemia. [20]. While obesity and insulin resistance were prevented in PAI-1-deficient mice on a high-fat diet [21], overexpression of Pai-1 attenuated obesity in mice fed a high-fat diet [22]. Although less is known about the role of PAI-1 in the renal complications of diabetes, progression of diabetic nephropathy is possibly associated with PAI-1 production and activity [1, 2]. In patients with diabetes, albuminuria is associated with increased plasma PAI-1 levels [23, 24]. Intrarenal PAI-1 levels are increased in glomeruli and the interstitium, and PAI-1 is prominent in fibrotic areas and vessels in diabetic nephropathy [4, 5]. In experimental diabetic nephropathy, PAI-1 is increased in streptozotocin-induced diabetes and Otsuka Long-Evans Tokushima Fatty rats [25]. Moreover, in early streptozotocin-induced diabetes (4 weeks) PAI-1-deficient mice were protected from renal changes [26].
The current studies explored the role of PAI-1 in long-term diabetic nephropathy. Experimental diabetes was induced by administration of streptozotocin in PAI-1-deficient mice to determine whether PAI-1 plays a pathogenetic role both in albuminuria and in glomerular and tubulointerstitial injury. Diabetes was induced in mice that had been transgenically engineered to overexpress Pai-1 (Pai-1 TG mice) to test the hypothesis that these mice would develop more severe renal injury than genetically normal mice.
Materials and methods
Experimental mice
Mice deficient in Pai-1 (Serpine1 gene) [27] (C57BL/6 × 129/SvJ, backcrossed once onto a C57BL/6 background; 75% C57BL/6, 25% 129/SvJ) were bred in a specific pathogen-free facility (Monash Medical Centre, Melbourne, VIC, Australia). For ease of understanding, mice are described as PAI-1-deficient or PAI-1 wild-type (WT) mice. Mice of the same background (75% C57BL/6, 25% 129/SvJ) from the backcross were used as PAI-1 WT controls. Pai-1 TG mice ([11], C57BL/6 background, expressing a murine Serpine1 minigene under a cytomegalovirus promoter [D. Ginsburg, Howard Hughes Medical Institute, Ann Arbor, MI, USA]) were bred at Monash Medical Centre. Genetically normal C57BL/6 mice were used as the appropriate control mice. Studies were approved by the animal ethics committees of the Baker Heart Research Institute (Melbourne, VIC, Australia) and Monash University, in accordance with the Principles of Laboratory Animal Care (NIH). After breeding, animals were housed at the Baker Institute. Mice were randomised into non-diabetic and diabetic groups. Diabetes was induced by five daily i.p. streptozotocin injections (55 mg/kg in citrate buffer; Boehringer-Mannheim, Mannheim, Germany) [28] in 6-week-old male mice. Low-dose streptozotocin was used to avoid non-specific streptozotocin toxicity [29]. Only animals with blood glucose levels >15 mmol/l 2 days after diabetes induction were included in studies. Control mice received buffer alone. Animals had unrestricted access to water, with standard mouse chow (Barastoc, Pakenham, VIC, Australia), and were maintained on a 12-h light–dark cycle.
For PAI-1-deficient mice, the following groups were studied: non-diabetic PAI-1 WT mice (PAI-1 WT control study group [Ctrl], n = 13); diabetic PAI-1 WT mice (PAI-1 WT diabetic study group [DM], n = 15); non-diabetic PAI-1-deficient mice (n = 16); and diabetic PAI-1-deficient (n = 14).
For Pai-1 TG mice, the following groups were studied: non-diabetic C57BL/6 mice (C57BL/6 Ctrl, n = 8); diabetic C57BL/6 mice (C57BL/6 DM, n = 10); non-diabetic Pai-1 TG mice (Pai-1 TG Ctrl, n = 10); and diabetic Pai-1 TG mice (Pai-1 TG DM, n = 15).
PCR was used to confirm the genotype of all experimental mice. The Serpine1 transgene was detected as previously described [11]. PAI-1-deficient mice were genotyped using primers for the wild-type Serpine1 gene, which is deleted in PAI-1-deficient mice, and the neomycin resistance gene for the mutant Serpine1 allele [27]. Genotyping results were confirmed by intrarenal PAI-1 RT-PCR. After 24 weeks, animals were anaesthetised by i.p. pentobarbitone (55 mg/kg; Boehringer Ingelheim, Artarmon, NSW, Australia). Kidneys were dissected, frozen and stored at −80°C or in buffered formalin (10%). Blood was collected, centrifuged and plasma and erythrocytes were stored at −20°C. HbA1c was determined in erythrocyte lysates by HPLC (Biorad, Richmond, CA, USA).
