
Abstract
Aims/hypothesis
Type 2 diabetes is a complex disorder with strong heritability. The aim of our study was to investigate whether common polymorphisms in the genes regulating the early insulin signalling pathway (insulin; A-23T, insulin-like growth factor 1 receptor [IGF-1R]; GAG1013GAA, plasma cell membrane glycoprotein 1 [PC-1]; K121Q, insulin receptor substrate [IRS-1]; G972R, insulin receptor substrate 2 [IRS-2]; G1057D and phosphatidylinositol 3-kinase p85α [PI3K]; M326I) affect the weight change and development of Type 2 diabetes in the Finnish Diabetes Prevention Study.
Methods
We screened for the polymorphisms in 490 overweight subjects with impaired glucose tolerance whose DNA was available from the Finnish Diabetes Prevention Study. These subjects were randomly allocated into a control group and an intervention group characterised by intensive, individualised diet and exercise.
Results
In carriers of the GAA1013GAA genotype of IGF-1R, the R972 allele of IRS-1 and the D1057D genotype of IRS-2, lifestyle intervention did not lead to significant differences in weight loss between the intervention and control groups, implying a role of these risk genotypes in the regulation of body weight. We observed a statistically significant difference in the conversion rate from IGT to diabetes between the genotypes of the IGF-1R gene (GAG1013GAG: 18.6%, GAG1013GAA: 10.4%, GAA1013GAA: 19.5%, p=0.033). Common polymorporphisms in the insulin, PC-1 and PI3K genes did not regulate weight change or conversion to diabetes.
Conclusions/interpretation
The common polymorphisms of the IGF-1R, IRS-1 and IRS-2 genes may modify the weight change response to a lifestyle intervention but not the conversion from IGT to Type 2 diabetes, whereas IGF-1R may also regulate the risk of developing Type 2 diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus is characterised by peripheral insulin resistance in liver, skeletal muscle and adipose tissue, as well as impaired insulin secretion [1]. Known major risk factors for the disease include obesity, central obesity, low physical activity, high intake of saturated fat and low intake of dietary fibre [2, 3]. Genetic factors also play a key role in the development of the disease. However, the genetic basis of this disease has remained unclear, although some promising susceptibility genes for Type 2 diabetes have been identified including calpain-10 [4], PPARγ2 [5], and Kir6.2 [6].
The insulin signalling pathway begins with the binding of insulin to the α-subunit of the insulin receptor and ends with the biological effects of insulin in multiple tissues. One of the most important effects of insulin is to stimulate glucose transport. Upon insulin stimulation, glucose transporter 4 translocates to the plasma membrane, allowing glucose import into the cell. Even though other pathways stimulate glucose uptake [7], the major mediators regulating glucose transport are insulin receptor substrates and phosphatidylinositol 3-kinase (PI3K). Variants in these genes have been associated with insulin resistance and Type 2 diabetes.
Genes regulating the proximal insulin signalling pathway could contribute to insulin resistance and are therefore potential candidate genes for Type 2 diabetes. The insulin variable number of tandem repeat (VNTR) locus, upstream of the insulin gene, has been linked to alterations in insulin secretion, depending on the number of repeats (class I: 26 to 63 repeats vs class III: 141 to 209 repeats). The T-23 allele of the insulin gene promoter is in complete linkage disequilibrium with the class I VNTR allele, and the A-23 allele is in linkage disequilibrium with the class III VNTR allele [8, 9].
The insulin receptor is an important mediator between the extracellular and intracellular insulin signalling pathway. Plasma cell membrane glycoprotein-1 (PC-1) affects insulin signalling by direct interaction with insulin receptor α-subunit blocking insulin action [10]. Carriers of the Q121 allele of the PC-1 gene are insulin-resistant and they have increased glucose and insulin levels independent of whether they suffer from obesity or not [11, 12].
Insulin receptor substrates 1 and 2 (IRS-1 and IRS-2) are important mediators in the insulin signalling pathway and transfer information from insulin receptor to PI3K. The R972 allele of the IRS-1 gene inhibits IRS-1 binding to PI3K. Both IRS-1 and IRS-2 have been shown to impair beta cell function in knockout mice [13]. In addition, mice deficient in IRS-2 developed Type 2 diabetes due to the lack of beta cell compensation [14], whereas the results in humans have been inconsistent [15, 16, 17, 18]. The R972 allele of the IRS-1 gene has been associated with increased prevalence of Type 2 diabetes, insulin resistance and impaired insulin secretion in cultured pancreatic beta cells [19, 20, 21, 22, 23]. Insulin-like growth factor 1 receptor (IGF-1R) influences insulin signalling by activating IRS proteins independent of insulin receptor activation [24]. Although there is no previous evidence that variants in the IGF-1R gene cause diabetes in humans, animal models have shown that alterations in IGF-1R activity can lead to impaired insulin secretion and even Type 2 diabetes [24, 25].
