
Abstract
Aims/hypothesis
Activation of apoptosis in embryos is thought to be a key event in the pathogenesis of diabetes-induced embryopathies such as early embryonic death and inborn structural anomalies. TNF-α can activate apoptotic and anti-apoptotic signalling cascades, indicating its ability to contribute to and counteract diabetes-induced maldevelopment. To investigate how TNF-α regulates the response of embryos to diabetes-induced embryopathic stress, we used streptozotocin-induced diabetic TNF-α knockout mice.
Materials
To evaluate the reproductive performance, mated diabetic female mice were examined on days 4 and 8 of pregnancy for the presence of blastocysts or embryos in uterine horns. To evaluate the teratogenic effect, the female mice were killed on day 18 of pregnancy and fetuses were examined for gross external anomalies. In addition, apoptotic nuclei were localised by the TUNEL assay and DNA-binding activity of the transcription factor NF-κB was evaluated by electrophoretic mobility shift assay in 10- and 11-day-old embryos respectively.
Results
Severely diabetic TNF-α+/+ female mice had a much greater decrease in pregnancy rate but a lower incidence of malformed fetuses in litters than severely diabetic TNF-α−/− female mice. Also, the intensity of excessive apoptosis was higher, but the amount of active NF-κB complexes was lower in malformed TNF-α−/− embryos than in TNF-α+/+ embryos.
Conclusions/interpretation
TNF-α contributes to death of peri-implantation embryos and possibly protects postimplantation embryos exposed to diabetes-induced teratogenic stimuli via activation of NF-κB-mediated anti-apoptotic signalling. It seems that TNF-α prevents the birth of malformed offspring in severely diabetic mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Tumour necrosis factor-α, a multifunctional cytokine, is expressed by uterine, placental and embryonic cells practically at all stages of development [1, 2]. In the last decade convincing evidence has been collected that TNF-α plays a role in mediating diabetes-induced adverse effects on early embryos. Thus the amount of TNF-α released into the culture supernatant by uterine explants dissected from diabetic rats at the time of implantation was four times higher than in uterine explants dissected from non-diabetic rats [3]. Blastocysts incubated in media preconditioned with uterine explants from diabetic rats developed less well than those incubated in control media [4]. Finally various studies [5, 6, 7] showed that the pregnancy rate of chemically induced diabetic mice is lower than in their non-diabetic counterparts. This decrease in pregnancy rate was accompanied by increased expression of TNF-α in uterine cells from the beginning of implantation onwards [8].
Other data suggest that TNF-α could be involved in the formation of teratogen-induced structural anomalies. We, for example, observed increased expression of TNF-α mRNA and protein in mouse embryos exposed to a teratogen such as cyclophosphamide at doses which induce structural anomalies [9]. Also, the expression of TNF-α protein was higher in embryos cultured in a media with teratogenic concentrations of glucose [10].
Teratogens usually initiate maldevelopment by activating the apoptosis program in target embryonic structures [11]. It is conceivable that TNF-α, with its potential to activate apoptotic signalling cascades [12], acts as a mediator of stimuli inducing embryonic maldevelopment. However, TNF-α has also been shown to activate anti-apoptotic signalling cascades, the most effective of which is associated with activation of the transcription factor NF-κB, which in turn activates a number of genes encoding anti-apoptotic proteins [12, 13]. Our studies [14, 15] suggest that NF-κB activity in embryos responding to a teratogenic stress correlate directly with the embryos’ resistance to the stress.
Due to the ability of TNF-α to activate apoptotic and anti-apoptotic signalling cascades, its functional role in the regulation of embryo response to diabetes-induced embryopathic stress is not clear.
Together, the above results prompted us to do a comparative study in chemically induced diabetic TNF-α−/− and TNF-α+/+ mice. The aim was to evaluate the role of TNF-α in mediating diabetes-induced embryopathies like early embryonic death and gross structural inborn anomalies. We also compared the intensity of apoptosis and NF-κB DNA-binding activity in postimplantation embryos which had undergone a diabetes-induced teratogenic insult.
Materials and methods
Animal model
We used TNF-α−/− (B6;129S-Tnftm1Gkl) and the wild-type B6129SF2/J mice (recommended as an appropriate control for TNF-α−/− mice), which were purchased from Jackson Laboratories (Bar Harbor, Me., USA). The mice were maintained on a light and dark cycle of 14 h and 10 h respectively with free access to food and tap water. The female mice were caged with males for 3 hours, from 7 to 10 a.m. (dark period) and examined for the presence of a vaginal plug at 11 a.m. The day on which a vaginal plug was found was designated as day 1 of pregnancy.