Assessment of albuminuria and morphological changes
At weeks 12 and 24, animals were housed in metabolic cages for 24 h to collect urine for albumin measurement by a modified radioimmunoassay for measuring rat albumin [30] using a rabbit anti-mouse albumin antibody ([31]; 1:6,000; Cappel, Aurora, OH, USA). Plasma urea and creatinine concentrations were measured by autoanalyser (Beckman, Fullerton, CA, USA). For renal pathology, 2-μm sections were stained with periodic acid-Schiff’s reagent. Assessment of glomerular injury was performed semi-quantitatively in a masked fashion. At least 40 glomeruli from each mouse were graded individually as follows: 0, intact glomerulus; 1, expansion or accumulation of material positive for periodic acid-Schiff reagent or sclerosis involving <25% of the glomerular tuft; 2, as for 1, but involving 25–50% of the glomerulus; 3, as for 1, but involving 50–75% of the glomerulus; 4, as for 1, but involving >75% of the glomerulus. The index of glomerular injury was calculated using the following formula: \( {\text{GI}} = {{\left( {1 \times n_{1} } \right)} + {\left( {2 \times n_{2} } \right)} + {\left( {3 \times n_{3} } \right)}{\left( {4 \times n_{4} } \right)}} \mathord{\left/ {\vphantom {{{\left( {1 \times n_{1} } \right)} + {\left( {2 \times n_{2} } \right)} + {\left( {3 \times n_{3} } \right)}{\left( {4 \times n_{4} } \right)}} {{\left( {n_{0} + n_{1} + n_{2} + n_{3} + n_{4} } \right)}}}} \right. \kern-\nulldelimiterspace} {{\left( {n_{0} + n_{1} + n_{2} + n_{3} + n_{4} } \right)}} \), where GI is glomerular injury and n x = number of glomeruli in each grade. The cortical interstitial area was assessed by point-counting, on the basis of 100 points counted on a 1-cm2 eyepiece graticule per field. Twelve high-power fields (×400) were counted per animal. Each high-power field was 0.076 mm2 and the total area counted per slide was 0.91 mm2. The interstitial area was defined as being equal to the number of interstitial grid intersections/total number of intersections.
Immunohistochemistry
We used 2-μm paraffin kidney sections for immunohistochemistry, performed with a monoclonal mouse anti-human α-smooth muscle actin (α-SMA, also known as actin, alpha 2, smooth muscle, aorta [ACTA2]) antibody (1:50; Dako, Copenhagen, Denmark) or a polyclonal rabbit anti-TGFB1 antibody (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA). For α-SMA immunostaining, sections were dewaxed, hydrated and endogenous peroxidase quenched (3% H2O2/PBS), followed by incubation for 30 min at room temperature in protein blocking agent (PBA; Lipshaw-Immunon, Pittsburgh, PA, USA). Sections were then incubated with the primary antibody (overnight, 4°C). Sections for TGFB1 were heated, dewaxed, microwaved (0.01 mol/l citrate buffer, pH 6.0) and incubated with 3% H2O2/PBA and anti-TGFB1 antibody (1 h, room temperature). Next we applied biotinylated horse anti-mouse immunoglobulin (1:250, Vector Laboratories, Burlingame, CA, USA) for α-SMA and biotinylated goat anti-rabbit immunoglobulin (1:250, Vector Laboratories) for TGFB1, followed by the ABC Elite Kit (Vectastain; Vector Laboratories). Peroxidase conjugates were visualised using 3,3′-diaminobenzidine tetrahydrochloride (0.08% H2O2/PBS; Sigma-Aldrich, Castle Hill, NSW, Australia) and counterstained with Mayer’s haematoxylin. Sections were examined under light microscopy and digitised. To quantitate the proportional area of staining, 20 views (×400) were randomly located in the renal cortex (6.2-Video Pro-32; Optimas, Bedford Park, SA, Australia). TGFB1 was analysed by separately assessing glomerular, tubular and interstitial compartments.
Protein extraction and gelatin zymography
Protein isolation for MMP2 and MMP9 was performed by heat extraction [32]. Frozen cortex was lysed in protein extraction buffer (50 mmol/l Tris-HCl pH 7.6, 0.2 mol/l NaCl, 5 mmol/l CaCl2, 0.02% Brij-35). Lysates were centrifuged (3,300 g, 30 min, 4°C), pellets re-suspended (0.1 mol/l CaCl2), mixed, heated (60°C, 5 min), chilled, then centrifuged at 16,000 g for 40 min, concentrated by centrifugation filtration (Microcon YM-10; Millipore, Bedford, MA, USA) and stored at −80°C. Protein content was determined [33] and pro- and active MMP2 and MMP9 were analysed by gelatin zymography [34], using 8% SDS/polyacrylamide gels (SDS-PAGE) containing 1 mg/ml gelatin (Labchem, Auburn, NSW, Australia), non-reducing conditions (no boiling or addition of dithiothreitol or β-mercaptoethanol). Protein (30 μg per lane) was resolved in Laemmli-buffer (final concentration: 50 mmol/l Tris-HCl, pH 6.5, 10% glycerol, 2% SDS, 0.01% bromophenol blue), gels washed three times (15 min, room temperature) with renaturing buffer (final concentration: 50 mmol/l Tris-HCl, pH 7.5, 10 mmol/l CaCl2, 1 μmol/l ZnCl2, 2.5% Triton X-100, 0.02% NaN3) to remove SDS, incubated with developing buffer (1% Triton-X, >16 h, 37°C), then stained with Coomassie Brilliant Blue R-250 (0.5%, 1 h) and destained with Coomassie Blue solution (10% acetic acid, 40% methanol). Gelatinase bands were determined using the LabScan system (Amersham, Castle Hill, NSW, Australia) and quantified using Photoshop (Adobe, San Jose, CA, USA).
