
Abstract
Aims/hypothesis
The renin angiotensin system is emerging as a potential therapeutic target for diabetic retinopathy. This study examines the effects of angiotensin-converting-enzyme inhibition by captopril and angiotensin AT1 receptor antagonism using candesartan-cilexetil on retinal blood flow and acetylcholine-stimulated vasodilatation in normotensive diabetic rats.
Methods
Non-diabetic or streptozotocin-induced diabetic rats were treated for 2 weeks with captopril (100 mg/kg/day) or candesartan cilexetil (2 mg/kg/day). Retinal haemodynamics were measured using video fluorescein angiography. Effects of exogenous acetylcholine on retinal haemodynamics were examined following intravitreal injection. Total retinal diacylglycerol was labelled using diacylglycerol kinase, separated by thin-layer chromatography, and quantified using autoradiography.
Results
Diabetic rats had prolonged retinal mean circulation time and decreased retinal blood flow compared with non-diabetic rats. Treatment of diabetic rats with either captopril or candesartan blocked the development of these blood flow abnormalities. Intraviteral injection of acetylcholine (10−5 mol/l) in non-diabetic rats increased retinal blood flow by 53.9±22.0% relative to baseline whereas this response to acetylcholine was blunted in diabetic rats (4.4±19.6%, p<0.001). Candesartan treatment of diabetic rats restored the acetylcholine-stimulated retinal blood flow response to 60.0±18.7% compared with a 56.2+20.1% response in candesartan-treated non-diabetic rats. Total retinal diacylglycerol levels were increased in diabetic rats (3.75±0.98 nmol/mg, p<0.05) compared with non-diabetic rats (2.13±0.25 nmol/mg) and candesartan-treatment of diabetic rats normalized diacylglycerol levels (2.10±0.25 nmol/mg, p<0.05).
Conclusion/interpretation
This report provides evidence that angiotensin-converting enzyme inhibition and AT1 receptor antagonism ameliorates retinal haemodynamic dysfunctions in normotensive diabetic rats.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The pathogenesis of diabetic retinopathy includes a spectrum of functional and histological abnormalities in the retinal microvasculature, which frequently leads to loss of vision. Although tight glycaemic control has been shown to reduce or delay the onset of this complication of diabetes [1], additional proactive therapies are needed. Recent studies have shown that inhibition of the renin-angiotensin system (RAS) can reduce the progression of diabetic retinopathy [2, 3]. The EURODIAB Controlled trial of Lisinopril in Insulin-dependent Diabetes (EUCLID) study group reported that the angiotensin-converting enzyme (ACE) inhibitor, lisinopril, decreased retinopathy progression in normotensive patients with Type 1 diabetes with little or no nephropathy [3]. These findings suggested that inhibition of the RAS can provide protective effects against retinopathy progression in the absence of hypertension, a factor that exacerbates the progression of this disease [2]. The ongoing Diabetes Retinopathy Candesartan Trial (DIRECT) programme includes three randomized, double-masked placebo control trials, which will examine whether treatment with the angiotensin AT1 receptor blocker candesartan can reduce the incidence and progression of diabetic retinopathy in both Type 1 and Type 2 diabetic patients [4]. A primary objective of this programme will determine whether AT1 antagonism provides protective effects against the incidence of retinopathy in a group of Type 1 diabetic patients without pre-existing retinopathy, nephropathy, or hypertension. While the use of AT1 receptor antagonism to prevent early retinal vasculopathies in normotensive diabetic patients is currently under investigation, the mechanisms that could mediate beneficial effects of RAS inhibition on early changes in diabetic retinopathy, prior to the onset of proliferative disease, are not fully understood.
Recent reports on the role of the RAS on the retina have examined its effects on the expression of VEGF and its receptors in retinal cells and tissues. Treatment of spontaneously hypertensive streptozotocin (STZ)-induced diabetic rats with an ACEI or an AT1 receptor antagonist reduced VEGF and VEGF-R2 expression [5, 6]. In addition, ACE inhibition has been shown to reduce retinal VEGF expression in normotensive STZ-induced diabetic rats [7]. These findings suggest that the angiotensin II/AT1 pathway contributes to increased retinal VEGF expression in diabetes and hypertension and thereby could contribute to increased vascular permeability and neovascularization. While there is considerable evidence that RAS inhibition can reduce retinal VEGF expression in diabetes and hypertension, the roles of the RAS in retinal haemodynamic dysfunctions are unknown.
A reduction in retinal blood flow (RBF) is one of the earliest retinal vascular abnormalities detected in diabetes. This haemodynamic change has been described in patients with Type 1 diabetes [8, 9, 10] and in rodent models of diabetes [11]. The decrease in RBF caused by diabetes occurs prior to the development of histological abnormalities and is the result of increased microvascular resistance, which reduces the velocity of blood perfusion across the retinal arterioles and capillaries [9, 11]. The mechanisms by which diabetes causes microvascular resistance in the retina are not fully understood. Moreover, the role of the RAS on this vascular abnormality has not yet been described.
Another early diabetic vascular dysfunction, which has been described for a number of vascular tissues [12, 13], is the impairment of acetylcholine-induced (endothelium-dependent) vasodilatation and blood flow. A limited number of studies have shown that ACE inhibitors and AT1 antagonist can improve endothelial-dependent vasodilator function in brachial and femoral arteries in patients with Type 1 or Type 2 diabetes [14, 15, 16]. While endothelium-dependent vasodilatory response induced by acetylcholine (ACh) has been shown in isolated retinal arteries [17], the effects of diabetes and RAS inhibition on vascular autoregulation in the retina in vivo have not yet been described.
This report investigates the effects of RAS inhibition by captopril, an ACE inhibitor, or candesartan, an AT1 receptor antagonist, on RBF and ACh-induced vasodilatation in normotensive rats with STZ-induced diabetes. These studies examine whether RAS inhibition can ameliorate retinal haemodynamic abnormalities in diabetes.