To induce diabetes, mice were injected i.p. with 240 mg/kg streptozotocin (STZ) (Sigma, St Louis, Mo., USA) in buffer acetate (pH 4.2) immediately after detection of vaginal plugs. Two types of control mice were used in this study: age-matched intact pregnant females and mated female mice injected with STZ but without hyperglycaemia at the tested time points. Animal experiments were approved by the Ethics Committee for Animal Use of Tel Aviv University and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23 revised 1996). They were carried out according to the Declaration of Helsinki as revised in 2000.
Assessment of diabetic status
Glucose concentrations in the maternal blood were measured with a commercially available glucometer (MediSense, Abbott Laboratories, Bedford, Mass., USA) in blood samples obtained from the orbital sinus. In preliminary experiments we found that the blood glucose concentration (BGC) in intact TNF-α+/+ and TNF-α−/− female mice is 7.7±2.2 mmol/l (means ± 3 SD, n=8) and 7.9±2.0 mmol/l (n=9) respectively. Based on these data, STZ-treated female mice with a BGC of 10 mmol/l or more at the tested time points were considered to be diabetic.
In all STZ-treated mated females, the BGC was measured 2 days after the STZ injection and then on days 4, 8 or 9 of pregnancy. Only diabetic female mice with the same BGC at both measurements or an increased BGC at the second measurement were used in the study. Accordingly, STZ-treated female mice with a BGC of less than 10 mmol/l at both tested time points were considered to be non-diabetic and used as control mice.
Evaluation of the reproductive performance and teratological testing
The mated female mice were divided into three groups according to tested time points, i.e. days 4, 8 and 18 of pregnancy. On day 4 of pregnancy mice were tested for the presence of blastocysts in the uterus as described elsewhere [16] and blastocysts were visually categorised as normal or fragmented by light microscopy. On day 8 of pregnancy the mice were examined for the presence of embryos in the uterus. On day 18 of pregnancy, implantation sites and resorptions were recorded and all live fetuses were examined for gross external malformations and weighed and fixed in Bouin’s solution. To verify the existence of anomalies like micro- and anophthalmia and to reveal such an anomaly as cleft palate, the heads of these embryos were analysed using Wilson’s sectioning technique [5]. The BGC in these mice was measured on day 9 of pregnancy, which is thought to be the day of the highest sensitivity of murine embryos to diabetes-induced gross structural anomalies [17].
Apoptosis assay
Three 10-day-old embryos with open neural folds and three normal embryos were obtained from different litters of diabetic TNF-α+/+ or TNF-α−/− mice. Also, three normal embryos were obtained from different litters of control non-diabetic TNF-α+/+ and TNF-α−/− female mice. The embryos were fixed in formaldehyde, dehydrated in alcohol and xylene and embedded in paraffin. Transversal tissue sections of the head (7 µmol/l) were prepared and analysed by the TUNEL assay to localise apoptotic nuclei as described elsewhere [15].
Electrophoretic mobility shift assay (EMSA)
The DNA-binding activity of NF-κB was measured in nuclear extracts isolated from the head of 11-day-old embryos. The BGC was measured on day 9 of pregnancy. Embryonic tissue samples collected from four to five litters were pooled and used to prepare a sample of the nuclear extract [18]. The protein concentration was measured using a DC Protein Assay kit (Bio-Rad Laboratories, Calif., USA) and a 550 Microplate Reader (Bio-Rad Laboratories) according to the manufacturer’s instructions. Samples were frozen in aliquots in liquid nitrogen and stored at −70°C until use.