Phosphatidylinositol 3-kinase regulatory subunit p85α (PI3K p85α) plays a major role in the insulin signalling pathway, as it mediates the message from the insulin receptor substrates to the catalytic subunit p110γ of PI3K [26, 27]. Mice lacking PI3K p85α subunit have increased insulin sensitivity and hypoglycaemia [28]. In addition, the I326I genotype carriers have been reported to have lower prevalence of Type 2 diabetes than carriers of the M326 allele of PI3K p85α [29]. However, results suggesting the opposite have also been reported [30, 31].
To investigate how genes regulating the proximal insulin signalling pathway determine the risk of Type 2 diabetes, we studied six common polymorphisms in the insulin (T-23A), IGF-1R (GAG1013GAA), PC-1 (K121Q), IRS-1 (G972R), IRS-2 (G1057D) and PI3K p85α subunit (M326I) genes in relation to weight change and the development of Type 2 diabetes in participants of the Finnish Diabetes Prevention Study (DPS) [30]. Because the Finnish DPS had a lifestyle intervention group with a marked lower risk of diabetes than a control group, it also offered a possibility to investigate gene–lifestyle interaction.
Subjects and methods
Subjects and research design
A detailed description of subjects, methods and study design has already been reported [32, 33]. The Finnish DPS is a prospective intervention and follow-up study carried out in five participating centres (Helsinki, Kuopio, Oulu, Tampere and Turku). The main objective of the study was to investigate whether lifestyle intervention influences the cumulative incidence of Type 2 diabetes. Altogether 522 middle-aged (40 to 65 years) and overweight (BMI ≥25 kg/m2) subjects with impaired glucose tolerance (fasting plasma glucose <7.8 mmol/l; 120-min plasma 7.8–11.0 mmol/l in an OGTT [34]) participated in the study. At baseline mean age was 55±7 years and mean BMI 31.2±4.6 kg/m2. Exclusion criteria were a previous diagnosis of diabetes mellitus or another chronic disease, or characteristics that made survival until the end of the study improbable. Subjects enrolled in the study were randomised into one of the two study groups: the control group or the intervention group. The control group was given general advice about benefits of reducing weight, healthy food choices and increasing physical activity, whereas the intervention group was given intensive and individualised nutritional counselling, as well as individual advice to increase physical activity by an individualised exercise programme. An OGTT was performed at each annual follow-up visit and the diagnosis of diabetes was confirmed with a second test. The study protocol was approved by the Ethics Committee of the National Public Health Institute in Helsinki and all the study subjects gave written informed consent.
Measurements
Physical examinations were performed and the medical history was recorded at baseline and at each annual follow-up visit. Height, weight, BMI, waist and hip circumference, and glucose and insulin levels before (0 min) and at 120 min in the OGTT (75-g glucose load after a 12-hour overnight fast [32]) were determined at baseline and at the annual follow-up examinations. Plasma glucose concentration was measured by a glucose oxidase method (Glucose Auto & Stat, Model GA-110; Daiichi, Kyoto, Japan) and plasma insulin by a radioimmunoassay method (Phadeseph Insulin RIA 100; Pharmacia Diagnostica, Uppsala, Sweden). Weight change was calculated from the baseline value to the last weight measurement available, which varied from 1 to 3 years in the event of diabetes being newly diagnosed before the 3-year follow-up visit. For subjects who did not convert to diabetes, weight change was calculated as difference in weight between baseline and 3 years. Changes in fasting and 120-min glucose in an OGTT were calculated to achieve variables that indicate changes in glucose tolerance more sensitively.
