
Abstract
Aims/hypothesis.
Poly(ADP-ribose) polymerase activation depletes NAD+ and high-energy phosphates, activates protein kinase C, and affects gene expression in various tissues. This study was designed to characterise the effects of the potent, orally active poly(ADP-ribose) polymerase inhibitor PJ34 in the Wistar rat model of early diabetic neuropathy.
Methods.
Control and streptozotocin-diabetic rats were maintained with or without PJ34 treatment (30 mg·kg−1·day−1) for two weeks, after two weeks without treatment. Endoneurial blood flow was assessed by hydrogen clearance; metabolites and high-energy phosphates were assayed by enzymatic spectrofluorometric methods; and poly(ADP-ribose) was detected by immunohistochemistry.
Results.
Blood glucose concentrations were increased to a similar extent in untreated and PJ34-treated diabetic rats compared with controls. Intense poly(ADP-ribose) immunostaining was observed in the sciatic nerve of diabetic rats, but not in other groups. Final sciatic motor nerve conduction velocity and digital sensory nerve conduction velocity were reduced by 24% and 22% respectively in diabetic rats compared with controls (p<0.01 for both), and both were 98% corrected by PJ34 (p<0.01 vs diabetic group for both). In contrast, with PJ34 treatment, nerve blood flow showed a modest (17%) increase, and vascular conductance showed a tendency to increase. Free mitochondrial and cytosolic NAD+:NADH ratios, assessed from the glutamate and lactate dehydrogenase systems, phosphocreatine concentrations, and phosphocreatine:creatine ratios were decreased in diabetic rats and essentially normalised by PJ34. In both untreated and PJ34-treated diabetic rats, nerve glucose, sorbitol and fructose were increased to a similar extent. PJ34 did not affect any variables in control rats.
Conclusions/interpretation.
Short-term poly(ADP-ribose) polymerase inhibitor treatment reverses functional and metabolic abnormalities of early diabetic neuropathy. Complete normalisation of nerve blood flow is not required for correction of motor or sensory nerve conduction velocities, provided that a therapeutic agent can restore nerve energy state via direct action on Schwann cells.
Similar content being viewed by others

Introduction
Activation of poly(ADP-ribose) polymerase (PARP) (EC 2.4.2.30), a downstream effector of free radical and oxidant-induced DNA single-strand breakage, plays an important role in a number of pathological conditions associated with oxidative stress, i.e. cardiovascular and neurodegenerative diseases, cancer and inflammation [1]. Growing evidence indicates that PARP activation is an early pivotal mechanism in the pathogenesis of autoimmune diabetes mellitus [2] and chronic diabetic complications [3, 4, 5]. Activation has been documented in beta cells and tissue sites for diabetic complications in streptozotocin (STZ)-diabetic rats and mice [2, 3, 4, 5], as well as in skin vessels of patients with diabetes mellitus [6], and it may lead to diabetic complications via several mechanisms. On the one hand, activation of PARP, which cleaves NAD to form nicotinamide and ADP-ribose residues, depletes NAD and ATP (or other high-energy phosphates), which are utilised for NAD regeneration from nicotinamide [1, 3, 5]. NAD depletion results in the inhibition of the glyceraldehyde 3-phosphate dehydrogenase reaction of glycolysis, and in the diversion of the glycolytic flux towards the formation of diacylglycerol, an activator of protein kinase C (PKC), and the formation of methylglyoxal, a precursor of advanced glycation end products [7]. On the other hand, PARP controls activation of a number of transcription factors, i.e. nuclear factor-kappa B, activator protein-1, signal transducer and activator of transcription-1, octamer-1 and p53 protein via direct binding, poly(ADP-ribosyl)ation or both [8, 9]. This, in turn, up-regulates numerous genes [9, 10], including those implicated in the pathogenesis of peripheral diabetic neuropathy (PDN), such as endothelin-1 [11], cyclooxygenase-2 [12, 13] and inflammatory genes [14]. Thus, PARP activation can be a trigger of multiple mechanisms implicated in diabetes-associated neuropathic changes.