The EMSA was done with a DIG Gel Shift Kit (Roche Molecular Biochemicals, Mannheim, Germany) as described elsewhere [15], using a double stranded oligonucleotide containing the NF-κB site (5′AGTTGAGGGGACTTTCCCAGGC3′, 3′TCAACTCCCCTGAAAGGGTCCG5′, Santa Cruz Biotechnology, Santa Cruz, Calif., USA) and labelled with DIG-11-ddUTP (Roche Molecular Biochemicals). For NF-κB binding specificity, a 100-fold excess of unlabelled NF-κB or Oct2A oligonucleotides was added to the binding reaction. The NF-κB complexes were identified by chemiluminescent detection and membranes were exposed to X-ray film (Kodak, Hemel Hempstead, UK). Films were scanned on a UMAX Power Look III scanner (UMAX Technologies, Dallas, Tex., USA) attached to a PC computer. A Bio Image System 202D densitometer (Rhenium, Jerusalem, Israel) and TINA v.2.07d Software (Raytest Isotopen Meßgeräte, Straubenhardt, Germany) were used to measure the density of bands of interest and a background in a given gel and to calculate final values. The results of the EMSA were reproduced in three independent experiments with different samples isolated from embryos of the control and experimental groups.
Statistical analysis
Statistical analysis of the teratological data was done on a litter basis. The proportion of resorptions and malformed fetuses per litter were transformed to arcsine values by Freeman-Tukey’s method [19]. The number of implantation sites, the resorption rate, the fetal weight and the proportion of malformed fetuses were analysed statistically by the GT-2 method for multiple (a posteriori) comparisons [20]. The pregnancy rate (proportion of pregnant mice) and the proportion of litters with malformed fetus were analysed by Fisher’s exact test. The two-tailed significance of differences was considered to be given by a p value of 0.05 or less.
Results
Reproductive performance of diabetic mice tested on day 18 of pregnancy
TNF-α+/+ female mice
In diabetic female mice indices like the number of implantation sites and fetal weight did not differ statistically from those in non-diabetic female mice, regardless of whether they were intact or treated with STZ (Table 1). The pregnancy rate was lower in diabetic than in non-diabetic female mice (approximately 23% vs 52 to 53%) (Table 1). The resorption rate in these diabetic mice (17.7%) was not statistically different from that in the concurrent control groups of non-diabetic female mice (7.4 to 5.4%; Table 1), but as in our previous studies, did exceed the rate seen in our “historical” control mice (12 to 13%).
TNF-α−/− female mice
These mice also had a lower pregnancy rate than non-diabetic TNF-α−/− female mice (approximately 40% vs 59.1 to 61.5%), but it was higher (p<0.05) than the pregnancy rate in TNF-α+/+ diabetic mice (approximately 23%; Table 1). Moreover, the weight of fetuses of TNF-α−/− diabetic mice was lower (0.44 g, p<0.05) than that of fetuses of TNF-α+/+ diabetic (0.69 g) and non-diabetic mice (0.69 to 0.74 g; Table 1). The resorption rate in TNF-α−/− diabetic mice (25.8%) was comparable with that in TNF-α+/+ diabetic mice (17.7%), and was also higher than that in non-diabetic TNF-α−/− mice (12.3 to 12.5%; Table 1), although this difference was statistically insignificant.
Differences between TNF-α+/+ and TNF-α−/− female mice became more distinct when populations of diabetic mice were divided into three groups according to the severity of diabetes.
Severely diabetic female mice (BGC 19.4 mmol/l or higher)
In severely diabetic TNF-α+/+ mice the pregnancy rate was only 11.6% (Fig. 1a). In severely diabetic TNF-α−/− mice it was lower than in the control non-diabetic mice (approximately 35%) (Fig. 1a), but still higher than in diabetic TNF-α+/+ female mice (Fig. 1a). These TNF-α−/− and TNF-α+/+ mice also differed with regard to the weight of their fetuses (Fig. 1b). However, whereas the decrease in the pregnancy rate was much more prominent in severely diabetic TNF-α+/+ female mice, the weight of their fetuses was significantly higher than in TNF-α−/− mice (approximately 0.56 g vs 0.4 g respectively, p<0.05; Fig. 1b). Remarkably, the resorption rate was practically identical in severely diabetic TNF-α+/+ and TNF-α−/− female mice (approximately 28% and 24% respectively).

Pregnancy rate and weight of fetuses of diabetic mice. Pregnancy rate (a) tested on day 18 of pregnancy. Weight of fetuses (b) tested on day 18 of pregnancy. Pregnancy rate tested on day 4 (c) and day 8 (d) of pregnancy. Values in bars represent the number of mated female mice. Data for pregnancy rate were analysed statistically by Fisher’s exact test (*p<0.05). Results representing fetal weight were analysed by the GT2 method and are presented as 95% comparison limits for the means. Means with limits that do not overlap are significantly different
Moderately and weakly diabetic female mice
Moderately diabetic TNF-α−/− and TNF-α+/+ female mice (BGC between 13.9 mmol/l and 19.4 mmol/l) had an equally decreased pregnancy rate (approximately 38% and 40% respectively; Fig. 1a), but the weight of TNF-α−/− fetuses was lower (p<0.05) than that of TNF-α+/+ fetuses (Fig. 1b). The weight of fetuses was also lower (p<0.05) in weakly diabetic (BGC between 10 mmol/l and 13.9 mmol/l) TNF-α−/− mice, but not in weakly diabetic TNF-α+/+ mice (Fig. 1b).