Materials and methods
Instrumentation
The video fluorescein angiography (VFA) system used for these studies has been described previously [11, 18, 19]. Briefly, the system consists of an imaging camera, a video digitizing system, and an archival unit. For the captopril experiments, the imaging system consisted of a Nikon NFC-50 Fundus camera (Nikon, Tokyo, Japan) connected via an adapter to a DAGE-MTI Silicon Intensified Target low-light level video camera (Michigan City, Ind., USA). The fundus was imaged using the 50° field of the fundus camera. For the candesartan experiments, a scanning laser ophthalmoscope (SLO, Rodenstock Instrument, Munich, Germany) was used to image the fundus. The SLO provides a better signal to noise ratio and a higher image resolution than the Nikon/DAGE-MTI system. For the SLO, the argon blue laser (488 nm) was used for illumination with the filter setting for fluorescein angiography in the 40° field. The laser power and image gain levels were set to 3. The video output (NTSC) from either camera was digitized at 30 frames/s by a frame grabber board (Targa 2000, Pinnacle Systems, Mountain View, Calif., USA) and stored as an AVI sequence. The angiograms are digitized with a 640×480 pixel 8-bit format providing a 256-level gray scale. The analog video signal was recorded on a Sony Umatic video recorder (Sony, Tokyo, Japan) for archival purposes.
Animals
We used 92 male Sprague-Dawley rats (Taconic Farms, Germantown, N.Y., USA) with initial weights between 240 and 270 g. All experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Animal Care and Use Committee of the Joslin Diabetes Center. The rats were housed under standard conditions with free access to water and standard food. Diabetes was induced in 46 rats with an intraperitoneal injection of 55 mg/kg of streptozotocin (STZ) (Sigma, St. Louis, Mo., USA) in 10 mmol/l citrate buffer, pH 4.5 after a 12-h fast. Diabetes was confirmed with blood glucose measurements (>14 mmol/l) 24 h after STZ injection. All animals were maintained for 2 weeks before retinal blood flow measurements. Blood glucose concentrations and body weights were monitored every other day. Insulin (2 U Humulin NPH, Eli Lilly) was provided to diabetic rats every other day as needed to maintain stable body weight and blood glucose less than 28 mmol/l. Diabetic rats were not given insulin on the day of retinal haemodynamic analysis.
All animals, 24 h before retinal blood flow measurements, (under anaesthesia, 50 mg/kg pentobarbital sodium; Abbott Laboratories, North Chicago, Ill., USA) underwent catheterization with a polyvinyl catheter inserted into the right jugular vein as previously described [11, 18, 19]. The catheter was flushed with 0.1 ml of 1000 U sodium heparin before and after implantation. It was positioned subcutaneously along the shoulder, and the distal end was externalized to the back of the neck.
VFA procedure
Immediately before VFA measurements, each rat was anaesthetized, the left eye was dilated (1% tropicamide, Mydriacyl: Alcon, Fort Worth, Tex., USA), and a 100 µl syringe (Hamilton, Reno, Nev., USA) containing 10% sodium fluorescein was connected to the externalized jugular vein catheter. The rats were positioned on a platform attached to the imaging camera. The optic disc was centred and focused in the field of view, the VFA recording sequence was initiated, and a 5 µl bolus of fluorescein dye was rapidly injected into the jugular vein catheter [11, 18, 19]. The injection time was marked on the video recording.
Baseline angiograms were recorded from each rat before intravitreal injection with ACh or vehicle alonen. A further series of angiograms was then recorded at selected time points after the intravitreal injection.
Intravitreal injections were carried out by inserting a 27-gauge needle, attached to a 10 µl syringe (Hamilton), into the vitreous from a site 1 mm posterior to the limbus, as previously described [11, 18, 19, 20]. Injections were carried out directly over the optic disc region under direct visualization, and a timer was immediately started. VFA recordings were obtained at selected times after injection. The effective final concentrations of the injected agents were estimated knowing that the rat vitreous volume is approximately 120 µl [21]. Thus the retina would be exposed to a 12-fold lower concentration than the injected concentration.
Time course and dose response of intravitreal injections of acetylcholine
Intravitreal injections in STZ-induced diabetic rats and non-diabetic rats were done over a range of concentrations from 10−6 to 10−3 mol/l of ACh (Sigma, St. Louis, Mo., USA) dissolved in vehicle of phosphate-buffered saline (PBS). Rats injected intravitreally with vehicle alone served as control subjects.
VFA recordings were obtained before and at 2, 5, and 10 min after intravitreal injection. Blood pressures and heart rates were monitored using a non-invasive tail-cuff sensor and monitoring system (Ueda Electronics, Tokyo, Japan). Animals were maintained on a heated pad during the course of the measurements.
Data analysis
The recorded fluorescein angiograms were digitized on a frame-by-frame basis and analyzed densitometrically to determine retinal vessel diameters and retinal mean circulation times (MCTs) [11, 18, 19].
Sample sites were chosen using primary retinal vessels at a fixed (1 optic disc diameter) radial distance from the centre of the optic disc. Vessel diameters in units of pixels were determined during peak fluorescein arterial and venous filling times at the defined vessel sample sites using a boundary-crossing algorithm. The average diameter for each vessel was measured for each sample site. The average vessel diameters for each eye represent the average of the individual vessel diameters for that eye.
At the fixed vessel sites, the average vessel fluorescence within a sample area defined by the vessel width was measured on a frame-by-frame basis to generate temporal fluorescence intensity or dye dilution curves. The resultant artery and vein fluorescence data were fit to a log normal distribution function from which average arterial and venous circulation times were calculated [11, 22, 23]. The arterial appearance time (AT) of the dye bolus, defined as the time between dye injection and the first detectable appearance (vessel fluorescence intensity greater than background level by 2 times the standard deviation of the average background intensity) of dye in the retinal artery, represents an assessment of systemic circulation times. The average MCT was calculated as the difference between the average retinal mean arterial and venous filling times for all primary arteries and veins. Retinal blood flow was calculated by dividing the sum of the squares of the arterial and venous diameters by the MCT [24]. Data establishing the sensitivity of this technique has been reported previously [25].
