
Abstract
Aims/hypothesis
The purpose of this study was to assess the therapeutic implication of leptin in insulin-deficient diabetes.
Methods
Insulin-deficient diabetes was induced by streptozotocin (STZ) in transgenic skinny mice overexpressing leptin. Plasma concentrations of glucose, insulin, and leptin were measured. The effects on body weight, food intake, and hypothalamic gene expressions were analyzed. After diabetes was induced, graded doses of insulin ranging from 0.4 to 51.2 mU·g–1·day–1 were injected. Co-administration of leptin and insulin was also carried out using osmotic pumps.
Results
After STZ injection, both transgenic and non-transgenic littermates developed marked hyperglycaemia as a result of severe hypoinsulinaemia [termed diabetic transgenic skinny mice overexpressing leptin (diabetic TGM) and diabetic non-transgenic littermates (diabetic WT) respectively], although diabetic TGM were more sensitive to exogenously administered insulin than diabetic WT. Diabetic WT were hypoleptinaemic and hyperphagic relative to non-diabetic WT, whereas diabetic TGM, which remained hyperleptinaemic, were less hyperphagic than diabetic WT. After STZ injection, hypothalamic expressions of orexigenic and anorexigenic peptide mRNAs were up-regulated and down-regulated, respectively, in diabetic WT, whereas they were unchanged in diabetic TGM. Diabetic TGM became normoglycaemic, when treated with insulin at such doses that did not improve hyperglycaemia in diabetic WT. We found that a sub-threshold dose of insulin that does not affect glucose homeostasis is effective in improving the diabetes in normal mice rendered diabetic by STZ injection, when combined with leptin.
Conclusions/interpretation
This study suggests that leptin could be used as an adjunct of insulin therapy in insulin-deficient diabetes, thereby providing an insight into the therapeutic implication of leptin as an anti-diabetic agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Insulin plays an essential role in the treatment of insulin-dependent or insulin-deficient diabetes. The goal of insulin therapy for this pathological condition is to eliminate the clinical symptoms associated primarily with hyperglycaemia (i.e. diabetic ketoacidosis and hyperosmolar coma) and to delay or prevent the progression of the long-term microvascular complications of diabetes (i.e. retinopathy, neuropathy, and nephropathy) [1]. This can be achieved with intensive insulin therapy that involves multiple injections or boluses of insulin each day or continuous subcutaneous insulin infusion to meet the physiological demands of the patient for an insulin response to increased postprandial blood glucose levels [2].
Leptin is an adipocyte-derived hormone that is involved in the regulation of food intake and energy expenditure [3, 4]. It has been reported that plasma leptin concentrations are increased in obese subjects in proportion to the degree of adiposity [5, 6, 7], suggesting that leptin is a signal informing the body of the size of its adipose stores. We previously created transgenic skinny mice overexpressing leptin under the control of the liver-specific human serum amyloid P component promoter [8]. The "hyperleptinaemia" of these skinny mice causes the disappearance of lipids from the adipose tissue and provides a unique experimental system to investigate the long-term in vivo effects of chronic hyperleptinaemia [8, 9, 10]. The skinny mice have increased glucose metabolism and insulin sensitivity, accompanied by the activation of insulin signalling for glucose disposal in the skeletal muscle and liver [8]. These findings support the idea that leptin acts as an anti-diabetic hormone on a long-term basis. We observed that transgenic overexpression of leptin accelerates the recovery from diabetes in calorically-restricted lethal yellow agouti mice (KKA y mice) [11]. In addition, transgenic overexpression of leptin has rescued the insulin resistance and diabetes in a mouse model of lipoatrophic diabetes [12]. We, therefore, postulate that leptin could be used therapeutically in the treatment of non-insulin-dependent diabetes (i.e. Type 2 diabetes and lipoatrophic diabetes). There are several reports showing a marked decrease in plasma leptin concentrations in insulin-deficient diabetic rats and humans [13, 14, 15, 16, 17, 18, 19, 20], suggesting the therapeutic implication of leptin in insulin-deficient diabetes. However, whether or not leptin is therapeutically beneficial for insulin-dependent or insulin-deficient diabetes and, if so, how it should be used for this class of diabetes has not been examined rigorously.