Reproductive performance of mice tested on days 4 and 8 of pregnancy
Neither implantation sites nor resorptions were found in uteri of mated diabetic mice considered not to be pregnant on day 18 of pregnancy. To find out whether this outcome of mating resulted from early embryonic lethality or if it was due to peri-implantation death of embryos, we assessed the pregnancy rate of mice tested on days 4 (pre-implantation period) and 8 (end of implantation period) of pregnancy.
Day 4 of pregnancy
The pregnancy rate was practically identical in all groups of diabetic and non-diabetic TNF-α+/+ and TNF-α−/− mice (Fig. 1c) and did not differ from that in non-diabetic mice tested on day 18 of pregnancy (Fig. 1a). Also, the proportion of blastocysts with morphologically degenerated blastomeres (“fragmented” blastocytes) in groups of STZ-treated female mice did not differ from that seen in intact non-diabetic female mice (single blastocysts with morphologically degenerated blastomeres, data not presented).
Day 8 of pregnancy
The pregnancy rate of diabetic mice tested at this time point (Fig. 1d) was comparable with that observed in mice tested on day 18 of pregnancy (Fig. 1a). Remarkably, the pregnancy rate was approximately 13% in severely diabetic TNF-α+/+ diabetic mice tested on day 8 of gestation (Fig. 1d) and approximately 12% in those tested on day 18 of gestation (Fig. 1a).
The teratogenic response of diabetic female mice
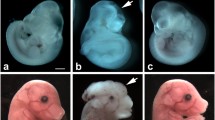
No fetuses with gross structural anomalies except single ones with microphthalmia were registered in intact and STZ-treated non-diabetic female mice (Table 2). Maternal diabetes in TNF-α+/+ and TNF-α−/− mice was associated with the occurrence of fetuses with structural anomalies such as micro- and anophthalmia, encephalocele, cleft face, cleft palate and gastroschisis.
TNF-α+/+ female mice
The proportion of malformed fetuses reached 24% in severely diabetic mice, whereas in other groups it did not exceed 3.8% (Table 2). Most malformed fetuses had micro-and anophthalmia. Single embryos of severely and moderately diabetic mice had encephalocele, cleft face and gastroschisis. Remarkably, the greater proportion of malformed fetuses in litters of severely diabetic TNF-α+/+ mice was accompanied by a substantial increase in the proportion of litters with malformed fetuses (60% vs 0 to 18.2% in other groups of TNF-α+/+ female mice; Table 2).
TNF-α−/− female mice
The proportion of malformed fetuses in severely diabetic TNF-α−/− mice (approximately 53%) was higher (p<0.05) than that in severely diabetic TNF-α+/+ female mice (Table 2). Moreover, practically the same incidence of malformed fetuses (48.5%) was seen in moderately diabetic TNF-α−/− mice. As in TNF-α+/+ female mice, this increase in the proportion of malformed fetuses was accompanied by an increase in the proportion of litters with malformed fetuses, which reached approximately 90% in severely diabetic TNF-α−/− mice (Table 2).
Apoptosis in embryos of diabetic mice
In most tested sections of normal embryos no apoptotic nuclei were observed (Fig. 2a). In embryos with open neural folds, obtained either from TNF-α+/+ or TNF-α−/− diabetic mice, apoptotic nuclei were observed in the neuroepithelium of the rhombencephalon (Fig. 2c, d) and prosencephalon (data not presented), as well as in the surface ectoderm (Fig. 2b–d). To compare the intensity of apoptosis in malformed TNF-α+/+ and TNF-α−/− embryos, we counted the number of apoptotic nuclei in five sections of the rhombencephalon of these embryos. The average number of TUNEL-positive nuclei in malformed TNF-α+/+ and TNF-α−/− embryos was estimated to be 11.8±1.9 and 18.6±2.3 respectively (means ± SEM, p<0.05), suggesting that diabetes-triggered excessive apoptosis could be more intensive in the brain of TNF-α−/− than in the brain of TNF-α+/+ embryos.