Support for the important role of PARP in PDN was generated by our recent study using PARP-deficient mice and two structurally diverse PARP inhibitors, 3-aminobenzamide and 1,5-isoquinolinediol, in the rat model [5]. However, neither of these are orally active compounds, which makes them unsuitable for chronic experiments. Among other PARP inhibitors, the potent orally active PARP inhibitor PJ34 (the hydrochloride salt of N-(-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide [3, 15]) has been that most extensively studied with regard to diabetic complications [3, 4, 7, 15, 16]. Long-term evaluation of the effect of PJ34 on diabetic retinopathy is in progress (T.S. Kern, personal communication). In contrast to other PARP inhibitors, i.e. nicotinamide and 3-aminobenzamide, PJ34 is highly potent and specific [3, 15], and does not have direct antioxidant properties in concentrations up to 10 mmol/l [3]. This study aimed to evaluate the effects of PJ34 on established functional and metabolic abnormalities of early PDN.
Materials and methods
Reagents
Unless otherwise stated, all chemicals were of reagent grade quality and were purchased from Sigma (St. Louis, Mo., USA). Methanol (HPLC grade), perchloric acid, hydrochloric acid and sodium hydroxide were obtained from Fisher Scientific (Pittsburgh, Pa., USA). Ethyl alcohol (200 proof dehydrated alcohol, USP punctilious) was purchased from McCormick Distilling (Weston, Mo., USA). We purchased β-d-glucose, sorbitol (NF) and d-fructose (USP) from Pfanstiehl Laboratories (Waukegan, Ill., USA). Reagents for immunohistochemistry were purchased from Vector Laboratories (Burlingdale, Calif., USA).
Animals
Model.
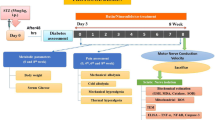
Male Wistar rats (Charles River, Wilmington, Mass., USA), with body weights of 250 to 300 g, were fed a standard rat chow (PMI Nutrition, Brentwood, Mo., USA) and had free access to drinking water. STZ-diabetes was induced as described previously [5, 17, 18]. Blood samples for measurements of glucose were taken from the tail vein approximately 48 h after the STZ injection, and the day before the animals were killed. The rats with blood glucose concentrations of 13.8 mmol/l or more were considered diabetic. The experimental groups comprised control and diabetic rats with or without PJ34 treatment (30 mg·kg body weight−1·day−1 in the drinking water). The treatment began after the initial two weeks without treatment. The duration of treatment was also two weeks. The experiments were performed in accordance with regulations specified by the National Institutes of Health “Principles of Laboratory Animal Care, 1985 Revised Version” and the University of Michigan Protocol for Animal Studies.
Anaesthesia.
We have previously reported [19, 20] that a number of anaesthetics distort the profile of peripheral nerve metabolites, whereas sedation by a short exposure to CO2 (approximately 15–20 s) with immediate cervical dislocation preserves high-energy phosphate levels in the range of those obtained after decapitation without any narcosis. For this reason, two different sets of animals were used for functional and metabolic studies. The functional studies measured nerve blood flow (NBF), motor nerve conduction velocity (MNCV) and sensory nerve conduction velocity (SNCV). Measurements of MNCV and SNCV were taken at the onset (prior to induction of diabetes) and at the 2-week time point (beginning of interventions) in rats anaesthetised with a mixture of ketamine and xylazine (45 mg/kg body weight and 15 mg/kg body weight respectively, both i.p.). For final NBF, MNCV and SNCV measurements, rats were anaesthetised with inactin (65–85 mg/kg body weight, i.p.). Sciatic MNCV and digital SNCV measurements were taken before the assessment of NBF on the contralateral nerve. In all measurements, body temperature was monitored by a rectal probe and maintained at 37 °C with a warming pad. Hind limb skin temperature was also monitored by a thermistor and maintained between 36 and 38 °C by radiant heat. For metabolic studies, the animals were sedated by CO2 in a specially designed chamber [19] and immediately killed by cervical dislocation. The left nerves were rapidly dissected, carefully blotted with fine filter paper to remove any accompanying blood, and frozen in liquid nitrogen for subsequent measurements of glycolytic and tricarboxylic acid cycle intermediates and variables of energy state. The right nerves were divided into two parts. Segments of approximately 50 mg were frozen in liquid nitrogen for subsequent measurements of the sorbitol pathway intermediates. The remaining parts were fixed in 10% formalin and later used for immunohistochemistry.