Real-time quantitative RT-PCR
Frozen cortical kidney sections (6–9 mice per group) were lysed (Polytron; Kinematica, Lucerne, Switzerland) in TRIzol (Invitrogen, Carlsbad, CA, USA), RNA extracted and the RNA concentration measured. RNA (3.6 μg) was treated with RNase-free DNase (Ambion, Austin, TX, USA), and DNase-treated RNA was used to synthesise cDNA with a synthesis system (Superscript First Strand; Invitrogen). For details on primers and probes, see Electronic supplementary material (ESM) Table 1. TaqMan minor groove binder probes (Applied Biosystems, Foster City, CA, USA) were linked to 6-carboxyfluorescein (FAM, 5′ end). Gene expression was analysed by real-time quantitative RT-PCR (TaqMan; Applied Biosystems), with 18S ribosomal RNA expression being assessed (rRNA, 18S rRNA TaqMan Control Reagent Kit; Applied Biosystems) as a control. Amplifications used the following time course: 50°C, 2 min and 10 min at 95°C; and 40 cycles of 94°C, 20 s, 60°C, 1 min. Results are expressed proportional to non-diabetic (Ctrl) genetically normal mice.
Statistical analysis
Data were analysed by ANOVA, followed by Tukey’s test for multiple comparisons or by t test. Data are shown as the mean±SEM.
Results
Intrarenal Pai-1 gene expression
Pai-1 mRNA was detected in kidneys of genetically normal mice without diabetes (Ctrl), but no Pai-1 mRNA was present in PAI-1-deficient mice. Increased renal Pai-1 expression was observed in both groups of genetically normal diabetic mice when compared with non-diabetic groups (PAI-1 WT mice: Ctrl 1.00 ± 0.07, DM 1.27 ± 0.14 arbitrary units [AU], p < 0.05 [WT without DM normalised to 1]; C57BL/6 mice: Ctrl 1.01 ± 0.07, DM 1.83 ± 0.13 AU, p < 0.001 [C57BL/6 without DM normalised to 1]). Non-diabetic and diabetic Pai-1 TG groups exhibited increased renal Pai-1 expression (Pai-1 TG Ctrl 1.49 ± 0.11 p < 0.01 vs C57BL/6 Ctrl mice, Pai-1 TG DM 2.09 ± 0.47 vs C57BL/6 Ctrl mice), but Pai-1 mRNA expression was not further increased in kidneys of Pai-1 TG mice after 24 weeks of diabetes.
Functional and structural data
Streptozotocin-induced diabetic mice weighed less than non-diabetic mice (Tables 1 and 2). In diabetic mice, PAI-1 deficiency resulted in somewhat higher body weights and in genetically modified diabetic mice PAI-1 overproduction resulted in lower body weights than weights of genetically unmodified diabetic mice. Diabetes was associated with similar increases in HbA1c levels in all groups. Systolic blood pressure was unaffected by diabetes after 24 weeks, with the exception of diabetic PAI-1 WT mice, which had reduced systolic blood pressure compared with the non-diabetic control group. Diabetes resulted in an increase in kidney weight to body weight ratio compared with control mice in each group. PAI-1-deficient non-diabetic (Ctrl) mice were heavier and had increased kidney weight to body weight ratios compared with PAI-1 WT mice without diabetes, but an altered kidney weight to body weight ratio was not observed between groups of diabetic mice. PAI-1 deficiency did not affect metabolic parameters, but vehicle-injected 30-week-old Pai-1 TG mice developed higher plasma glucose and HbA1c levels than C57BL/6 mice (albeit still much lower than seen in the streptozotocin groups) suggesting that PAI-1 overproduction may, over time, have deleterious effects on glucose tolerance.
PAI-1 deficiency protects mice from diabetic glomerular injury and albuminuria
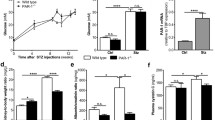
The induction of diabetes resulted in increased urinary albumin excretion at week 12, with a further increase by week 24 (Fig. 1a,b). Diabetes was associated with development of glomerular injury, characterised by matrix expansion and occasional glomerulosclerosis in genetically unmodified mice, with a mean glomerular injury index of approximately 3 (Fig. 1c). PAI-1 WT diabetic mice were protected from the progression of albuminuria after 12 weeks and developed less glomerular histological injury, demonstrating that endogenous PAI-1 plays a pathogenetic role in diabetic glomerular injury. However, PAI-1 overproduction did not result in increased histological or functional glomerular injury. TGFB1 protein was detected in glomeruli of diabetic kidneys from genetically intact mice (Fig. 2a,d,e). In mice with diabetes, TGFB1 protein was not significantly altered by PAI-1 deficiency or overproduction (Fig. 2h,i). However, increased TGFB1 production was observed in glomeruli of 30-week-old non-diabetic PAI-1 WT and Pai-1 TG mice (Fig. 2a,f,g) compared with TGFB1 in PAI-1 WT and C57BL/6 mice, respectively.