Representative TUNEL-stained transversal sections of the brain of 10-day-old embryos. TUNEL-positive nuclei (arrowheads) appear as darkly brown or black stained nuclei. The sections shown are: a normal TNF-α+/+ embryo (×99, scale bar: 50.5 µm) (a); a TNF-α+/+ embryo (b) with an open neural tube (×99, scale bar: 50.5 µm); high magnification (c) of the rhombencephalic region of a TNF-α+/+ embryo presented in (b) (×396, scale bar: 12.6 µm); high magnification (d) of the rhombencephalic region of a TNF-α−/− embryo with an open neural tube (×396, scale bar: 12.6 µm)
NF-κB DNA-binding activity
The DNA-binding activity of NF-κB was evaluated in nuclear extracts obtained from the heads of 11-day-old embryos of intact non-diabetic mice and diabetic mice with blood glucose concentrations between >10 and ≤13.9 mmol/l (“weak” diabetes) or concentrations of 19.4 mmol/l or more (“severe” diabetes). We found that in embryos of severely diabetic TNF-α−/− mice with a BGC higher than 19.4 mmol/l, the level of NF-κB complex formation was somewhat lower than in other tested samples (Fig. 3).

NF-kB complex formation in nuclear extracts isolated from the heads of 11-day-old TNF-α−/− and TNF-α+/+ embryos of diabetic mice. Bands were quantified by optical densitometry and the graphical data are presented as percentages of the maximum band density obtained. The data are representative of three independent experiments done with different samples. Control of the chemiluminescence detection reaction (a) was done by evaluating Oct2A binding to its specific oligonucleotide and evaluating the specificity of NF-kB binding by competition with 100-fold excess of NF-kB or Oct2A unlabelled oligonucleotide. Nuclear extracts (b) are from embryos of diabetic mice with blood glucose concentrations between 10 and 13.9 mmol/l (weak diabetes) or concentrations of 19.4 mmol/l or more (severe diabetes)
Discussion
As in other studies in rodent models, STZ-induced diabetes in TNF-α+/+ and TNF-α−/− mice led to the occurrence of fetuses with gross structural anomalies. The pregnancy rate was also decreased in diabetic mice. As the proportion of fetuses with gross structural anomalies in STZ-treated non-diabetic and intact non-diabetic mice was near to zero and the pregnancy rate was also not lower, the above disturbances in reproductive performance of diabetic female mice can be attributed to STZ-induced maternal diabetes and not to an embryotoxic effect of STZ itself.
Two main observations indicating a mechanistic role for TNF-α in diabetes-induced embryopathies were made in this study.
The first is a much greater decrease in the pregnancy rate in severely diabetic TNF-α+/+ mice than in TNF-α−/− mice. The comparative analysis of results obtained in mice tested on days 4, 8 and 18 of pregnancy suggests that TNF-α can induce the death of peri-implantation embryos in severely diabetic mice.The death of peri-implantation embryos could be due to disturbances in the function of uterine cells or to disturbances in the development of pre-implantation embryos. Evidence that TNF-α can induce these disturbances in severely diabetic mice comes from data showing an increased expression of cytokine in the embryonic environment of diabetic mice [8], as well as the ability of TNF-α to exert a cytotoxic effect on uterine epithelial cells [21] and to increase the frequency of apoptotic cells in murine blastocysts [4]. Moreover, TNF-α receptors are expressed by rodent blastocysts [22] and cells of the reproductive tract [1], indicating that the death-receptor-mediated signalling pathway [12] could be engaged by TNF-α to activate apoptosis in early embryos and uterine cells of severely diabetic mice.
Our observation that TNF-α mediates death of peri-implantation embryos does not contradict data showing the involvement of Bax and p53 in mediating hyperglycaemia-induced excessive apoptosis in murine blastocysts and consequently diabetes-induced early embryonic death [23, 24]. Thus the pregnancy rate in severely and moderately diabetic TNF-α−/− mice (approximately 35% and 38% respectively) was lower than in the non-diabetic control mice, indicating that TNF-α is not the only mediator of diabetes-induced embryotoxic stimuli. On the other hand, data suggesting that TNF-α can activate the mitochondria-mediated signalling pathway via activation of Bid and Bax proteins and the involvement of p53 in TNF-α-dependent apoptosis [12] do not rule out the possibility that Bax and p53 are engaged by TNF-α to induce death of early embryos in severely diabetic mice.