Specific methods
Functional studies.
Sciatic endoneurial nutritive NBF was assessed by microelectrode polarography and hydrogen clearance [21] as we have already described [5]. Nutritive NBF was taken as the slow component of the curve. An average of two measurements at different sites was used to determine nutritive NBF. Sciatic MNCV and hind limb digital SNCV were measured as previously described [5].
Metabolic studies.
The steady-state concentrations of glutamate, α-ketoglutarate, ammonia, pyruvate, lactate, ATP, phosphocreatine (PCr), creatine (Cr), glucose, sorbitol and fructose were assayed spectrofluorometrically by enzymatic procedures as already described [17, 18, 19, 20]. Free mitochondrial and cytosolic NAD+:NADH ratios were calculated from the steady-state metabolite concentrations and equilibrium constants of the glutamate dehydrogenase and lactate dehydrogenase systems [17, 19].
Assessment of poly(ADP-ribose) immunoreactivity.
All immunohistochemical samples were coded and examined by a single investigator in a blinded fashion. Paraffin-embedded, 5-micron-thick longitudinal sections of sciatic nerve were deparaffinised in xylene and rehydrated in decreasing concentrations of ethanol followed by a 5-min incubation period in PBS. Sections were treated with 0.3% hydrogen peroxide for 15 min to block endogenous peroxidase activity and then rinsed briefly in PBS. Non-specific binding was blocked by incubating the slides for 1 h in 0.25% Triton/PBS containing 2% horse serum. To detect poly(ADP-ribose), a routine histochemical procedure was applied as previously described [5]. Mouse monoclonal anti-poly(ADP-ribose) antibody and isotype-matched control antibody were applied in a dilution of 1:400 for 1 h at room temperature.
Statistical analysis
The results are expressed as means ± SEM. Data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance was attained, individual between-group comparisons were made using the Student-Newman-Keuls multiple range test. Results were considered significant at a p value of 0.05 or less. When between-group variance differences could not be normalised by log transformation (datasets for body weight, plasma glucose and some metabolic parameters), the data were analysed by non-parametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni-Dunn test for multiple comparisons.
Results
Final body weights were lower in diabetic rats than in controls (p<0.01, Table 1). They were slightly, but significantly, lower in the PJ34-treated diabetic rats than in the corresponding untreated group (p<0.05). PJ34 did not affect weight gain in control rats. Blood glucose concentrations were 4.7-fold higher in diabetic rats than in controls (p<0.01). PJ34 did not affect blood glucose concentrations in control or diabetic rats.
Sciatic nerve poly(ADP-ribose) immunoreactivity was not detected in control rats with or without PJ34 treatment (Fig. 1). It was clearly manifest in diabetic rats. Several nuclei stained for poly(ADP-ribose) were present in sciatic nerves of PJ34-treated diabetic rats, although the immunoreactivity was essentially suppressed compared with the untreated diabetic group.

Representative microphotographs of nuclear poly(ADP-ribose) immunostaining in the sciatic nerves of control and diabetic rats with and without PJ34 treatment. Magnification ×400; C, control group; D, diabetic group
The onset MNCV and SNCV values (prior to induction of STZ-diabetes) for all experimental groups, as well as the MNCV and SNCV values at the beginning of interventions, i.e. the 2-week time point, are given in Table 2. Final MNCV and SNCV (Fig. 2a and b) were reduced by 24% and 22% in diabetic rats compared with controls. Both MNCV and SNCV deficits were 98% corrected by PJ34 treatment. PJ34 had no effect on MNCV or SNCV in non-diabetic rats.