Albumin excretion rates (a, b) and glomerular injury at 24 weeks (c) in mice groups as specified. PAI-1 deficiency protected mice from increasing albuminuria (a) and diminished glomerular histological changes (c), but Pai-1 TG mice developed similar injury to genetically normal mice with diabetes. Key: a dashed/dotted line, PAI-1 WT Ctrl; thin continuous line, PAI-1 WT DM; bold dashed line, PAI-1-deficient Ctrl; bold continuous line, PAI-1-deficient DM; b dashed/dotted line, C57BL/6 Ctrl; thin continuous line, C57BL/6 DM; bold dashed line, Pai-1 TG Ctrl; bold continuous line, Pai-1 TG DM. *p < 0.01; ***p < 0.001

TGFB1 immunostaining in glomeruli in PAI-1-intact and -deficient mice, genetically normal C57BL/6 mice and Pai-1 TG mice with diabetes mellitus. The TGFB1 signal was minimal in glomeruli of control kidneys from genetically intact mice (a–c). TGFB1 was produced in diabetic mice (d, e) and was present in control non-diabetic PAI-1-deficient (f) and Pai-1 TG mice (g). In diabetic PAI-1-deficient (h) or Pai-1 TG mice (i) TGFB1 was not significantly reduced compared with genetically normal diabetic mice. *p < 0.05. b–i Medium power views of immunoperoxidase staining with haematoxylin counterstain
Interstitial area and TGFB1 production
Tubulointerstitial lesions in diabetic mice were characterised by tubulointerstitial fibrosis and occasional tubular cell dilatation and/or atrophy. Non-diabetic mice with either PAI-1 deficiency or PAI-1 overproduction exhibited an increase in interstitial area (Fig. 3a). Diabetes did not result in a significant increase in interstitial area in genetically normal mice, but the interstitial area was increased in diabetic Pai-1 TG mice compared with diabetic C57BL/6 mice. Assessment of tubulointerstitial TGFB1 staining revealed similar findings (Fig. 3b,c). In non-diabetic mice, tubular and interstitial TGFB1 protein was increased both in the absence of PAI-1 and in the context of PAI-1 overproduction. Diabetes resulted in increased TGFB1 protein in genetically normal mice, but TGFB1 production was not altered in PAI-1-deficient or Pai-1 TG mice with diabetes compared with diabetic genetically normal mice.

Tubulointerstitial changes and TGFB1 in mice. In genetically normal mice diabetes was associated with a consistent trend to increased tubulointerstitial area (a), but both PAI-1-deficient and Pai-1 TG non-diabetic 30-week-old mice developed an increased interstitial area, which was increased even more in the Pai-1 TG DM group. Tubular (b) and interstitial (c) TGFB1 protein was increased in genetically normal mice with diabetes and also increased in 30-week-old PAI-1-deficient and Pai-1 TG mice without diabetes. *p < 0.05
Renal cortical immunostaining for α-SMA
In non-diabetic genetically normal mice, immunostaining for α-SMA was found in the vasculature (Fig. 4a,b). In kidneys from diabetic genetically normal PAI-1 WT and C57BL/6 mice, α-SMA was increased in the tubulointerstitium (Fig. 4e,f,i). Compared with diabetic genetically normal mice, α-SMA protein was reduced in PAI-1-deficient mice with diabetes (Fig. 4g,i), but unchanged in diabetic Pai-1 TG mice (Fig. 4h). However, in non-diabetic 30-week-old PAI-1-deficient or PAI-1 over-producing mice, cortical immunostaining for α-Sma was increased compared with genetically normal mice (Fig. 4e,f,i). Changes in α-Sma renal mRNA expression demonstrated equivalent increases in PAI-1 WT and PAI-1-deficient mice with diabetes compared with control PAI-1 WT mice, an increase in control PAI-1-deficient mice compared with PAI-1 WT control mice and no significant changes in the Pai-1 TG mice (Fig. 4j).