Streptozotocin (STZ) administration in rodents leads to the destruction of pancreatic beta cells and thus provides the useful experimental model of insulin-deficient diabetes. In this study, to explore the therapeutic implication of leptin in insulin-deficient diabetes, we tried to induce uncontrolled diabetes by STZ in both transgenic skinny mice (TGM) and non-transgenic littermates (WT). Here we show that TGM developed marked hyperglycaemia similar to that induced in WT after the STZ treatment (termed diabetic TGM and diabetic WT, respectively). Furthermore, diabetic TGM responded to insulin administered at such doses that did not improve the glucose metabolism in diabetic WT. We also found that a sub-threshold dose of insulin was sufficient to improve the glucose metabolism in STZ-induced diabetic mice, when combined with leptin. These findings suggest that leptin could be used as an adjunct of insulin therapy in insulin-deficient diabetes.
Materials and methods
Animals
We used 10- to 14-week-old male transgenic skinny mice (TGM) and non-transgenic littermates (WT) on the C57BL/6 background in this study [8]. In this study 8-week-old male C57BL/6 mice were purchased from CLEA, Tokyo, Japan. They were maintained in a temperature-, humidity- and light-controlled room (12 h light/12 h dark cycle) and allowed free access to standard rat chow (CE-2, 352 kcal/100 g, CLEA), except for the pair-feeding experiment. The care of the animals and all experimental procedures were conducted in accordance with the institutional ethical guidelines of Kyoto University Graduate School of Medicine Committee on Animal Research.
Induction of diabetes and transgenic study protocols
Diabetes was induced in WT and TGM by a single i.p. injection of STZ (180~240 mg/kg body weight in 200 µl of 10 mmol/l sodium citrate buffer, pH 4.0) (Sigma, St. Louis, Mo., USA) or vehicle. Control non-diabetic groups of each genotype received an equal volume of vehicle (termed non-diabetic WT and non-diabetic TGM). Blood glucose concentrations were monitored after the STZ treatment. The STZ-treated mice of each genotype developed severe hyperglycaemia accompanied by marked hypoinsulinaemia (termed diabetic WT and diabetic TGM, respectively).
Diabetic WT and TGM received i.p. injections of graded amounts of NPH insulin 3 weeks after the STZ treatment, (Humulin N, Eli Lilly Japan, Kobe, Japan) twice daily over 36 days; 0.2×2 mU·g–1·day–1 for the first 3 days, 0.8×2 mU·g–1·day–1 for 6 days, 1.6×2 mU·g–1·day–1 for 12 days, 12.8×2 mU·g–1·day–1 for 6 days, and up to 25.6×2 mU·g–1·day–1 for 9 days (Fig. 5). For the pair-feeding experiment, a group of diabetic WT was provided with the amount of food consumed the previous day by diabetic TGM. To prevent prolonged fasting in the pair-fed animals, food was divided in two portions, and each portion of the food was given soon after every insulin injection [12]. Blood glucose concentrations were monitored every 3 days from the starting day of insulin administration, and each measurement was made 8 to 10 h after the previous insulin injection. Daily food intake and body weight were measured on the indicated days during the time period after the blood glucose measurement and before the insulin injection during the experiments.
Immunostaining of insulin
The animals were exsanguinated via the abdominal vena cava under ether anaesthesia. The whole pancreas of each animal was removed and fixed in 10% neutral buffered formalin. After proper fixation, the pancreas was routinely processed, embedded in paraffin, sectioned, stained, and examined microscopically. Immunohistochemical staining was carried out to detect insulin-positive pancreatic beta cells. Briefly, after dehydration through graded series of ethanol, endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in absolute methanol for 30 min. For insulin immunohistochemical staining, sections were blocked with non-immune serum (Zymed Laboratories, South San Francisco, Calif., USA) and incubated with guinea pig polyclonal anti-porcine insulin antibody (DAKO, Carpinteria, Calif., USA) at 1:500 dilution for 30 min. Immunolocalization was demonstrated using a Histostain SP Kit (Zymed Laboratories).