Final motor (a) and sensory (b) nerve conduction velocities, nerve blood flow (c), blood pressure (d) and vascular conductance (e) in control and diabetic rats with and without PJ34 treatment. Data are means ± SEM; n=6–14; **p<0.01 vs control group; ## p<0.01 vs untreated diabetic group; C, control group; D, diabetic group; MNCV, motor nerve conduction velocity; SNCV, sensory nerve conduction velocity; NBF, nerve blood flow; VC, vascular conductance
The diabetes-induced 50% reduction in sciatic endoneurial nutritive NBF was reversed by 67% through PJ34 treatment (Fig. 2c). The diabetes-associated 80% decrease in mean systemic blood pressure was corrected by PJ34 to a level that was not significantly different from control or diabetic rats (Fig. 2d). The diabetes-induced 37% deficit in endoneurial vascular conductance (Fig. 2e), i.e. NBF normalised for blood pressure, tended to increase with PJ34 treatment, but the difference from the untreated group was not statistically significant.
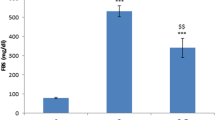
Nerve glutamate concentrations were higher in diabetic rats than in the control group (p<0.01, Table 3). PJ34 treatment normalised glutamate concentrations in diabetic rats, but did not affect them in control rats. Concentrations of α-ketoglutarate were reduced in diabetic rats. This variable was not affected by PJ34 treatment in control rats but was increased by PJ34 in diabetic rats. Ammonia concentrations were similar in all groups. Lactate concentrations were higher, and pyruvate concentrations lower in diabetic rats than in controls. Lactate concentrations were reduced to below the control level in PJ34-treated diabetic rats (p<0.01 vs untreated diabetic group and p<0.05 vs control group), whereas pyruvate concentrations remained in the range found in the untreated diabetic group. Treatment with PJ34 reduced lactate concentrations in control rats (p<0.01 vs control group).
Free mitochondrial and cytosolic NAD+:NADH ratios were lower in diabetic rats than in the control group (p<0.01 for both). The decrease in both ratios was essentially corrected by PJ34 (p<0.01 vs untreated diabetic group). PJ34 did not affect the redox state of mitochondrial or cytosolic NAD couples in control rats.
Nerve ATP concentrations were similar in all groups (Table 4). PCr concentration and PCr:Cr ratio, but not Cr concentration, were higher in diabetic rats than in controls (p<0.01 and p<0.05 respectively). PJ34 treatment did not affect PCr and Cr concentrations and PCr:Cr ratio in control rats. In diabetic rats, PJ34 increased PCr concentration (p<0.05 vs untreated diabetic group) and essentially normalised PCr:Cr ratio (p<0.01 vs untreated diabetic group), but did not affect Cr concentrations.
Nerve glucose, sorbitol and fructose concentrations were 4.6-fold, 12.7-fold and 5.1-fold higher respectively in diabetic rats than in the control group (p<0.01 for all three comparisons, Table 5). These concentrations were not affected by PJ34 treatment in control or diabetic rats.
Discussion
“The neuropathic changes are reversible until they affect the phenotype of neurons and Schwann cells” (D. R. Tomlinson, personal communication). The validity of this postulate is supported by numerous reversal studies with inhibitors of aldose reductase [17] and PKC [22], with antioxidants [23] and with other agents, all indicating that consequences of diabetic hyperglycaemia such as increased sorbitol pathway activity, oxidative stress and PKC activation do not cause irreversible changes in the peripheral nervous system, at least in the early stage of PDN. More recently, it has been shown that intervention even further downstream than hyperglycaemia, at the level of oxidative-stress-initiated, mitogen-activated protein kinase signalling cascade, successfully restores normal nerve function [24]. The reversal of functional and metabolic abnormalities of early PDN has been achieved in this study and in our previous study [5] with several structurally diverse inhibitors of the enzyme PARP, a downstream effector of DNA oxidative damage [1].