Captopril and candesartan treatment in rats
Rats were treated with captopril (100 mg/kg/day, Sigma), candesartan cilexetil (TCV-116, 2 mg/kg/day, provided by Dr. Peter Morsing, Astra Hassle AB, Mölndal, Sweden), or vehicle in drinking water for 2 weeks [26]. The treatment was started immediately after diabetes was diagnosed. The vehicle for candesartan consisted of 10% polyethylene glycol (PEG400), 2% chemophor El, and 5% ethyl alcohol in distilled water (Sigma). Vehicle was mixed in the drinking water to match the volume of candesartan.
Retinal diacylglycerol assay
The rats were killed by inhalation of carbon dioxide after VFA measurements. Retinas were dissected and frozen immediately in liquid nitrogen. Retinal DAG was assayed as described previously [27, 28]. Briefly, the frozen retinal samples were thawed with 2.5 ml cold PBS and homogenized with a Polytron (Teramar, N.J., USA) for 20 s. A small aliquot of 5 µl was used for protein assay, using spectrophotometer (DU-530, Beckman, Fullerton, Calif., USA). After the addition of cold methanol and chloroform, total lipids were extracted twice and total DAG was measured using a DAG Assay kit (Amersham, Arlington Heights, Ill., USA) and DAG kinase (Calbiochem, San Diego, Calif., USA). The resulting radiolabelled phosphatidic acid, derived from DAG, was separated by thin-layer chromatography. Levels of total DAG were visualized and quantified by PhosphorImager analysis (Molecular dynamics, Sunnyvale, Calif., USA). The results for total retinal DAG were normalized by protein.
Statistical analysis
All values are reported as the means±standard deviation. Statistical analysis software (SigmaStat; Jandel Scientific, San Rafael, Calif., USA) was used for statistical comparisons. Statistical analysis was conducted using the paired Student’s t-test to compare baseline values with the values determined after injection in each group. Comparisons between two groups were carried out using the unpaired Student’s t-test. For multiple comparisons, data were analyzed using Students-Newman-Keuls method for normal distributions or the Kruskal-Wallis one way analysis on ranks using Dunn’s Method for unequal variance. A p value of less than 0.05 was considered statistically significant.
Results
Effects of ACE inhibition and AT1 receptor antagonism on retinal haemodynamics
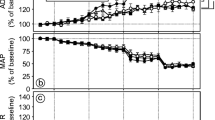
Retinal blood flow (RBF) was measured in STZ-induced diabetic (DM) and non-diabetic (NDM) rats in the absence or presence of treatment with the ACE inhibitor captopril. Group comparisons show that DM rats and captopril-treated DM rats had increased blood glucose concentrations and gained less weight than their NDM counterparts (Table 1). There were no differences in haematocrit and heart rate between the untreated- and treated- DM and NDM rats. Captopril treatment resulted in small decreases in systolic and mean blood pressure for both the DM and NDM groups. The mean circulation time (MCT) from primary retinal arteries to veins was measured using video fluorescein angiography. MCT in untreated NDM and DM groups were 0.69±0.18 s and 0.99±0.27 s (p<0.005), respectively (Fig. 1A). Captopril normalized the MCT in DM rats in a dose-dependent manner to 0.73±0.20 s at 100 mg/kg/day captopril (p<0.05 vs untreated DM rats). The diameter of primary retinal arteries and veins and the fluorescein appearance time (AT) in DM and NDM groups were similar (Table 1). RBF was reduced in DM rats compared with NDM rats (64.7±18.7% vs 100±33.4%, respectively, p<0.005). Treatment of DM rats with captopril at 50 and 100 mg/kg/day ameliorated this RBF abnormality (Fig. 1B). Captopril did not alter MCT, vessel diameters, or RBF in NDM rats.

Effects of captopril on retinal mean circulation time (MCT) and retinal blood flow (RBF) in non-diabetic (NDM) and STZ-induced diabetic (DM) rats. NDM and DM rats were treated with the indicated doses of captopril for 2 weeks. MCT (a) and vessel diameters were measured by video fluorescence angiography. RBF (b) was calculated from MCT and vessels diameters. Results are expressed as means±SD and statistical differences are indicated as *p<0.05, **p<0.01, ***p<0.005 (ANOVA)
The role of the AT1 receptor in the retinal haemodynamic abnormalities in diabetes was examined in rats treated with the angiotensin AT1 receptor antagonist candesartan cilexetil at the dose of 2 mg/kg/day. Candesartan-treatment did not affect blood glucose, body weight, haematocrit, retinal appearance time, or artery and vein diameters in either DM or NDM groups (Table 2). Systolic and mean blood pressure of candesartan-treated NDM and DM rats was lower than vehicle-treated control rats (p<0.05). Measurement of retinal haemodynamics using a scanning laser ophthalmoscope (SLO) revealed prolonged MCT in DM rats (1.33±0.32 s) as compared with NDM rats (0.84±0.15 s) and treatment of DM rats with candesartan reduced MCT to 1.03±0.20 s (p<0.005)(Fig. 2A). Candesartan also ameliorated the RBF abnormality in DM rats from 67±24 to 86±15% (p<0.05) relative to untreated NDM control rats (Fig. 2B). Candesartan treatment of NDM rats did not affect MCT or RBF compared to vehicle-treated NDM controls.