Co-administration study protocols
Recombinant mouse leptin (Amgen, Thousand Oaks, Calif., USA) (40 ng·g–1·h–1) or a equal volume of saline solution (vehicle) was started to be infused into 10-week-old male C57BL/6 mice rendered diabetic by the STZ treatment (a single i.p. injection of STZ at a dose of 180 mg/kg body weight), using an osmotic micropump (Alzet model 1002D, Alzet Pharmaceuticals, Palo Alto, Calif., USA) delivering 0.5 µl/h [9, 12]. Mice were anaesthetized with diethylether, and an osmotic micropump was implanted s.c. in the midscapular region. They were treated with or without 5 mU·g–1·day–1 human regular insulin (Humulin R, Eli Lilly Japan, Kobe, Japan) delivered s.c. using an osmotic micropump. These animals were divided into four groups: vehicle-treated mice (V group), mice treated with insulin alone (I group), mice treated with leptin alone (L group), and mice treated with both insulin and leptin (I + L group) (n=6). During the 12-day infusion, body weight, food intake, blood glucose, plasma insulin and leptin concentrations were measured.
Blood glucose, plasma insulin and leptin measurements
Blood was sampled from the retro-orbital sinus of mice at 7:00 p.m., when fed ad libitum. Blood glucose and plasma insulin concentrations were determined by the glucose oxidase method with a reflectance glucometer (One Touch II, Lifescan, Milpitas, Calif., USA) and the enzyme immunoassay with Glazyme Insulin-EIA TEST (Wako Pure Chemical Industries, Osaka, Japan), respectively. In this study, the detection limit of the EIA was 0.10 ng/ml. Plasma leptin concentrations were determined using the RIA for mouse leptin (Linco Research Immunoassay, St. Louis, Mo., USA).
Insulin tolerance test
For insulin tolerance test, both diabetic and non-diabetic groups of each genotype were treated with an i.p. injection of 0.8 mU/g body weight human regular insulin (Humulin R, Eli Lilly Japan, Kobe, Japan) [8]. Blood was sampled from the mouse tail vein before and 30, 60, 90, and 120 min after the insulin injection. Insulin tolerance tests were carried out 2 days before and 14 days after the STZ treatment.
Northern blot analysis
The whole hypothalami were obtained from TGM and WT mice 14 days after the STZ treatment. Total RNA was extracted and Northern blot analysis was done using the mouse proopiomelanocortin (POMC), neuropeptide Y (NPY), and agouti-related protein (AGRP) cDNA fragments [21, 22]. The mRNA levels were normalized to the β-actin mRNA levels in each sample to correct for differences in the amount of total RNA applied.
Statistical analysis
All values were expressed as means ± SEM. Comparisons among genotypes and effect of STZ or insulin treatment were assessed by two-way ANOVA and completed by post hoc test. The main effect of treatment (i.e., co-administration study) was assessed by one-way ANOVA with repeated measures and completed by post hoc test, as required. A Student's t test was carried out, where applicable. Statistical analyses were done by utilizing StatView 4.02 (Abacus Concepts).
Results
Induction of diabetes by STZ in WT and TGM
The size of the pancreatic islets was markedly reduced in non-diabetic TGM relative to non-diabetic WT. The number of insulin-positive pancreatic beta cells was substantially reduced in both diabetic WT and diabetic TGM (Fig. 1).

Insulin immunohistochmical study of pancreatic islets from WT and TGM 3 weeks after treatment with vehicle (non-diabetic) or STZ (diabetic). (A) Non-diabetic WT (B) Diabetic WT (C) Non-diabetic TGM (D) Diabetic TGM. Arrowheads denote the edge of pancreatic islets. Original magnification ×100. Bar=50 µm
Plasma insulin concentrations in non-diabetic TGM were lower than those in non-diabetic WT without the STZ treatment (0.48±0.53 and 1.20±0.19 ng/ml, respectively, n=18, p<0.01) (Fig. 2A). Plasma insulin concentrations were remarkably reduced in both WT and TGM 2 weeks after the STZ treatment (<0.10 ng/ml, n=8) (Fig. 2A). Consequently, both WT and TGM developed pronounced hyperglycaemia 2 weeks after the STZ treatment (536±33 and 444±31 mg/dl, respectively, n=18) (Fig. 2B). Plasma leptin concentrations in non-diabetic TGM were higher than those in non-diabetic WT (27.6±10.5 and 4.0±0.6 ng/ml, respectively, n=18, p<0.01) (Fig. 2C). Plasma leptin concentrations were reduced in WT 2 weeks after the STZ treatment (2.4±0.3 ng/ml, n=12), whereas TGM remained hyperleptinaemic (38.8±6.9 ng/ml, n=12) (Fig. 2C).