DNA analysis
DNA samples were available from 490 subjects. The polymorphisms of the insulin, IGF-1R, PC-1, IRS-1 and IRS-2 genes were screened by PCR, followed by restriction fragment length polymorphism analysis. The primers for screening for PC-1 and IRS-1 polymorphisms have been published [10, 35]. Other primers used in this study were: (i) for the insulin gene: forward 5′-CGTCAGGTGGGCTCAGGGTT-3′ and reverse 5′-ACAAAGGCTGCGGCTGGGTC-3′; (ii) for the IGF-1R gene: forward 5′-TGCTTTAATTACGGTTTCTTC-3′ and reverse 5′-GCTTTTCAGGAACTTTCTCTT-3′; and (iii) for the IRS-2 gene: forward 5′-AGCTGTACCGCCTGCCCCC-3′ and reverse 5′-CCGACACCCACGCCGCCCT-3′. All amplifications were carried out in a total volume of 10 µl (the final volume for PI3K was 25 µl) containing 50 ng genomic DNA, 5 pmol of each primer, 10 mmol/l Tris-HCl (pH 8.8), 50 mmol/l KCl, 1.5 mmol/l MgCl, 0.1% Triton X-100, 0.25 units of DNA polymerase (Finnzymes, Espoo, Finland) and 100 µmol/l dNTP. Restriction enzymes used in digestion were HphI (insulin gene), MnlI (IGF-1R), Eco47I (PC-1), MvaI (IRS-1) and Bsp143II (IRS-2). The M326I polymorphism of the PI3K gene was determined by PCR (forward primer: 5′-CCTCCTAAACCACCAAAAC-3′ and reverse primer: 5′-TGGAAGAGAACCAACTATGC-3′), followed by direct sequencing (ABI prism 3100 Genetic Analyzer; Applied Biosystems, Hitachi, Japan).
Statistical analysis
All data were analysed with the SPSS/Win programs (version 10.0, SPSS, Chicago, Ill., USA). Results are given as means ± SD or percentages. Variables, which were not normally distributed and skewed towards high values, were logarithmically transformed before statistical analyses. Analysis of variance (ANOVA) was used to compare three groups and Student’s t test for independent samples. The chi square test was used to compare two groups. Non-parametric Mann-Whitney U tests and Kruskal-Wallis H tests were applied to compare weight change (%) and changes (%) between 0-min and 120-min glucose. We considered p values of less than 0.05 to be statistically significant.
Results
In the entire study population the genotype distributions were as follows: Insulin gene: A-23A 62.9%, A-23T 32.2%, T-23T 4.9%; IGF-1R: GAG1013GAG 33.7%, GAG1013GAA 49.0%, GAA1013GAA 17.3%; PC-1: K121K 77.0%, K121Q 21.6%, Q121Q 1.4%; IRS-1: G972G 93.8%, G972R 5.8%, R972R 0.4%; IRS-2: G1057G 44.6%, G1057D 43.7%, D1057D 11.7%; and PI3K: M326M 81.6%, M326I 17.1%, I326I 1.2%. There was no difference in frequencies between the intervention and the control group (data not shown). All genotype frequencies were in Hardy-Weinberg equilibrium. Since the number of homozygous subjects for the rare allele in the insulin, PC-1, IRS-1 and PI3K genes was small (24, 7, 2 and 6 subjects respectively), they were not analysed separately. Baseline characteristics (age, sex, weight, BMI, waist-to-hip ratio, fasting and 120-min levels of glucose and serum insulin) did not differ between the genotypes of any of the polymorphisms investigated (data not shown).
We did not find any difference between the genotypes in relative weight change (Table 1). Subjects in the intervention group lost weight regardless of their genotype for the insulin, PC-1 and PI3K genes. In contrast, the difference in weight change between the intervention and control groups was not significantly different in carriers of (i) the GAA1013GAA genotype of the IGF-1R gene, (ii) the R972 allele of the IRS-1 gene, and (iii) the D1057D genotype of the IRS-2 gene (p=0.148, 0.346 and 0.434 respectively, compared with alternative genotypes, Table 1). These results remained non-significant even after adjustment for baseline weight and for achievement of the intervention goals (weight loss >5%, reduction in fat intake >30% of energy, reduction of saturated fat intake >10% of energy, increase in fibre intake >15 g/4200 kJ, physical exercise >4 h/week).