Production and expression of α-SMA in mice. α-SMA protein was minimal in PAI-1 WT mice (a) and C57BL/6 mice (b) without diabetes (i). Induction of diabetes resulted in increased expression of α-SMA in both PAI-1 WT (c) and C57BL/6 mice (d). Non-diabetic 30-week-old PAI-1-deficient (e) and Pai-1 TG mice (f) developed increased expression of α-Sma. Diabetic PAI-1-deficient mice had reduced α-SMA staining (g) compared with diabetic PAI-1 WT mice (c), but α-SMA in Pai-1 TG diabetic mice was unchanged (h). Quantitation of immunostaining (i) and mRNA expression (j) showed similar findings, although mRNA changes did not at times reach significance. a–h Medium power views of immunoperoxidase staining with haematoxylin counterstain. *p < 0.05; **p < 0.01; ***p < 0.001
Mmp2 and Mmp9 expression and activity
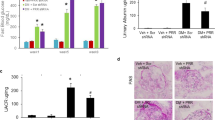
Diabetic renal disease resulted in increased renal Mmp2 expression (Fig. 5a) and increases in both pro-MMP2 (Fig. 5b,d) and active MMP2 protein levels (Fig. 5c,d), assessed by zymography (Fig. 5d). Compared with PAI-1 WT mice, diabetic PAI-1-deficient mice demonstrated decreased Mmp2 mRNA levels and decreased pro- and active MMP2 protein levels. Diabetic Pai-1 TG mice had MMP2 levels (mRNA and protein) similar to those of diabetic C57BL/6 mice. While Mmp2 was unchanged in control PAI-1-deficient mice, in non-diabetic TG mice, there was a significant increase in Mmp2 gene expression and protein (pro- and active MMP2) levels. Diabetes was associated with decreased Mmp9 expression (Fig. 6a) and in C57BL/6 mice with reduced pro-MMP9 (Fig. 6b,d) and active MMP9 protein (Fig. 6c,d). MMP9 in diabetic PAI-1-deficient and transgenic mice was similar to genetically normal diabetic mice and in non-diabetic Pai-1 TG mice was similar to levels in non-diabetic C57BL/6 mice. Mmp9 mRNA and active MMP9 levels in non-diabetic PAI-1 WT and PAI-1-deficient mice were similar, but increased levels of pro-MMP9 levels in PAI-1-deficient mice without diabetes were observed.

Mmp2 mRNA expression and MMP2 protein levels and activity in kidneys. Diabetes resulted in the induction of renal Mmp2 mRNA (a) that was coupled with increased pro-MMP2 (b) and active MMP2 (c) measured by zymography. d Illustrative zymograms. PAI-1 deficiency resulted in attenuated Mmp2 expression and activity. Non-diabetic Pai-1 TG mice also exhibited increased Mmp2 mRNA expression and protein activity. *p < 0.05; ** p < 0.01; *** p < 0.001

Mmp9 mRNA expression and MMP9 protein levels and activity in kidneys. Mmp9 mRNA expression (a) was decreased in genetically normal mice and PAI-1-deficient mice with diabetes. Pro-MMP9 (b) and active MMP9 (c) levels were significantly decreased in C57BL/6 mice with diabetes. Pro-MMP9 was increased in PAI-1-deficient mice without diabetes, and decreased with diabetes, but in the same mouse groups active MMP9 levels were not significantly altered. Diabetes reduced pro-MMP9 levels in Pai-1 TG mice. d Illustrative zymograms. *p < 0.05; **p < 0.01; ***p < 0.001
Discussion
It has been hypothesised that PAI-1 plays a profibrotic role in diabetic nephropathy. The current studies assessed the role of PAI-1 in longer term diabetes focusing on the effects of both PAI-1 deficiency and overproduction on diabetic renal injury. The findings of these studies are: (1) diabetes-induced albuminuria and glomerular injury were attenuated in PAI-1-deficient mice; (2) transgenic overproduction of PAI-1 did not result in a major increase in glomerular pathology but did affect the tubulointerstitium; and (3) in aged mice without diabetes, both PAI-1 deficiency and PAI-1 overproduction resulted in alterations in potential key markers and mediators of tubulointerstitial disease. While most, but not all studies have implicated PAI-1 as pathogenetic in renal disease [6–10, 26, 35], the present study suggests that PAI-1 plays complex and potentially divergent roles in normal ageing and in renal pathology.
The pathogenetic effects of endogenous PAI-1 suggest that PAI-1-dependent pathways play a role in long-term diabetic renal injury, in particular glomerular injury. The increased PAI-1 levels observed in diabetic kidneys in the present study is consistent with earlier findings in rat models and in diabetic patients [36]. Nicholas et al. reported that PAI-1-deficient mice were protected from albuminuria and increased fibronectin and TGFB production after only 4 weeks of streptozotocin-induced diabetes [26]. In our study, the beneficial effect of PAI-1 deficiency on albuminuria in diabetes was not seen by 12 weeks, but only at a later time-point (24 weeks). However, we cannot exclude an additional, but very early effect of PAI-1 on albuminuria, which was not measured at 4 weeks, as pathological changes are not observed early in this model. Glomerular 24-week histological changes were ameliorated in the absence of PAI-1, but a number of other renal parameters, including tubulointerstitial area, TGFB1 and α-SMA protein were either clearly unaffected or non-significantly altered in diabetes by PAI-1 deficiency. Transgenic overexpression of Pai-1 did not result in increased renal injury, although the tubulointerstitial area was increased in Pai-1 TG mice with diabetic renal disease. This modest effect in Pai-1 TG mice could be attributed to the lack of a significant increase in renal Pai-1 mRNA in comparison with genetically normal mice with diabetes, which already have an increase in Pai-1 mRNA as a result of diabetes per se. In other models of renal injury, anti-glomerular basement membrane glomerulonephritis [7] and fibrosis induced by unilateral ureteric obstruction [10], intrarenal PAI-1 levels were increased in Pai-1 TG mice, potentially reflecting the more overt fibrin-dependent [37, 38] and/or inflammatory nature of these lesions.