Plasma parameters, food intake, and body weight in non-diabetic and diabetic WT and TGM 2 weeks after the STZ treatment. (A) Plasma insulin concentrations (B) Blood glucose concentrations (C) Plasma leptin concentrations (D) 24 h food intake (E) Body weight. *p<0.05, **p<0.01. NS, not significant; Closed bars indicate TGM
Daily food intake was decreased in non-diabetic TGM relative to non-diabetic WT (Fig. 2D). The diabetic WT was more hyperphagic than non-diabetic WT (6.3±0.5 and 3.5±0.2 g/24 h, respectively, n=18, p<0.01). Daily food intake of diabetic TGM was increased, although significantly suppressed, relative to non-diabetic TGM (4.0±0.1 and 2.1±0.2 g/24 h, respectively, n=18, p<0.01). Body weight of non-diabetic TGM was reduced relative to non-diabetic WT (23.6±0.4 and 26.4±0.5 g, respectively, n=18, p<0.01) (Fig. 2E). The diabetic WT was smaller in weight than non-diabetic WT (22.7±0.4 and 26.4±0.5 g, respectively, n=18, p<0.01). Body weight of diabetic TGM tended to be reduced relative to non-diabetic TGM (20.7±0.6 and 23.6±0.4 g, respectively, n=18).
Hypothalamic neuropeptide mRNA expression in diabetic WT and TGM
Northern blot analysis showed that NPY and AGRP mRNA expression is decreased in non-diabetic TGM relative to non-diabetic WT (Fig. 3B,C). However, POMC mRNA expression was increased in non-diabetic TGM relative to non-diabetic WT (Fig. 3D). Expression of NPY and AGRP mRNAs was up-regulated (approximately two-fold, n=5, p<0.05), whereas POMC mRNA expression was down-regulated (approximately 50%, n=5, p<0.05), in diabetic WT relative to non-diabetic WT (Fig. 3B–D). In this study, NPY, AGRP, and POMC mRNA expression was unchanged in TGM even after the STZ treatment.

Hypothalamic expression of NPY, AGRP, and POMC mRNAs in non-diabetic and diabetic WT and TGM 2 weeks after the STZ treatment. (A) Northern blot analysis of NPY, AGRP, and POMC mRNAs in the hypothalami from WT and TGM. 10 µg of total RNA was used per lane. Quantification of (B) NPY mRNA (C) AGRP mRNA (D) POMC mRNA levels. The mRNA levels of non-diabetic WT was defined as 1.0 U. *p<0.05 vs non-diabetic WT. NS, not significant. Closed bars indicate TGM
Insulin sensitivity of diabetic WT and TGM
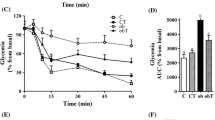
Insulin tolerance test showed exaggerated hypoglycaemic response to a single i.p. injection of insulin in non-diabetic TGM relative to non-diabetic WT (Fig. 4A). In this study, even after the STZ treatment, TGM had a marked hypoglycaemic response to exogenously administered insulin compared with WT (Fig. 4B).