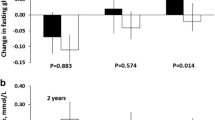
Altogether 69 of 490 subjects included in this analysis converted from IGT to Type 2 diabetes during the 3-year follow-up. The GAG1013GAA genotype of the IGF-1R gene was associated with a lower incidence of Type 2 diabetes than were other genotypes in all DPS subjects (GAG1013GAG 18.6%, GAG1013GAA 10.4% and GAA1013GAA 19.5%, p=0.033) (Fig. 1). The trend was similar in the intervention and control groups, but not statistically significant (p=0.272 and p=0.083 respectively). No other risk genotype predicted conversion from IGT to Type 2 diabetes (Figs. 1, 2). We also calculated the change (%) in 120-min plasma glucose in the OGTT in order to evaluate if a given risk genotype had a significant effect on glucose levels during the follow-up. In the intervention group, in fact, a larger decrease in 2-h glucose was found in subjects carrying the I326 allele than in subjects with the M326M genotype of the PI3K p85α gene (−7.90±30.58 vs −1.38±27.81%, p=0.031). All other comparisons between the genotypes were insignificant both in all subjects and in the intervention and control groups separately (data not shown).

Three-year incidence of Type 2 diabetes in the Finnish Diabetes Prevention Study according to the T-23A polymorphism of the insulin gene (a), the GAG1013GAA polymorphism of the IGF-1R gene (b) and the K121Q polymorphism of the PC-1 gene (c). Results are shown for the entire study population, the intervention group and the control group. Unless otherwise indicated, p values are non-significant. p=0.033 in Total population (b)

Three-year incidence of Type 2 diabetes in the Finnish Diabetes Prevention Study according to the G972R polymorphism of the IRS-1 gene (a), the G1057D polymorphism of the IRS-2 gene (b) and the M326I polymorphism of the PI3K p85α gene (c). Results are shown for the entire study population, the intervention and the control group. Unless otherwise indicated, p values are non-significant
Discussion
Genes regulating the insulin signalling pathway have been associated with insulin resistance or impaired insulin secretion. Therefore, they are promising candidate genes for Type 2 diabetes [1, 5, 36]. To our knowledge there are no previous studies on the effects of common polymorphisms in genes regulating the proximal insulin signalling pathway in IGT subjects with regard to their conversion to Type 2 diabetes.
Overall, the subjects in the intervention group lost more weight than the subjects in the control group, but no difference in weight change within the intervention group or within the control group was found between any of the genotypes compared. However, when we compared weight change between the intervention group and the control group, we found that subjects in the intervention group did not show significant weight loss, if they possessed the risk genotypes for diabetes in the IGF-1R gene (GAA1013GAA genotype), the IRS-1 gene (R972 allele) or the IRS-2 gene (D1057D genotype). This implies that lifestyle intervention was not very successful if the subjects were carrying these risk genotypes for Type 2 diabetes. Thus, risk genes for Type 2 diabetes may also regulate the ability to lose body weight. This conclusion is consistent with our previous study in the Finnish DPS on the PPARγ2 gene. In that study, carriers of the Pro12Pro genotype (a risk genotype for Type 2 diabetes) were not able to lose as much weight as carriers of the 12Ala allele of the PPARγ2 gene in the intervention group [37].
The R972 allele of the IRS-1 gene tended to be associated with a higher conversion rate to Type 2 diabetes than the G972G genotype, particularly in the intervention group. This suggests a possible gene–lifestyle interaction, because the R972 allele responded poorly to lifestyle changes, whereas the incidence of diabetes was significantly lower among carriers of the G972G genotype in the intervention group than among their counterparts in the control group. Due to the small number of subjects, the difference between the genotypes was not statistically significant among the groups.
A previous study showed significantly lower levels of fasting serum insulin and C-peptide among young and healthy heterozygous carriers of a silent GAG1013GAA polymorphism of the IGF-1R gene [38]. However, the authors were not able to repeat their findings in another population. In this study, we did not find any difference in fasting insulin levels between the genotypes of the IGF-1R gene, but in all study subjects the conversion from IGT to diabetes during the 3-year follow-up was significantly lower in heterozygotes, which is consistent with previous findings [38]. We do not have a compelling explanation for our findings, but it is possible that this polymorphism is in linkage disequilibrium with some other locus in the IGF-1R gene or that it interacts with another gene. In our present study it was not possible to evaluate such putative effects.
We were not able to find any association between the variants of the insulin, PC-1, IRS-2 and PI3K p85α genes and weight change or conversion from IGT to diabetes. However, the incidence of diabetes tended to be higher in carriers of the D1057D genotype of the IRS-2 gene and in the carriers of the M326 allele of the PI3K gene than it was in non-carriers of these polymorphisms. This is in accordance with previous findings [16, 29]. Although carriers of the Q121 allele of the PC-1 gene are insulin-resistant and potentially at high risk of developing diabetes [11, 12], there was no difference in incidence of diabetes between carriers and non-carriers of the Q121 allele.