Most, but not all, studies of the functional role of PAI-1 in experimental renal disease indicate a pathogenetic role for PAI-1. In experimental crescentic glomerulonephritis, PAI-1 deficiency protected from (and PAI-1 overproduction promoted) glomerular injury and renal collagen accumulation [7]. A mutant, non-inhibitory form of PAI-1 protected rats from glomerulosclerosis in mesangial proliferative glomerulonephritis [9] and in aldosterone-induced glomerular injury endogenous PAI-1 was pathogenetic [8]. However, in another model of glomerulonephritis PAI-1-deficient mice developed enhanced injury mediated by increased TGFB1 production [35]. In tubulointerstitial fibrosis mediated by unilateral ureteric obstruction, PAI-1 deficiency was protective [6], while PAI overproduction promoted fibrosis [10].
It is important to appreciate that the present studies used two different background strains as genetically unmodified mice, in as far as genetic background is relevant to the development of experimental diabetic nephropathy [29, 39]. For studies in PAI-1-deficient mice, both PAI-1-deficient mice and PAI-1 WT mice had 25% of the 129/SvJ strain in their predominantly C57BL/6 (75%) background. PAI-1-overexpressing mice and controls were on a 100% C57BL/6 background. Measurement of some parameters (albuminuria, glomerular injury and tubulointerstitial expansion) were similar in both genetically unmodified strains, but other parameters, including the relative increase in Pai-1 gene expression, TGFB1 immunostaining and the reduction in MMP9 activity with diabetes were higher in the pure C57BL/6 background. While 129/Sv mice are prone to glomerulosclerosis after 5/6 nephrectomy, this was associated with a degree of hypertension [40]. Hypertension is not a feature of the model employed in the current studies. No studies in streptozotocin-induced diabetes have been performed using a pure 129/Sv strain [29].
The mechanisms behind the pathogenetic effects of endogenous PAI-1 in experimental diabetic nephropathy remain unclear. Diabetic kidneys displayed increased numbers of α-SMA positive cells. In vivo studies have suggested links between PAI-1 and α-SMA [6, 10]. In our study, TGFB1 production, assessed by immunostaining, was increased in genetically normal mice with diabetes. While other studies, potentially, have shown a lesser increase in TGFB1 protein production in experimental diabetes [26], differences in the streptozotocin dose, timing of measurements and methods (e.g. semi-quantitation by western blotting vs assessing area of tissue affected by immunostaining) may be responsible for these differences. Although immunostaining suggested reductions in TGFB1 in diabetic PAI-1-deficient mice in glomerular and tubular compartments, these changes did not reach statistical significance. However, changes in PAI-1 levels did affect the expression and activation of MMPs. While increased MMP activity has the potential to limit ECM accumulation, which is a feature of diabetic renal disease, increased MMP2 or MMP9 activity may accelerate epithelial-to-myofibroblast transition [41, 42]. In the present study, streptozotocin-induced diabetes led to increased Mmp2 expression and enzymatic activity but to a decrease in Mmp9 expression and enzymatic activity in renal cortex. PAI-1-deficient mice with diabetes had less MMP2 activity (with lower mRNA and pro-MMP2 levels). While effects on MMP9 were less consistent, diabetes suppressed Mmp9 mRNA and, at least in C57BL/6 mice, suppressed active MMP9. While in diabetes little effect of PAI-1 deficiency or overproduction on MMP9 was seen, non-diabetic PAI-1-deficient mice did have increased MMP9 protein (via significant increases in pro-MMP9). Collectively, these results imply that in diabetic nephropathy, MMP2 may be more important in epithelial-to-myofibroblast transition and PAI-1 may limit its activity, while falls in MMP9 may be relevant to ECM accumulation. The importance of epithelial-to-myofibroblast transition in the progression of diabetic nephropathy remains controversial; it appears, however, to be linked to TGFB1 levels [43].
PAI-1-deficient and Pai-1 TG mice that did not receive streptozotocin but were left as aged control mice developed renal abnormalities by 30 weeks. PAI-1-deficient non-diabetic mice developed increased immunostaining for TGFB1, increased cortical α-Sma mRNA and protein, as well as increased interstitial area. While these results were unexpected given the protection that PAI-1 deficiency conferred on diabetic mice, aged PAI-1-deficient mice develop cardiac fibrosis [44], despite being protected from pathogenetic fibrosis post cardiac ischaemia [12]. PAI-1 participates both as a key effector molecule in fibrotic processes, including those driven by TGFB1, [45], and potentially as a negative regulator of TGFB1 [46]. Recent developments underlining the potential for beneficial and harmful effects of matrix degradation in renal and vascular disease [41, 47, 48], together with varying pro-fibrotic stimuli in various renal diseases, support a model whereby PAI-1 may have opposing effects in different situations. Some mediators, for example α-SMA, were increased in genetically unmodified PAI-1-deficient mice and reduced in the presence of diabetes. These findings are not easy to reconcile, but they may result from the effects of PAI-1 inhibiting interstitial matrix turnover. In the absence of injurious stimuli, the actions of PAI-1 in stabilising matrix may contribute to optimal maintenance of tubulointerstitial integrity. In the diabetic milieu, however, increased PAI-1 expression is pathogenetic and facilitates injury, including α-SMA production. The current studies emphasise the potential importance of PAI-1, not only in diabetes but also in non-diabetic conditions, including ageing.