Effects of candesartan on retinal haemodynamics in NDM and DM rats. Rats were treated 2 weeks with candesartan-cilexetil or vehicle. MCT (a) and RBF (b) were measured by video fluorescein angiography. Results are means±SD. Statistical differences are indicated as *p<0.05, ***p<0.005 (ANOVA)
The difference between the MCT times (Figs. 1A, 2A) and pixel diameters (Tables 1, Table 2) reported for the captopril and candesartan studies are due to the use of different optical magnifications provided by the fundus and SLO cameras. These two methods detected similar decreases in RBF in untreated DM rats 64.6±18.7% (Fig. 1B) and 67±24% (Fig. 2B) compared with untreated NDM rats. To further characterize the retinal vessels being studied, fluorescent microspheres with a diameter of 15 µm±<5% were infused and entrapped in the retinal vasculature. These microspheres were 7.75 pixels in diameter using a 40° field for the SLO, resulting in the conversion between pixel and metric unit of 1.94 µm per pixel for the rat retina visualized by this method. Using this conversion factor, the diameters of artery (12.1 pixels) and veins (16.7 pixels) from NDM rats (Table 2) were estimated at 23.5 µm and 32.4 µm, respectively.
Effect of acetylcholine on retinal haemodynamics
The effects of acetylcholine (ACh) on retinal haemodynamics in DM and NDM rats were assessed. MCT and RBF responses to intravitreal injections of ACh at concentrations of 10−6 to 10−3 mol/l or vehicle alone in NDM rats were determined (Fig. 3A,B). ACh (10−6 to 10−3 mol/l) decreased MCT at 2 min post injection (p<0.01) in NDM rats. The maximum decrease in MCT was obtained at 10−4 mol/l ACh (0.86±0.09 vs 0.63±0.07 s, baseline and 2 min after the injection, respectively p<0.01). MCT returned to the baseline 10 min after the injection. Intravitreal injection of 10−4 mol/l ACh increased retinal artery diameter compared to the baseline measurement (12.6±0.9 vs 13.0±1.0 pixel, baseline and 2 min after the injection, respectively, p<0.01). The retinal vein diameters were also increased at 2 min after the injection of the 10−6 mol/l to 10−3 mol/l of ACh (18.1±1.6 pixel at baseline and 20.5±1.8 pixel at 2 min after the injection of 10−4 mol/l ACh, p<0.01). Increases in RBF were observed 2 min after intravitreal injections of 10−6 to 10−3 mol/l of ACh (589.6±135.3 vs 960.1±165.6 pixel2/s, baseline and 2 min after 10−4mol/l ACh injection, respectively) (p<0.01) compared with the vehicle alone (617.3±83.9 vs 610.4±75.9 pixel2/s, baseline and 2 min, respectively) (Fig. 3B). The RBF returned to the baseline 10 min after the intravitreal injections of ACh. Intravitreal injection of vehicle alone did not alter MCT, primary retinal vessel diameters, or RBF.

Time course response of acetylcholine (ACh) on retinal haemodynamics in NDM and DM rats. Video fluorescein angiography was recorded before injection and 2, 5, 10 min after intravitreal injection of 10 µl of the indicated concentrations of ACh (10−6 to 10−3 mol/l) or vehicle. (a) and (b) show the MCT and RBF responses in non-diabetic rats. (c) and (d) show the MCT and RBF responses in rats with 2 weeks of STZ-induced diabetes. Each group includes six rats. “*” and “†” indicate significant difference compared to the baseline measurement using paired Student’s t-test (p<0.01 and p<0.05, respectively)
In contrast to results obtained with NDM rats, intravitreal injections of 10−6 mol/l or 10−5 mol/l of ACh did not shorten MCT (Fig. 3C). Injection of the 10−4 and 10−3 mol/l of ACh in the vitreous of DM rats decreased MCT at 2 min after injection compared with baseline values (p<0.05) and compared with MCT at 2 min after vehicle injection (p<0.01). There was no change in artery diameter in DM rats injected with ACh at 10−6 to 10−3 mol/l. An increase in vein diameter was observed at the highest concentration (10−3 mol/l) of ACh (16.1±0.9 vs 17.1±1.3 pixels, baseline and 2 min after 10−4 mol/l ACh injection, respectively, p<0.05). No difference was observed in RBF in diabetic rats at 10−6 mol/l (n=6) and 10−5 mol/l (n=6) ACh or vehicle alone. However, there was an increase in RBF 2 min after intravitreal injections of the 10−4 and 10−3 mol/l concentrations of ACh (453.8±61.5 vs 636.8±127.1 pixel2/s, baseline and 2 min after the injection of 10−4 mol/l ACh, p<0.01).
There was a dose-dependent increase in the percentage RBF change 2 min after ACh injection with the maximum at 10−4 mol/l ACh (65.0±15.4%, p<0.01) (Fig. 4). In diabetic rats, there were no differences in the RBF at 10−6 or 10−5 mol/l ACh compared to vehicle. An increase in RBF in DM rats was observed at 10−4 ACh (39.8±15.3%, p<0.01 compared to vehicle) and the magnitude of this increase was less at higher (10−3 mol/l) ACh concentration. Additionally, the magnitude of the per cent RBF change in diabetic rats was reduced compared to non-diabetic rats over the range of 10−6 to 10−3 mol/l ACh, (p<0.02).

Dose response effect of ACh on retinal blood flow in NDM and DM rats. Bar graph shows the percentage change in RBF at 2 min after intravitreal injection with the indicated concentrations of ACh compared with vehicle injection. Black bars and grey bars show the change in RBF in NDM and DM rats, respectively. Results are presented as means±SD. Statistical differences are indicated as *p<0.05 vs vehicle and p values for NDM vs DM comparisons are provided
Since the volume of the rat vitreous is approximately 120 µl, the final ACh concentration in the vitreous with a 10-µl injection would be diluted by a factor of 12. The half maximal effective concentration (EC50) of ACh 2 min after the injection in NDM rats was approximately 10−6 mol/l, which resulted in a final vitreous concentration of 80 nmol/l. The EC50 of ACh on RBF in DM rats was between 800 nmol/l and 8 µmol/l.