We conclude that common polymorphisms in the IGF-1R, IRS-1 and IRS-2 genes may modify weight change, but none of them predicted conversion from IGT to Type 2 diabetes in the Finnish Diabetes Prevention Study. We did not observe any effect of the common polymorphisms in the insulin, PC-1 and PI3K genes on weight change or conversion to diabetes. This does not, however, exclude the possibility that genes regulating the early insulin signalling pathway could have an effect on the risk of Type 2 diabetes in other populations.
Abbreviations
- DPS:
-
Finnish Diabetes Prevention Study
- IGF-1R:
-
insulin-like growth factor 1 receptor
- PC-1:
-
plasma cell membrane glycoprotein-1
- PI3K:
-
phosphatidylinositol 3-kinase
- PI3K p85α:
-
phosphatidylinositol 3-kinase regulatory subunit p85α
- PPARγ:
-
peroxisome proliferator-activated receptor γ
- VNTR:
-
variable number of tandem repeat
References
Groop LC (1997) The molecular genetics of non-insulin-dependent diabetes mellitus. J Intern Med 241:95–101
Knowler WC, Barrett-Connor E, Fowler SE et al. (2002) Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 346:393–403
Pan XR, Li GW, Hu YH et al. (1997) Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care 20:537–544
Weedon MN, Schwarz PE, Horikawa Y et al. (2003) Meta-analysis and a large association study confirm a role for calpain-10 variation in type 2 diabetes susceptibility. Am J Hum Genet 73:1208–1212
Altshuler D, Hirschorn JN, Klannemark M et al. (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26:76–80
Gloyn AL, Weedon MN, Owen KR et al. (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52:568–572
Saltiel AR, Pessin JE (2002) Insulin signaling pathways in time and space. Trends Cell Biol 12:65–71
Ong KK, Phillips DI, Fall C et al. (1999) The insulin gene VNTR, type 2 diabetes and birth weight. Nat Genet 21:262–263
Le Stunff C, Fallin D, Schork NJ, Bougnères P (2000) The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet 26:444–446
Maddux BA, Goldfine ID (2000) Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes 49:13–19
Frittitta L, Baratta R, Spampinato D et al. (2001) The Q121 PC-1 variant and obesity have additive and independent effects in causing insulin resistance. J Clin Endocrinol Metab 86:5888–5891
Pizzuti A, Frittitta L, Argiolas A et al. (1999) A polymorphism (K121Q) of the human glycoprotein PC-1 gene coding region is strongly associated with insulin resistance. Diabetes 48:1881–1884
Burks DJ, White MF (2001) IRS proteins and beta-cell function. Diabetes 50 [Suppl 1]:S140–S145
Withers DJ, Gutierrez JS, Towery H et al. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
Almind K, Frederiksen SK, Bernal D et al. (1999) Search for variants of the gene-promoter and the potential phosphotyrosine encoding sequence of the insulin receptor substrate-2 gene: evaluation of their relation with alterations in insulin secretion and insulin sensitivity. Diabetologia 42:1244–1249
Mammarella S, Romano F, Di Valerio A et al. (2000) Interaction between the G1057D variant of IRS-2 and overweight in the pathogenesis of type 2 diabetes. Hum Mol Genet 9:2517–2521
Fritsche A, Madaus A, Renn W et al. (2001) The prevalent Gly1057Asp polymorphism in the insulin receptor substrate-2 gene is not associated with impaired insulin secretion. J Clin Endocrinol Metab 86:4822–4825
Stefan N, Kovacs P, Stumvoll M et al. (2003) Metabolic effects of the Gly1057Asp polymorphism in IRS-2 and interactions with obesity. Diabetes 52:1544–1550
Almind K, Bjørbaek C, Vestergaard H, Hansen T, Echwald S, Pedersen O (1993) Amino acid polymorphisms of insulin receptor substrate-1 in non-insulin-dependent diabetes mellitus. Lancet 342:828–832
Sigal RJ, Doria A, Warram JH, Krolewski AS (1996) Codon 972 polymorphism in the insulin receptor substrate-1 gene, obesity, and risk of noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 81:1657–1659
Almind K, Inoue G, Pedersen O, Kahn CR (1996) A common amino acid polymorphism in insulin receptor substrate-1 causes impaired insulin signaling. Evidence from transfection studies. J Clin Invest 97:2569–2575