The second main observation is that the proportion of malformed fetuses in severely diabetic TNF-α+ /+ mice (approximately 24%) was much lower than in severely diabetic TNF-α−/− mice (approximately 53%). The question therefore is: which mechanisms does TNF-α engage to diminish the teratogenic susceptibility of embryos of diabetic mice?
The exact mechanisms remain undefined. Nevertheless, this study showed that the number of live fetuses in severely diabetic TNF-α+/+ mice did not differ significantly from that in severely diabetic TNF-α−/− mice. Also, the number of implantation sites in severely diabetic TNF-α+/+ mice was practically identical to that in non-diabetic TNF-α+/+ mice, indicating that TNF-α, while inducing peri-implantation death in the former, acts in an “all-or-nothing” fashion. Therefore, the lower proportion of malformed embryos in severely diabetic TNF-α+/+ mice is hardly attributable to TNF-α-mediated death of postimplantation embryos with severe structural anomalies or to TNF-α-mediated death of peri-implantation embryos, an event which might decrease the proportion of extremely sensitive embryos in a population exposed to a diabetes-induced teratogenic insult taking place on days 8 to 9 of pregnancy [17]. If so, it is conceivable, that some TNF-α-mediated mechanisms which prevent the formation of structural anomalies operate in postimplantation embryos exposed to diabetes-induced teratogenic stress.
We observed excessive apoptosis in the brain of malformed embryos of diabetic mice, the intensity of which was somewhat higher in TNF-α−/− than in TNF-α+/+ embryos. In parallel, in samples isolated from malformed TNF-α−/− embryos, the level of active NF-κB complexes was lower than in samples isolated from TNF-α+/+ embryos.
These results are not easy to interpret. Activation of the apoptosis program in postimplantation embryos is thought to be an initial event in the formation of diabetes-induced structural anomalies [25, 26]. Also, TNF-α has repeatedly been shown to activate transcription factor NF-κB, a key molecule controlling the apoptosis process and acting mainly to prevent cell death [12, 13]. It is not clear, however, to what extent the excessive apoptosis seen in the brain of embryos with open neural folds reflects the apoptotic process which took place in these embryos when the anomaly was being formed. It also remains unclear whether this apoptosis serves as a marker of processes leading to the formation of other structural anomalies later in development. In addition it is not understood whether there is a causal link between events like excessive apoptosis and suppression of NF-κB binding in the embryonic brain. Thus we can only hypothesise that if NF-κB functions as a negative regulator of apoptosis in postimplantation embryos exposed to “diabetic teratogens” [27], then TNF-α could, while counteracting diabetes-induced suppression of NF-κB activity, act to protect these embryos against diabetes-induced teratogenic stress.
In conclusion, our work suggests that the essential function of TNF-α could be to prevent the birth of offspring with structural anomalies in severely diabetic mice. It does this while contributing to total peri-implantation death of embryos in some of these mice and also possibly stimulating protective mechanisms like NF-κB-mediated anti-apoptotic signalling in postimplantation embryos exposed to a diabetes-induced teratogenic insult.