Activation of PARP is a response to increased free radical and oxidant (peroxynitrite) generation [1] present in neural elements and vasa nervorum of the peripheral nervous system [5, 25]. A number of studies, including those from our group, provide evidence of increased lipid peroxidation [17, 18, 19, 26], impaired oxidative defence [17, 18, 19, 20, 26], and superoxide formation [27, 28] in the peripheral nerve. Furthermore, recent findings [27, 29] indicate increased formation of peroxynitrite, a product of the reaction between superoxide anion radicals and nitric oxide, in experimental models and human subjects with PDN. Studies with the potent peroxynitrite decomposition catalyst FP15 [30, 31] suggest that peroxynitrite is primarily responsible for enhanced poly(ADP-ribosyl)ation in a number of pathological conditions associated with oxidative stress. We have recently found that FP15 administered at the very low dose of 5 mg/kg for one week after two months of untreated STZ-diabetes completely reverses peripheral nerve nitrotyrosine and poly(ADP-ribose) accumulation, as well as MNCV and SNCV deficits of early PDN in the mouse model (I. G. Obrosova and C. Szabó, unpublished).
Poly(ADP-ribose) accumulation, the product of PARP activation, is found in all major tissue sites for diabetic complications, including in the vascular endothelium [3, 4], myocardium [4], retina [16, 32] and kidney [10] in rat and mouse models of STZ-diabetes. Recent findings, including those from our group, revealed the presence of diabetes-associated PARP activation in dorsal root ganglion neurons [25] and endothelial and Schwann cells of the peripheral nerve [5]. Growing evidence implicates PARP in the pathogenesis of chronic diabetic complications including endothelial and myocardial dysfunction [3, 4], peripheral [5] and autonomic [33] neuropathy, and early retinopathy [16, 32, 34]. Direct evidence that PARP activation plays a role in diabetic nephropathy is still missing, although we have recently demonstrated that PARP activation is responsible for the overexpression of endothelin-1 and endothelin A and B receptors [10], the important players in diabetic kidney disease [35].
In our study, the PARP inhibitor PJ34 essentially normalised MNCV and SNCV deficits and important metabolic abnormalities of early PDN such as free mitochondrial and cytosolic NAD+:NADH ratios, and energy deficiency. All these effects were specific to diabetic animals. In contrast, the effect of PARP inhibitor on nerve perfusion, manifested by a 17% increase in NBF and the absence of a significant increase in vascular conductance, was modest. While studies from several groups [17, 22, 23, 27, 28, 36], including one from our laboratory [17], suggest that vascular factor plays an important role in MNCV and SNCV deficits in PDN, growing evidence indicates that the aetiology of diabetes-induced nerve dysfunction is far more complex, and involves both vascular and non-vascular mechanisms. In particular, in diabetic and galactosaemic transgenic mice overexpressing aldose reductase (AR) specifically in the Schwann cells of the peripheral nerve under the control of the rat myelin protein zero promoter, there was a significantly greater reduction in MNCV than in the corresponding groups of non-transgenic mice with normal AR content [37]. Nerve function, but not NBF, has been preserved in the normal range in diabetic rats treated with the neurotrophic factor, prosaposin [38], or with sonic hedgehog protein [39]. All of these effects [37, 38, 39] have been observed in rats and mice with relatively early PDN and diabetes-like galactose-induced neuropathy lacking the signs of nerve demyelination, and are probably related to reversible modifications of the metabolic and neurotrophic state of the neural elements of the peripheral nervous system, i.e. neurons and Schwann cells. In our previous study [17], use of the vasodilator α1-adrenoceptor antagonist prazosin