Porzio O, Federici M, Hribal ML et al. (1999) The Gly972→Arg amino acid polymorphism in IRS-1 impairs insulin secretion in pancreatic beta cells. J Clin Invest 104:357–364
Marchetti P, Lupi R, Federici M et al. (2002) Insulin secretory function is impaired in isolated human islets carrying the Gly(972)→Arg IRS-1 polymorphism. Diabetes 51:1419–1424
Xuan S, Kitamura T, Nakae J et al. (2002) Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J Clin Invest 110:1011–1019
Férnandez AM, Kim JK, Yakar S et al. (2001) Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev 15:1926–1934
Dey BR, Furlanetto RW, Nissley SP (1998) Cloning of human p55 gamma, a regulatory subunit of phosphatidylinositol 3-kinase, by a yeast two-hybrid library screen with the insulin-like growth factor-I receptor. Gene 209:175–183
Shepherd PR, Kahn BB (1999) Glucose transporters and insulin action—implications for insulin resistance and diabetes mellitus. N Engl J Med 341:248–257
Terauchi Y, Tsuji Y, Satoh S et al. (1999) Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet 21:230–235
Baier LJ, Wiedrich C, Hanson RL, Bogardus C (1998) Variant in the regulatory subunit of phosphatidylinositol 3-kinase (p85alpha): preliminary evidence indicates a potential role of this variant in the acute insulin response and type 2 diabetes in Pima women. Diabetes 47:973–975
Hansen T, Andersen CB, Echwald SM et al. (1997) Identification of a common amino acid polymorphism in the p85alpha regulatory subunit of phosphatidylinositol 3-kinase: effects on glucose disappearance constant, glucose effectiveness, and the insulin sensitivity index. Diabetes 46:494–501
Hansen L, Zethelius B, Berglund L et al. (2001) In vitro and in vivo studies of a naturally occurring variant of the human p85alpha regulatory subunit of the phosphoinositide 3-kinase: inhibition of protein kinase B and relationships with type 2 diabetes, insulin secretion, glucose disappearance constant, and insulin sensitivity. Diabetes 50:690–693
Tuomilehto J, Lindström J, Eriksson JG et al. (2001) Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 344:1343–1350
Eriksson J, Lindström J, Valle T et al. (1999) Prevention of Type II diabetes in subjects with impaired glucose tolerance: the Diabetes Prevention Study (DPS) in Finland. Study design and 1-year interim report on the feasibility of the lifestyle intervention programme. Diabetologia 42:793–801
World Health Organization (1985) Diabetes mellitus. Report of a WHO Study Group. World Health Organ Tech Rep Ser 727:1–113
Laakso M, Malkki M, Kekäläinen P, Kuusisto J, Deeb SS (1994) Insulin receptor substrate-1 variants in non-insulin-dependent diabetes. J Clin Invest 94:1141–1146
Goldstein BJ (2002) Insulin resistance as the core defect in type 2 diabetes mellitus. Am J Cardiol 90:3G–10G
Lindi V, Uusitupa M, Lindström J et al.(2002) Association of the Pro12Ala polymorphism in the PPAR-gamma 2 gene with 3-year incidence of type 2 diabetes and body weight change in the Finnish Diabetes Prevention Study. Diabetes 51:2581–2586
Rasmussen SK, Lautier C, Hansen L et al. (2000) Studies of the variability of the genes encoding the insulin-like growth factor I receptor and its ligand in relation to type 2 diabetes mellitus. J Clin Endocrinol Metab 85:1606–1610
Acknowledgements
This study was financially supported by grants from the Academy of Finland (J. Tuomilehto: 38387 and 46558; M. Uusitupa: 40758), the EVO-fund of Kuopio University Hospital (M. Uusitupa: 5106; M. Laakso: 5194), the Ministry of Education, the Finnish Diabetes Research Foundation, the Juho Vainio Foundation, the Yrjö Jahnsson Foundation and the European Union (M. Laakso: QLRT-1999-00674). We are grateful to Päivi Kärkkäinen, Sami Salminen and Guijing Lu for their skilful technical assistance in genotyping the subjects.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Laukkanen, O., Pihlajamäki, J., Lindström, J. et al. Common polymorphisms in the genes regulating the early insulin signalling pathway: effects on weight change and the conversion from impaired glucose tolerance to Type 2 diabetes.. Diabetologia 47, 871–877 (2004). https://doi.org/10.1007/s00125-004-1395-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1395-6