Non-diabetic Pai-1 TG mice exhibited increased TGFB1 protein levels, increased interstitial area, increased α-SMA production by immunostaining and increased Mmp2 expression and activity. These abnormalities were complicated by the observation that Pai-1 TG mice developed increased plasma glucose and HbA1c levels. Although modest when compared with streptozotocin-induced diabetes, these increases do point towards a role for high PAI-1 levels in abnormal glucose tolerance. It is not clear whether any of the renal changes in non-diabetic Pai-1 TG mice were related to increased glucose. However, some changes were not dissimilar to diabetic C57BL/6 mice, suggesting that the modest elevation in glucose in non-diabetic Pai-1 TG mice is not in itself sufficient to account for the renal changes observed.
In conclusion, PAI-1 deficiency ameliorated diabetic renal injury, demonstrating that endogenous PAI-1 plays a pathogenetic role in diabetic nephropathy, particularly with respect to glomerular lesions. In non-diabetic ageing mice, both PAI-1-deficient and PAI-1-overproducing mice developed abnormalities in the tubulointerstitium, pointing towards complex roles for PAI-1 in the maintenance of renal structure.
Abbreviations
- Ctrl:
-
control study group (non-diabetic mice)
- DM:
-
diabetic study group
- ECM:
-
extracellular matrix
- MMP:
-
matrix metallopeptidase
- PAI-1:
-
plasminogen activator inhibitor-1
- Pai-1 TG:
-
mice transgenically engineered to overexpress Pai-1
- PAI-1 WT:
-
PAI-1 wild-type mice
- PBA:
-
protein blocking agent
- SERPINE1:
-
serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1
- α-SMA:
-
α-smooth muscle actin
References
Rerolle JP, Hertig A, Nguyen G, Sraer JD, Rondeau EP (2000) Plasminogen activator inhibitor type 1 is a potential target in renal fibrogenesis. Kidney Int 58:1841–1850
Eddy AA (2002) Plasminogen activator inhibitor-1 and the kidney. Am J Physiol Renal Physiol 283:F209–F220
Rondeau E, Mougenot B, Lacave R et al (1990) Plasminogen activator inhibitor 1 in renal fibrin deposits of human nephropathies. Clin Nephrol 33:55–60
Yamamoto T, Noble NA, Cohen AH et al (1996) Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int 49:461–469
Paueksakon P, Revelo MP, Ma LJ, Marcantoni C, Fogo AB (2002) Microangiopathic injury and augmented PAI-1 in human diabetic nephropathy. Kidney Int 61:2142–2148
Oda T, Jung YO, Kim HS et al (2001) PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int 60:587–596
Kitching AR, Kong YZ, Huang XR et al (2003) Plasminogen activator inhibitor-1 is a significant determinant of renal injury in experimental crescentic glomerulonephritis. J Am Soc Nephrol 14:1487–1495
Ma J, Weisberg A, Griffin JP et al (2006) Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int 69:1064–1072
Huang Y, Haraguchi M, Lawrence DA et al (2003) A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest 112:379–388
Matsuo S, Lopez-Guisa JM, Cai X et al (2005) Multifunctionality of PAI-1 in fibrogenesis: evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int 67:2221–2238
Eitzman DT, McCoy RD, Zheng X et al (1996) Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 97:232–237
Takeshita K, Hayashi M, Iino S et al (2004) Increased expression of plasminogen activator inhibitor-1 in cardiomyocytes contributes to cardiac fibrosis after myocardial infarction. Am J Pathol 164:449–456
Wang H, Vohra BP, Zhang Y, Heuckeroth RO (2005) Transcriptional profiling after bile duct ligation identifies PAI-1 as a contributor to cholestatic injury in mice. Hepatology 42:1099–1108
Maisonneuve P, Agodoa L, Gellert R et al (2000) Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis 35:157–165
Fisher EJ, McLennan SV, Yue DK, Turtle JR (1997) High glucose reduces generation of plasmin activity by mesangial cells. Microvasc Res 53:173–178
Lee HS, Moon KC, Song CY et al (2004) Glycated albumin activates PAI-1 transcription through Smad DNA binding sites in mesangial cells. Am J Physiol Renal Physiol 287:F665–F672
Lee EA, Seo JY, Jiang Z et al (2005) Reactive oxygen species mediate high glucose-induced plasminogen activator inhibitor-1 up-regulation in mesangial cells and in diabetic kidney. Kidney Int 67:1762–1771
Nakamura S, Nakamura I, Ma L, Vaughan DE, Fogo AB (2000) Plasminogen activator inhibitor-1 expression is regulated by the angiotensin type 1 receptor in vivo. Kidney Int 58:251–259
Tomooka S, Border WA, Marshall BC, Noble NA (1992) Glomerular matrix accumulation is linked to inhibition of the plasmin protease system. Kidney Int 42:1462–1469
Schafer K, Fujisawa K, Konstantinides S, Loskutoff DJ (2001) Disruption of the plasminogen activator inhibitor 1 gene reduces the adiposity and improves the metabolic profile of genetically obese and diabetic ob/ob mice. FASEB J 15:1840–1842