Intravitreal injections of ACh did not affect the retinal appearance time, which was 1.78±0.19 s at baseline and 1.79±0.20 s, 2 min after ACh injection. These results indicate that intravitreal injection of ACh does not affect the systemic circulation.
Effect of candesartan treatment on retinal haemodynamic ACh response
The ACh (10−5 mol/l) stimulated increase in RBF was impaired in DM rats (5.0±8.6%) compared with NDM rats (52.7±17.7%, p<0.05) (Fig. 3, Fig. 4). Therefore this dose of ACh was utilized to examine the effect of candesartan treatment on ACh-induced MCT, vessel dilatation, and RBF in NDM and DM rats. As similarly shown in Figure 3C, 10−5 mol/l ACh did not change MCT in DM rats (Fig. 5A). In contrast, this dose of ACh increased MCT in candesartan-treated DM rats by 22.0±10.7% (p<0.01), which was comparable to the ACh-induced MCT response in NDM rats (24.0±7.6%). Candesartan treatment did not affect the ACh-stimulated MCT in NDM rats (19.5±6.8% vs 22.0±7.6% for vehicle-treated NDM rats).

Effects of candesartan on ACh responses in the retinal vasculature in diabetic and non-diabetic rats. NDM and DM rats were treated for 2 weeks with Candesartan (+) or vehicle (−). Video fluorescein angiography was carried out before and 2 min after intravitreal injection with 10−5 mol/l ACh or vehicle. The effect of ACh on RBF, MCT, and arterial and venous diameters are shown. Results are expressed as means±SD and statistical differences are indicated as *p<0.05, **p<0.01, ***p<0.005 (ANOVA)
Intravitreal injection of 10−5 mol/l ACh increased the diameter of retinal arteries by 5.6±2.7% (p<0.05, compared with baseline) in NDM rats but did not dilate arteries in DM rats (−1.2±6.5%, Fig. 5B). Candesartan treatment preserved ACh-induced retinal artery vasodilatation in DM rats (5.4±3.6% increase, p<0.05 compared with baseline). The changes in retinal vein diameters in diabetes and candesartan-treated groups paralleled those observed in retinal arteries. ACh increased vein diameter by 10.9±5.9% in NDM rats compared with −0.7±4.3% and 13.9±2.2% for vehicle-treated and candesartan-treated DM rats, respectively (Fig. 5C).
As shown above, ACh increased RBF in NDM rats by 53.9±22.0% whereas ACh did not affect RBF in DM rats (4.4±19.6% relative to baseline, Fig. 5D). In contrast, ACh similarly increased RBF in candesartan-treated DM rats and candesartan-treated NDM rats by 60.0±18.7% and 56.2±20.1%, respectively. Thus candesartan treatment preserved ACh-induced MCT, retinal vessel dilatation, and RBF in DM rats.
Captopril treatment of DM rats similarly improved ACh-induced MCT to 23.3±6.9% (p<0.02), retinal artery dilatation to 7.4±4.8% (p=0.006), retinal vein dilatation to 10.8±4.4% (p<0.001), and RBF to 57.2±9.8% (p<0.02) (responses 2 min post ACh compared with baseline, data not shown). The effects of captopril and candesartan on these retinal haemodynamic parameters were not different.
Effect of candesartan treatment on diacylglycerol levels in the retina
Total retinal DAG levels were increased in DM rats (3.75±0.98 nmol/mg, p<0.05) compared with NDM rats (2.13±0.25 nmol/mg, Fig. 6). The increase in DAG levels in DM rats was prevented by candesartan treatment (2.01±0.59 nmol/mg, p<0.05 vs untreated DM rats). There was no difference between vehicle treatment and candesartan treatment in non-diabetic rats (2.13±0.25 vs 2.10±0.25 nmol/mg, respectively), and the levels of these rats were comparable to that of non-treated NDM rats (2.18±0.33 nmol/mg).

The effect of candesartan on total diacylglycerol (DAG) levels in the retina. NDM and DM rats were treated with candesartan (+) or vehicle (−) as indicated. Retinal DAG levels were quantitated and normalized to total retinal protein. Data is expressed as means±SD and significant differences are indicated as *p<0.05 (ANOVA)
Discussion
This report shows that inhibition of the RAS can ameliorate retinal blood flow abnormalities in diabetes. Treatment with either an ACE inhibitor (captopril) or an AT1 receptor antagonist (candesartan) blocked the prolongation of MCT, normalized RBF, and preserved ACh-induced vasodilatation in the diabetic retina. These effects of RAS inhibition occurred in diabetic rats in the absence of hypertension and without affecting blood glucose. Although both captopril and candesartan caused a small decrease in blood pressure in both NDM and DM rats, which were normotensive with similar blood pressures, these treatments only affected retinal haemodynamics in DM rats. Since the decrease in systemic blood pressure caused by RAS inhibition did not alter RBF in NDM rats it is unlikely that this small change in blood pressure contributed to the normalization of RBF in captopril- or candesartan-treated DM rats. Previous work has shown that RBF is mainly controlled by autoregulatory mechanisms and local factors [29]. These results suggest that RAS inhibition blocked a diabetes-induced abnormality in the retinal vasculature, which thereby preserved retinal haemodynamics.