to prevent diabetes-induced nerve perfusion deficit as manifested by normal vascular conductance failed to preserve normal mitochondrial oxidative capacity. The latter is quite understandable in light of recent findings suggesting the dependence of inner mitochondrial membrane potential on insulin-dependent neurotrophic support [40], which is likely to be unrelated to the state of perfusion, but which is affected by intracellular oxidative stress [26, 41]. Our present findings demonstrate that inhibition of diabetes-induced PARP activation, which is known to be responsible for NAD depletion [1, 3] and down-regulation of glyceraldehyde 3-phosphate dehydrogenase (or insufficient up-regulation as in the peripheral nerve) [7], as well as for decreased rates of glycolysis and mitochondrial oxidation [1], restores normal nerve energy state, the metabolic variable that correlates best with nerve conduction [5, 17, 18, 20]. Unfortunately, the specific mechanisms by which diabetes disrupts electric impulse transmission in the peripheral nerve are not understood, which impedes the investigation of the role of any neuronal or Schwann cell factor, e.g. oxidative stress or poly(ADP-ribosyl)ation in diabetes-associated nerve conduction slowing. Future studies with PARP-antisense oligodeoxynucleotides delivered in viral vectors to specific elements of the peripheral nervous system could further elucidate the role of vascular and non-vascular, i.e. neuronal and Schwann cell, PARP in the pathogenesis of PDN. PARP antisense oligodeoxynucleotides have already been successfully employed by several laboratories [42, 43].
The relation between PARP and the sorbitol pathway of glucose metabolism is interesting in light of some recent developments in the field of diabetic complications. According to the “unifying concept” of diabetic complications [44], oxidative stress, and in particular mitochondrial superoxide production, is responsible for increased AR activity, formation of advanced glycation end products and activation of PKC. Recently, it has been reported that hyperglycaemia-induced activation of at least two of these mechanisms, i.e. glycation and PKC, is mediated via PARP-activation-linked inhibition of glycolysis associated with increased formation of methylglyoxal and diacylglycerol [7]. The role of PARP in diabetes-induced sorbitol pathway hyperactivity has never been explored. At the same time, growing evidence generated in studies with aldose reductase inhibitors [18, 45, 46] and antioxidants [20], as well as AR-overexpressing [36, 47] and AR-knockout mice [48], indicates that AR activation precedes and is largely responsible for enhanced oxidative stress in tissue sites for diabetic complications. Furthermore, a recent study of endothelial cells exposed to high glucose concentrations [46], and the studies of our group on the peripheral nerve, retina and kidneys of diabetic rats (I. G. Obrosova and P. Pacher, unpublished) suggest that AR is also a key player in peroxynitrite-induced nitrosative stress, which is a major contributor to PARP activation. The fact that PJ34 had no effect on diabetes-associated accumulation of peripheral nerve sorbitol pathway intermediates is consistent with the localisation of PARP downstream from increased AR activity and oxidative-nitrosative stress in the pathogenesis of PDN and other diabetic complications.
In conclusion, this short-term study with the orally active PARP inhibitor, PJ34, saw the reversal of established functional and metabolic abnormalities of early PDN in the STZ-diabetic rat model. Chronic experiments are needed to establish whether PARP inhibitors are effective against demyelination, nerve fibre loss and other indices of advanced PDN, and whether or not long-term suppression of PARP, the enzyme involved in DNA repair [1], is associated with adverse side effects. If not, it could be a novel approach for the prevention and treatment of diabetic complications.