Abbreviations
- EMSA:
-
Electrophoretic mobility shift assay
- STZ:
-
streptozotocin
- BGC:
-
blood glucose concentration
References
Terranova PF, Hunter VJ, Roby KF, Hunt JS (1995) Tumor necrosis factor-alpha in the female reproductive tract. Proc Soc Exp Biol Med 209:325–342
Kohchi C, Noguchi K, Tanabe Y, Mizuno D, Soma G (1994) Constitutive expression of TNF-alpha and -beta genes in mouse embryo: roles of cytokines as regulator and effector on development. Int J Biochem 26:111–119
Pampfer S, Vanderheyden I, De Hertogh R (1997) Increased synthesis of tumor necrosis factor-alpha in uterine explants from pregnant diabetic rats and in primary cultures of uterine cells in high glucose. Diabetes 46:1214–1224
Pampfer S (2001) Dysregulation of the cytokine network in the uterus of the diabetic rat. Am J Reprod Immunol 45:375–381
Torchinsky A, Toder V, Carp H, Orenstein H, Fein A (1997) In vivo evidence for the existence of a threshold for hyperglycemia-induced major fetal malformations: relevance to the etiology of diabetic teratogenesis. Early Pregnancy 3:27–33
Torchinsky A, Toder V, Savion S, Shepshelovich J, Orenstein H, Fein A (1997) Immunostimulation increases the resistance of mouse embryos to the teratogenic effect of diabetes mellitus. Diabetologia 40:635–640
Machado AF, Zimmerman EF, Hovland DN Jr, Weiss R, Collins MD (2001) Diabetic embryopathy in C57BL/6J mice. Altered fetal sex ratio and impact of the splotch allele. Diabetes 50:1193–1199
Fein A, Kostina E, Savion S et al. (2001) Expression of tumor necrosis factor-alpha in the pregnant uterus of diabetic mice: effect of maternal immunopotentiation. Am J Reprod Immunol 46:161–168
Ivnitsky I, Torchinsky A, Gorivodsky M et al. (1998) TNF-alpha expression in embryos exposed to a teratogen. Am J Reprod Immunol 40:431–440
Torchinsky A, Brokhman I, Shepshelovich J et al. (2003) Increased TNF-alpha expression in cultured mouse embryos exposed to teratogenic concentrations of glucose. Reproduction 125:527–534
Toder V, Carp H, Fein A, Torchinsky A (2002) The role of pro- and anti-apoptotic molecular interactions in embryonic maldevelopment. Am J Reprod Immunol 48: 235–244
Baud V, Karin M (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11:372–377
Karin M, Lin A (2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3:221–227
Torchinsky A, Lishanski L, Wolstein O et al. (2002) NF-kappaB DNA-binding activity in embryos responding to a teratogen, cyclophosphamide. BMC Dev Biol 2:2
Torchinsky A, Shepshelovich J, Orenstein H et al. (2003) TNF-alpha protects embryos exposed to developmental toxicants. Am J Reprod Immunol 49:159–168
Fein A, Torchinsky A, Pinchasov M, Katz N, Toder V, Herkovits J (1997) Cadmium embryotoxicity: evidence of a direct effect of cadmium on early rat embryos. Bull Environ Contam Toxicol 59:520–524
Sadler TW (1997) Mouse embryos in culture: models for understanding diabetes-induced embryopathies and gene function. Int J Dev Biol 41:291–297
Schreiber E, Matthias P, Muller MM, Schaffner W (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 17:6419
Torchinsky A, Fein A, Toder V (1995) Immunoteratology: I. MHC involvement in the embryo response to teratogens in mice. Am J Reprod Immunol 34:288–298
Sokal RR, Rohlf FJ (1995) Biometry, 3rd edn. W.H. Freeman, New York
Pampfer S, Cordi S, Cikos S, Picry B, Vanderheyden I, Hertogh RD (2000) Activation of nuclear factor kappaB and induction of apoptosis by tumor necrosis factor-alpha in the mouse uterine epithelial WEG-1 cell line. Biol Reprod 63:879–886
Pampfer S, Wuu YD, Vanderheyden I, De Hertogh R (1994) Expression of tumor necrosis factor-alpha (TNF alpha) receptors and selective effect of TNF alpha on the inner cell mass in mouse blastocysts. Endocrinology 134:206–212
Moley KH, Chi MM, Knudson CM, Korsmeyer SJ, Mueckler MM (1998) Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways. Nat Med 4:1421–1424
Keim AL, Chi MM, Moley KH (2001) Hyperglycemia-induced apoptotic cell death in the mouse blastocyst is dependent on expression of p53. Mol Reprod Dev 60:214–224
Phelan SA, Ito M, Loeken MR (1997) Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes 46:1189–1197
Moley KH (2001) Hyperglycemia and apoptosis: mechanisms for congenital malformations and pregnancy loss in diabetic women. Trends Endocrinol Metab 12:78–82
Eriksson UJ, Cederberg J, Wentzel P (2003) Congenital malformations in offspring of diabetic mothers--animal and human studies. Rev Endocr Metab Disord 4:79–93
Acknowledgements
This work was supported by The Israel Science Foundation (grant No. 541/00) and The Israel Ministry of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Torchinsky, A., Gongadze, M., Orenstein, H. et al. TNF-α acts to prevent occurrence of malformed fetuses in diabetic mice. Diabetologia 47, 132–139 (2004). https://doi.org/10.1007/s00125-003-1283-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1283-5