Previous studies have shown that treatment of DM rats with an endothelin type A receptor antagonist (BQ123), a PKCβ inhibitor (LY333531), or vitamin E could also ameliorate RBF abnormalities [10, 27, 30, 31]. In addition, intravitreal injections of endothelin-1 or phorbol dibutyrate (an activator of PKC) rapidly induced retinal arteriole constriction and prolonged MCT, which mimicked diabetes [31, 32]. In contrast, intravitreal injection of angiotensin II, at doses up to 10−3 mol/l, did not increase MCT or inhibit RBF (data not shown). Moreover, while intraviteral injections of ETA receptor antagonist BQ-123 rapidly normalized RBF in diabetes, intravitreal injection of saralasin, an angiotensin receptor antagonist, did not acutely affect retinal blood flow [31]. The absence of effects of an immediate RBF response to intravitreal administration of angiotensin II or saralasin suggests that angiotensin II or AT1/AT2 receptors do not acutely modulate retinal haemodynamics. In contrast, our study shows that a 2-week treatment of diabetic rats with an ACE inhibitor or an AT1 antagonist normalized RBF. These results suggest that sustained effects of the AT1 pathway contribute to the RBF abnormalities in diabetes. The mechanisms that mediate these chronic effects of the AT1 pathway on RBF likely involve the local production of factors that cause microvascular vasoconstriction. The AngII/AT1 pathway has been shown to up-regulate a number of pathways that induced vasoconstriction, including endothelin-1 expression, superoxide ion production, and the DAG/PKC activation in a number of vascular tissues in diabetes [33, 34, 35, 36, 37]. Retinal DAG levels are increased in STZ-induced diabetic rats and elevation of DAG levels by treatment of NDM rats with a DAG kinase inhibitor reduces RBF in a manner similar to that observed in diabetes [28]. Since activation of the angiotensin AT1 receptor can lead to increased DAG [38], the effect of candesartan on retinal DAG levels was measured. In this report, we show that the AT1 receptor antagonism normalized retinal DAG levels in diabetes. Since RBF is regulated by resistance arteriole and capillary pericytes [31], we propose that AngII/AT1 effects on retinal haemodynamics are mediated by the up-regulation factors, such as endothelin-1 and/or DAG/PKC pathways, which induce sustained microvascular constriction [31, 32]. This conclusion is consistent with the results showing that treatment of DM rats with either captopril or candesartan normalized MCT and RBF without affecting primary retinal artery or vein diameters.
Several mechanisms could contribute to the enhanced actions of the AT1 receptor in the retina in diabetes. Increased expression of ACE expression in the retina in STZ-diabetes has been reported, which could cause an increase in the local generation of angiotensin II [39]. It has also been shown that both retinal microcapillary endothelial cells and pericytes express angiotensin AT1 receptors [40, 41]. Therefore increased production of angiotensin II within the retina might act in an autocrine or paracrine manner to increase factors, such as DAG, which leads to arteriole vasoconstriction [32]. Alternatively, diabetes could decrease activities of retinal-derived relating factors [42, 43], resulting in the enhanced vasoconstriction by normal levels of angiotensin II-activation of AT1 receptor activity.
While our group and others have reported that there is a decrease in RBF in diabetic animal models and in patients with Type 1 diabetes [8, 9, 10, 11], other groups have reported that diabetes increases RBF [44, 45, 46]. The different results from these groups could be related to the methodologies used (video fluorescein angiography and laser Doppler velocimetry), the duration of diabetes, whether or not clinically observable diabetic retinopathy was present, the blood glucose levels at the time of the measurement, and the level of glycaemic control or haemoglobin A1c in the studied patients [20, 47]. Previous studies have shown that RBF is reduced early in diabetes followed by a transition to increased RBF, which correlates with diabetes duration and level of diabetic retinopathy [20, 48]. A review article details a discussion on some of these issues affecting retinal haemodynamics measurement in early diabetes [49]. A reduction in RBF in early diabetes is consistent with existing results regarding the effects of PKC activation, increased production of vasoconstricting agents such as endothelin-1, and the attenuation in the action of vasodilating agents indicating that the expected net effect in the diabetic retina would be an increase in vascular resistance and a concomitant reduction in blood flow.
A previous study examined the effects of the ACE inhibitor perindopril and the beta-blocker atenolol on RBF in hypertensive diabetic subjects [50]. Although the perindopril-treated group in this study showed a trend for reduced RBF, which would reflect an improvement in this group, this change did not reach statistical significance compared to baseline. Interestingly, the RBF responses for the perindopril- and atenolol-treated groups were significantly different (p<0.05) [50], indicating differences on the effects of these blood pressure treatments on retinal haemodynamics.
This study also shows that diabetes impairs ACh induced blood flow and vasodilatation in the retina. ACh-induced vasodilatation of both retinal arteries and veins in NDM rats whereas both these responses were blunted in DM rats. In NDM rats, the magnitude of ACh-stimulated vasodilitation for veins was greater than that in arteries. These results could suggest that ACh induces vasodilatation of both primary retinal arteries as well as resistance microvessels, including arterioles. Microvascular dilation would increase venous blood flow. Alternatively, differences in artery and vein dilation could reflect differential effects of acetylcholine on arterial versus venous smooth muscle contractility. Previous studies have shown that the ACh-induced vasodilatation in isolated retinal arteries is mediated by endothelium-derived nitric oxide [17]. In addition, ACh could mediate endothelium-independent vasorelaxation of retinal arteries via the elaboration of nitric oxide from perivascular nerves [51, 52]. Intravitreal-injected ACh would have access to both neuronal tissue and the basolateral surface of the endothelium. While the physiological roles of retinal vasodilatation are not fully understood, retina-derived relaxation factor and/or nitric oxide have been proposed to mediate retinal vascular auto-regulation and its response to hypoxia [42, 43]. Thus RAS inhibition might improve vasodilatation in diabetes under conditions of hypoxia, and thereby decrease the subsequent hypoxia-induced stimulation of VEGF expression in retinal cells [53].
Our report shows that ACE inhibition and AT1 receptor antagonism prevent the development of retinal haemodynamic abnormalities in normotensive diabetic rats. These findings suggest that RAS inhibition can provide beneficial effects in preserving retinal blood flow in diabetes. This intervention could provide an additional proactive therapy for reducing the incidence of diabetic retinopathy.