Ma LJ, Mao SL, Taylor KL et al (2004) Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 53:336–346
Lijnen HR, Maquoi E, Morange P et al (2003) Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol 23:78–84
Gruden G, Cavallo-Perin P, Bazzan M et al (1994) PAI-1 and factor VII activity are higher in IDDM patients with microalbuminuria. Diabetes 43:426–429
Hirano T, Kashiwazaki K, Moritomo Y, Nagano S, Adachi M (1997) Albuminuria is directly associated with increased plasma PAI-1 and factor VII levels in NIDDM patients. Diabetes Res Clin Pract 36:11–18
Hagiwara H, Kaizu K, Uriu K et al (2003) Expression of type-1 plasminogen activator inhibitor in the kidney of diabetic rat models. Thromb Res 111:301–309
Nicholas SB, Aguiniga E, Ren Y et al (2005) Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int 67:1297–1307
Carmeliet P, Kieckens L, Schoonjans L et al (1993) Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest 92:2746–2755
Candido R, Jandeleit-Dahm KA, Cao Z et al (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106:246–253
Breyer MD, Bottinger E, Brosius FC et al (2005) Mouse models of diabetic nephropathy. J Am Soc Nephrol 16:27–45
Allen TJ, Cooper ME, O’Brien RC et al (1990) Glomerular filtration rate in streptozocin-induced diabetic rats. Role of exchangeable sodium, vasoactive hormones, and insulin therapy. Diabetes 39:1182–1190
Lassila M, Seah KK, Allen TJ et al (2004) Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol 15:2125–2138
Woessner JF Jr (1995) Quantification of matrix metalloproteinases in tissue samples. Methods Enzymol 248:510–528
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Rauch BH, Bretschneider E, Braun M, Schror K (2002) Factor Xa releases matrix metalloproteinase-2 (MMP-2) from human vascular smooth muscle cells and stimulates the conversion of pro-MMP-2 to MMP-2: role of MMP-2 in factor Xa-induced DNA synthesis and matrix invasion. Circ Res 90:1122–1127
Hertig A, Berrou J, Allory Y et al (2003) Type 1 plasminogen activator inhibitor deficiency aggravates the course of experimental glomerulonephritis through overactivation of transforming growth factor beta. FASEB J 17:1904–1906
Tarnow L, Stehouwer CD, Emeis JJ et al (2000) Plasminogen activator inhibitor-1 and apolipoprotein E gene polymorphisms and diabetic angiopathy. Nephrol Dial Transplant 15:625–630
Kitching AR, Holdsworth SR, Ploplis VA et al (1997) Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med 185:963–968
Drew AF, Tucker HL, Liu H et al (2001) Crescentic glomerulonephritis is diminished in fibrinogen-deficient mice. Am J Physiol Renal Physiol 281:F1157–F1163
Qi Z, Fujita H, Jin J et al (2005) Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes 54:2628–2637
Ma LJ, Fogo AB (2003) Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 64:350–355
Yang J, Shultz RW, Mars WM et al (2002) Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest 110:1525–1538
Cheng S, Lovett DH (2003) Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am J Pathol 162:1937–1949
Oldfield MD, Bach LA, Forbes JM et al (2001) Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest 108:1853–1863
Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA (2004) Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res 95:637–644
Krag S, Danielsen CC, Carmeliet P, Nyengaard J, Wogensen L (2005) Plasminogen activator inhibitor-1 gene deficiency attenuates TGF-beta1-induced kidney disease. Kidney Int 68:2651–2666
Hertig A, Rondeau E (2004) Plasminogen activator inhibitor type 1: the two faces of the same coin. Curr Opin Nephrol Hypertens 13:39–44
Edgtton KL, Gow RM, Kelly DJ, Carmeliet P, Kitching AR (2004) Plasmin is not protective in experimental renal interstitial fibrosis. Kidney Int 66:68–76
Luttun A, Lupu F, Storkebaum E et al (2002) Lack of plasminogen activator inhibitor-1 promotes growth and abnormal matrix remodeling of advanced atherosclerotic plaques in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 22:499–505
Acknowledgements
These studies were supported by Grants from the National Health and Medical Research Council of Australia (NHMRC). M. Lassila was supported by the Academy of Finland, the Einar och Karin Foundation and the Paavo Nurmi Foundation. K. Jandeleit-Dahm was supported by a National Heart Foundation/NHMRC Career Development Award.
Duality of interest
The authors have no relevant duality of interest in respect of this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Lassila and K. Fukami contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lassila, M., Fukami, K., Jandeleit-Dahm, K. et al. Plasminogen activator inhibitor-1 production is pathogenetic in experimental murine diabetic renal disease. Diabetologia 50, 1315–1326 (2007). https://doi.org/10.1007/s00125-007-0652-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0652-x