Abbreviations
- AR:
-
aldose reductase
- Cr:
-
creatine
- MNCV:
-
motor nerve conduction velocity
- NBF:
-
nerve blood flow
- SNCV:
-
sensory nerve conduction velocity
- PARP:
-
poly(ADP-ribose) polymerase
- PDN:
-
peripheral diabetic neuropathy
- PKC:
-
protein kinase C
- PCr:
-
phosphocreatine
- STZ:
-
streptozotocin
References
Virag L, Szabo C (2002) The therapeutic potential of poly (ADP-Ribose) polymerase inhibitors. Pharmacol Rev 54:375–429
Suarez-Pinzon Wl, Mabley JG, Power R, Szabo C, Rabinovitch A (2003) Poly (ADP-ribose) polymerase inhibition prevents spontaneous and recurrent autoimmune diabetes in NOD mice by inducing apoptosis of islet-infiltrating leukocytes. Diabetes 52:1683–1688
Garcia Soriano F, Virag L, Jagtap P et al. (2001) Diabetic endothelial dysfunction: the role of poly (ADP-ribose) polymerase activation. Nature Med 7:108–113
Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C (2002) The role of poly (ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes 51:514–521
Obrosova IG, Li F, Abatan OI et al. (2004) Role for poly (ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes 53:711–720
Szabo C, Zanchi A, Komjati K et al. (2002) Poly (ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation 106:2680–2686
Du X, Matsumura T, Edelstein D et al. (2003) Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112:1049–1057
Soldatenkov VA, Chasovskikh S, Potaman VN, Trofimova I, Smulson ME, Dritschilo A (2002) Transcriptional repression by binding of poly (ADP-ribose) polymerase to promoter sequences. J Biol Chem 277:665–670
Ha HC, Hester LD, Snyder SH (2002) Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA 99:3270–3275
Minchenko AG, Stevens MJ, White L et al. (2003) Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly (ADP-ribose) polymerase activation. FASEB J 17:1514–1516
Stevens EJ, Tomlinson DR (1995) Effects of endothelin receptor antagonism with bosentan on peripheral nerve function in experimental diabetes. Br J Pharmacol 115:373–379
Freshwater JD, Svensson CI, Malmberg AB, Calcutt NA (2002) Elevated spinal cyclo-oxygenase and prostaglandin release during hyperalgesia in diabetic rats. Diabetes 51:2249–2255
Pop-Busui R, Marinescu V, Van Huysen C et al. (2002) Dissection of metabolic, vascular, and nerve conduction interrelationships in experimental diabetic neuropathy by cyclooxygenase inhibition and acetyl-L-carnitine administration. Diabetes 51:2619–2628
Vinik AI (1999) Diabetic neuropathy: pathogenesis and therapy. Am J Med 107:17S–26S
Soriano FG, Pacher P, Mabley J, Liaudet L, Szabo C (2001) Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res 89:684–691
Zheng L, Peachey N, Szabo C, Kern T (2003) PARP inhibitor corrects diabetes-induced alterations in retinal function and leukostasis. Diabetes 52 [Suppl 1]:A205 (Abstract)
Obrosova IG, Van Huysen C, Fathallah L, Cao X, Stevens MJ, Greene DA (2000) Evaluation of alpha(1)-adrenoceptor antagonist on diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J 14:1548–1558
Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ (2002) An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J 16:123–125
Obrosova IG, Fathallah L, Lang HJ, Greene DA (1999) Evaluation of a sorbitol dehydrogenase inhibitor on diabetic peripheral nerve metabolism: a prevention study. Diabetologia 42:1187–1194
Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA (2000) Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes 49:1006–1105
Tuck RR, Schmelzer JD, Low PA (1984) Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain 107:935–950
Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC (1999) Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia 42:1120–1130
Cameron NE, Tuck Z, McCabe L, Cotter MA (2001) Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia 44:1161–1169
Agthong S, Tomlinson DR (2002) Inhibition of p38 MAP kinase corrects biochemical and neurological deficits in experimental diabetic neuropathy. Ann NY Acad Sci 973:359–362
Cheng C, Zochodne DW (2003) Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes 52:2363–2371
Obrosova IG, Fathallah L, Stevens MJ (2001) Taurine counteracts oxidative stress and nerve growth factor deficit in early experimental diabetic neuropathy. Exp Neurol 172:211–219
Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA (2001) Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 50:1927–1937
Coppey LJ, Gellett JS, Davidson EP, Yorek MA (2003) Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium-dependent vasodilation. Free Radic Res 37:33–40
Hoeldtke RD, Bryner KD, McNeill DR et al. (2002) Nitrosative stress, uric acid, and peripheral nerve function in early type 1 diabetes. Diabetes 51:2817–2825
Szabo C, Mabley JG, Moeller SM et al. (2002) Part I: Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med 8:571–580
Pacher P, Liaudet L, Bai P et al. (2003) Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation 107:896–904
Obrosova I, Seigel G, Frank R et al. (2003) Early diabetes-induced changes in the retina: comparison of STZ-induced models in rat and mouse. Diabetes 52 [Suppl 1]:A202 (Abstract)
Cameron NE, Gibson TM, Cotter MA (2003) Effects of poly(ADP-ribose) polymerase inhibition on large and small nerve fibre function in experimental diabetes. Diabetologia 46 [Suppl 2]:A315 (Abstract)
Obrosova I, Minchenko A, Kennedy A, Szabo C, Frank R, Stevens M (2002) Poly(ADP-ribose) synthetase inhibitors counteract diabetes- and hypoxia-induced retinal VEGF formation. Ophthalm Res 34 [Suppl 1]:128 (Abstract)
Cooper ME (2001) Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 44:1957–1972
Low PA, Nickander KK, Tritschler HJ (1997) The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes 46 [Suppl 2]:S38–42
Song Z, Fu DTW, Chan YS, Leung S, Chung SSM, Chung SK (2003) Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol Cell Neurosci 23:638–647
Mizisin AP, Steinhardt RC, O’Brien JS, Calcutt NA (2001) TX14(A), a prosaposin-derived peptide, reverses established nerve disorders in streptozotocin-diabetic rats and prevents them in galactose-fed rats. J Neuropathol Exp Neurol 60:953–960
Calcutt NA, Allendoerfer KL, Mizisin AP et al. (2003) Therapeutic efficacy of sonic hedgehog protein in experimental diabetic neuropathy. J Clin Invest 111:507–514
Huang TJ, Price SA, Chilton L et al. (2003) Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes 52:2129–2136
Hounsom L, Corder R, Patel J, Tomlinson DR (2001) Oxidative stress participates in the breakdown of neuronal phenotype in experimental diabetic neuropathy. Diabetologia 44:424–428
Simbulan-Rosenthal CM, Rosenthal DS, Ding R et al. (1998) Prolongation of the p53 response to DNA strand breaks in cells depleted of PARP by antisense RNA expression. Biochem Biophys Res Commun 253:864–868
Ullrich O, Diestel A, Eyupoglu IY (2001) Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol 3:1035–1042
Nishikawa T, Edelstein D, Du XL et al (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Obrosova IG, Minchenko AG, Vasupuram R et al. (2003) Aldose reductase inhibitor fidarestat prevents retinal oxidative stress and vascular endothelial growth factor overexpression in streptozotocin-diabetic rats. Diabetes 52:864–871
El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB (2003) High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci 44:3135–3143
Lee AY, Chung SS (1999) Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 13:23–30
Chung SS, Ho EC, Lam KS, Chung SK (2003) Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 14 [Suppl 3]:S233–236
Acknowledgements.
The study was supported by the American Diabetes Association Research Grant and the National Institutes of Health Grant DK59809-01 (both to I. G. Obrosova), the Juvenile Diabetes Research Foundation Center for the Study of Complications of Diabetes Grant 4-200-421 (to I. G. Obrosova and M. J. Stevens), and the National Institutes of Health Grant HL/DK 71215-01 (to C. Szabó).
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Pacher’s present address is NIAAA, DICBR, Laboratory of Physiologic Studies, Section of Neuroendocrinology, 12420 Parklawn Drive, Rm 445, Rockville, Maryland 20852, USA. P. Pacher is on leave from the Department of Pharmacology, Semmelweis University, Budapest, Hungary.
Rights and permissions
About this article
Cite this article
Li, F., Szabó, C., Pacher, P. et al. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetologia 47, 710–717 (2004). https://doi.org/10.1007/s00125-004-1356-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1356-0