Abbreviations
- ACh:
-
acetylcholine
- AT:
-
appearance time
- DAG:
-
diacylglycerol
- DM:
-
diabetic
- MCT:
-
mean circulation time
- NDM:
-
non-diabetic
- RAS:
-
renin angiotensin system
- RBF:
-
retinal blood flow
- STZ:
-
streptozotocin
References
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study Group (1998) Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 317:713–720
Chaturvedi N, Sjolle AK, Stephenson JM, Abrahamian H, Keipes M, Castellarin A, Rogulja-Pepeonik Z, Fuller JH (1998) Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. Lancet 351:28–31
Chaturvedi N, Sjoelie AK, Svensson A (2002) The DIabetic Retinopathy Candesartan Trials (DIRECT) Programme, rationale and study design. J Renin Angiotensin Aldosterone Syst 3:255–261
Moravski CJ, Skinner SL, Stubbs AJ, Sarlos S, Kelly DJ, Cooper ME, Gilbert RE, Wilkinson-Berka JL (2003) The renin-angiotensin system influences ocular endothelial cell proliferation in diabetes: transgenic and interventional studies. Am J Pathol 162:151–160
Nagisa Y, Shintani A, Nakagawa S (2001) The angiotensin II receptor antagonist candesartan cilexetil (TCV-116) ameliorates retinal disorders in rats. Diabetologia 44:883–888
Gilbert RE, Kelly DJ, Cox AJ, Wilkinson-Berka JL, Rumble JR, Osicka T, Panagiotopoulos S, Lee V, Hendrich EC, Jerums G, Cooper ME (2000) Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia 43:1360–1367
Kawagishi T, Nishizawa Y, Emoto M, Konishi T, Maekawa K, Hagiwara S, Okuno Y, Inada H, Isshiki G, Morii H (1995) Impaired retinal artery blood flow in IDDM patients before clinical manifestations of diabetic retinopathy. Diabetes Care 18:1544–1549
Feke GT, Buzney SM, Ogasawara H, Fujio N, Goger DG, Spack NP, Gabbay KH (1994) Retinal circulatory abnormalities in type 1 diabetes. Invest Ophthalmol Vis Sci 35:2968–2975
Bursell SE, Clermont AC, Aiello LP, Aiello LM, Schlossman DK, Feener EP, Laffel L, King GL (1999) High-dose vitamin E supplementation normalizes retinal blood flow and creatinine clearance in patients with type 1 diabetes. Diabetes Care 22:1245–1251
Bursell SE, Clermont AC, Shiba T, King GL (1992) Evaluating retinal circulation using video fluorescein angiography in control and diabetic rats. Current Eye Res 11:287–295
Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA (1993) Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation 88:2510–2516
Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, King GL, LoGerfo FW, Horton ES, Veves A (1999) Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes 48:1856–1862
O’Driscoll G, Green D, Rankin J, Stanton K, Taylor R (1997) Improvement in endothelial function by angiotensin converting enzyme inhibition in insulin-dependent diabetes mellitus. J Clin Invest 100:678–684
O’Driscoll G, Green D, Maiorana A, Stanton K, Colreavy F, Taylor R (1999) Improvement in endothelial function by angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. J Am Coll Cardiol 33:1506–1511
Arcaro G, Zenere BM, Saggiani F, Zenti MG, Monauni T, Lechi A, Muggeo M, Bonadonna RC (1999) ACE inhibitors improve endothelial function in type 1 diabetic patients with normal arterial pressure and microalbuminuria. Diabetes Care 22:1536–1542
Benedito S, Prieto D, Nielsen PJ, Nyborg NC (1991) Role of the endothelium in acetylcholine-induced relaxation and spontaneous tone of bovine isolated retinal small arteries. Exp Eye Res 52:575–579
Bursell SE, Clermont AC, Oren B, King GL (1995) The in vivo effect of endothelins on retinal circulation in nondiabetic and diabetic rats. Invest Ophthalmol Vis Sci 36:596–607
Abiko T, Abiko A, Clermont AC, Shoelson B, Horio N, Takahashi J, Adamis AP, King GL, Bursell SE (2003) Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes 52:829–837
Clermont AC, Aiello LP, Mori F, Aiello LM, Bursell SE (1997) Vascular endothelial growth factor and severity of nonproliferative diabetic retinopathy mediate retinal hemodynamics in vivo: a potential role for vascular endothelial growth factor in the progression of nonproliferative diabetic retinopathy. Am J Ophthalmol 124:433–446
Hughes A (1979) A schematic eye for the rat. Vision Res 19:569–588
Riva CE, Feke GT, Ben Sira I (1978) Fluorescein dye-dilution technique and retinal circulation. Am J Physiol 234:H315–H322
Koyama T, Matsuo N, Shimizu K, Mihara M, Tsuchida Y, Wolf S, Reim M (1990) Retinal circulation times in quantitative fluorescein angiography. Graefes Arch Clin Exp Ophthalmol 228:442–446
Bulpitt CJ, Dollery CT (1971) Estimation of retinal blood flow by measurement of the mean circulation time. Cardiovasc Res 5:406–412
Takagi C, King GL, Clermont AC, Cummins DR, Takagi H, Bursell SE (1995) Reversal of abnormal retinal hemodynamics in diabetic rats by acarbose, an alpha-glucosidase inhibitor. Curr Eye Res 14:741–749
Chen HC, Bouchie JL, Perez AS, Clermont AC, Izumo S, Hampe J, Feener EP (2000) Role of the angiotensin AT(1) receptor in rat aortic and cardiac PAI-1 gene expression. Arterioscler Thromb Vasc Biol 20:2297–2302
Kunisaki M, Bursell SE, Clermont A, Ishii H, Ballas LM, Jirousek MR, Umeda F, Nawata H, King GL (1995) Vitamin E prevents diabetes-induced abnormal retinal blood flow via the diacylglycerol-protein kinase C pathway. Am J Physiol 269:E239–E246
Bursell SE, Takagi C, Clermont A, Takagi H, Mori F, Ishii H, King GL (1997) Specific retinal diacylglycerol and protein kinase C beta isoform modulation mimics abnormal retinal hemodynamics in diabetic rats. Invest Ophthal Vis Sci 38:2711–2720
Delaey C, Van de Voorde J (2000) Regulatory mechanisms in the retinal and choroidal circulation. Ophthalmic Res 32:249–256
Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL (1996) Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 272:728–731
Takagi C, Bursell SE, Lin YW, Takagi H, Duh E, Jiang Z, Clermont AC, King GL (1996) Regulation of retinal hemodynamics in diabetic rats by increased expression and action of endothelin-1. Invest Ophthalmol Vis Sci 37:2504–2518
Shiba T, Inoguchi T, Sportsman JR, Heath WF, Bursell SE, King GL (1993) Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am J Physiol 265:E783–E793
Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA (1995) An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol 268:H2274–H2280
Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS (2002) Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int 61:186–194
Itoh Y, Imamura S, Yamamoto K, Ono Y, Nagata M, Kobayashi T, Kato T, Tomita M, Nakai A, Itoh M, Nagasaka A (2002) Changes of endothelin in streptozotocin-induced diabetic rats: effects of an angiotensin converting enzyme inhibitor, enalapril maleate. J Endocrinol 175:233–239
Malhotra A, Reich D, Nakouzi A, Sanghi V, Geenen DL, Buttrick PM (1997) Experimental diabetes is associated with functional activation of protein kinase C epsilon and phosphorylation of troponin I in the heart, which are prevented by angiotensin II receptor blockade. Circ Res 81:1027–1033
Malhotra A, Kang BP, Cheung S, Opawumi D, Meggs LG (2001) Angiotensin II promotes glucose-induced activation of cardiac protein kinase C isozymes and phosphorylation of troponin I. Diabetes 50:1918–1926
Freeman EJ, Tallant EA (1994) Vascular smooth muscle cells contain AT1 angiotensin receptors coupled to phospholipase D activation. Biochem J 304:543–548
Okada Y, Yamanaka I, Sakamoto T, Hata Y, Sassa Y, Yoshikawa H, Fujisawa K, Ishibashi T, Inomata H (2001) Increased expression of angiotensin-converting enzyme in retinas of diabetic rats. Jpn J Ophthalmol 45:585–591
Otani A, Takagi H, Oh H, Suzuma K, Matsumura M, Ikeda E, Honda Y (2000) Angiotensin II-stimulated vascular endothelial growth factor expression in bovine retinal pericytes. Invest Ophthalmol Vis Sci 41:1192–1199
Otani A, Takagi H, Suzuma K, Honda Y (1998) Angiotensin II potentiates vascular endothelial growth factor-induced angiogenic activity in retinal microcapillary endothelial cells. Circ Res 82:619–628
Nagaoka T, Sakamoto T, Mori F, Sato E, Yoshida A (2002) The effect of nitric oxide on retinal blood flow during hypoxia in cats. Invest Ophthalmol Vis Sci 43:3037–3044
Delaey C, Boussery K, Van de Voorde J (2000) A retinal-derived relaxing factor mediates the hypoxic vasodilation of retinal arteries. Invest Ophthalmol Vis Sci 41:3555–3560
Grunwald JE, Dupont J, Riva CE (1996) Retinal haemodynamics in patients with early diabetes mellitus. Br J Ophthalmol 80:327–331
Patel V, Rassam S, Newsom R, Wiek J, Kohner E (1992) Retinal blood flow in diabetic retinopathy. BMJ 305:678–683
Tilton RG, Chang K, Pugliese G, Eades DM, Province MA, Sherman WR, Kilo C, Williamson JR (1989) Prevention of hemodynamic and vascular albumin filtration changes in diabetic rats by aldose reductase inhibitors. Diabetes 38:1258–1270
Bursell SE, Clermont AC, Kinsley BT, Simonson DC, Aiello LM, Wolpert HA (1996) Retinal blood flow changes in patients with insulin-dependent diabetes mellitus and no diabetic retinopathy. Invest Ophthalmol Vis Sci 37:886–897
Konno S, Feke GT, Yoshida A, Fujio N, Goger DG, Buzney SM (1996) Retinal blood flow changes in type I diabetes. A long-term follow-up study. Invest Ophthalmol Vis Sci 37:1140–1148
Grunwald JE, Bursell SE (1996) Hemodynamic changes as early markers of diabetic retinopathy. Curr Opin Endocrinol Diabetes 3:298–306
Patel V, Rassam SM, Chen HC, Jones M, Kohner EM (1998) Effect of angiotensin-converting enzyme inhibition with perindopril and beta-blockade with atenolol on retinal blood flow in hypertensive diabetic subjects. Metabolism 47:28–33
Toda N, Zhang JX, Ayajiki K, Okamura T (1995) Mechanisms underlying endothelium-independent relaxation by acetylcholine in canine retinal and cerebral arteries. J Pharmacol Exp Ther 274:1507–1512
Toda N, Kitamura Y, Okamura T (1995) Functional role of nerve-derived nitric oxide in isolated dog ophthalmic arteries. Invest Ophthalmol Vis Sci 36:563–570
Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA (1995) Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol 113:1538–1544
Acknowledgements
This work was supported in part by National Institutes of Health grants DK 60165 (to E.P.F.) and DK 36836 (Joslin’s Diabetes and Endocrinology Research Center Grant), and grants from the Aichi D.R.G. Foundation (to N.H.), Juvenile Diabetes Foundation International (to E.P.F.) and Massachusetts Lions Eye Research Fund (to S.E.B.)
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Horio, N., Clermont, A.C., Abiko, A. et al. Angiotensin AT1 receptor antagonism normalizes retinal blood flow and acetylcholine-induced vasodiliation in normotensive diabetic rats. Diabetologia 47, 113–123 (2004). https://doi.org/10.1007/s00125-003-1262-x
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1262-x