Insulin tolerance test in non-diabetic and diabetic WT and TGM. (A) Non-diabetic groups treated with 0.8 U/kg regular insulin (B) Diabetic groups treated with 0.8 U/kg regular insulin. *p<0.05 vs WT
Insulin administration in diabetic WT and TGM
Since diabetic TGM were more sensitive to exogenous insulin than diabetic WT, we hypothesized that hyperglycaemia should be normalized in diabetic TGM in response to insulin administered at low doses that do not improve the glucose homeostasis in diabetic WT. We, therefore, examined the effect of administration of graded doses of insulin in diabetic WT and diabetic TGM (Fig. 5A). Treatment with NPH insulin twice daily at doses ranging from 0.4 to 3.2 mU·g–1·day–1 did not improve the hyperglycaemia in diabetic WT. Eventually, they responded to insulin administered at a dose of 51.2 mU·g–1·day–1. In contrast, diabetic TGM became normoglycaemic in response to insulin administered at a dose of 3.2 mU·g–1·day–1, which is at least tenfold less than those required to improve the glucose homeostasis in diabetic WT. In this study, no differences in plasma leptin concentration were noted between insulin-treated and insulin-untreated diabetic WT (5.6±1.2 vs 3.2±0.6 ng/ml, n=5) and between insulin-treated and insulin-untreated diabetic TGM (22.0±4.2 vs 38.8±6.9 ng/ml, n=5) 21 days after the insulin administration (3.2 mU·g–1·day–1) (Fig. 5B). Plasma insulin concentrations in both insulin-untreated diabetic WT and diabetic TGM were <0.10 ng/ml (Fig. 5C). After 12 days of the insulin administration at a dose of 3.2 mU·g–1·day–1, plasma insulin concentrations were 0.22±0.13 and 0.11±0.09 ng/ml in insulin-treated diabetic WT and diabetic TGM, respectively. When treated with insulin at a dose of 3.2 mU·g–1·day–1, daily food intake was reduced in insulin-treated diabetic WT and diabetic TGM relative to insulin-untreated groups (WT: 4.8±0.2 vs 6.3±0.5 g/24 h, n=6, p<0.05; TGM: 2.9±0.4 vs 4.0±0.1 g/24 h, respectively, p<0.05) (Fig. 5D). There were no differences in body weight between insulin-treated and insulin-untreated diabetic WT and between insulin-treated and insulin-untreated diabetic TGM during the experiment (Fig. 5E).

Effect of i.p. administration of NPH insulin (0.4–51.2 mU·g–1·day–1) in diabetic WT and TGM. (A) Time course of blood glucose concentrations in diabetic WT fed ad libitum [WT (ad lib), ○], diabetic WT pair-fed to diabetic TGM [WT (pair-fed), □], and diabetic TGM fed ad libitum [TGM (ad lib), ●] during the insulin administration. Black bars indicate the periods of food intake record for (D). *p<0.005 vs TGM (ad lib) on day 0; **p<0.001 vs TGM (ad lib) on day 0; ## p<0.005 vs WT (ad lib) on day 0. (B) Plasma leptin concentrations in diabetic WT and TGM before (diabetic) and 21 days after the insulin administration (diabetic + insulin). **p<0.01. (C) Plasma insulin concentrations in diabetic WT and TGM before (diabetic) and 21 days after the insulin administration (diabetic + insulin). (D) 24-h food intake of diabetic WT and TGM before (diabetic) and after the insulin administration (diabetic + insulin). *p<0.05. (E) Body weight in diabetic WT and TGM before (diabetic) and 21 days after the insulin administration (diabetic + insulin). *p<0.05. NS, not significant. Closed bars indicate TGM
Since diabetic TGM are less hyperphagic than diabetic WT during the insulin administration, it is possible that diabetic TGM are more sensitive to insulin therapy than WT because of the hypophagic effect of leptin. However, pair-feeding diabetic WT to the amount of food consumed by diabetic TGM did not improve the diabetes (Fig. 5A), showing that the beneficial effect of leptin is not due to the decreased food intake. This is consistent with our previous observation that the leptin's anti-diabetic effect is not due to its hypophagic effect in a mouse model of lipoatrophic diabetes.
Co-administration of leptin and insulin in STZ-induced diabetic mice
To assess whether combination therapy of leptin and insulin might be beneficial for insulin-deficient diabetes, we also examined the effect of chronic co-administration of leptin and insulin in normal mice rendered diabetic by STZ (Fig. 6). No changes in blood glucose concentration were noted in STZ-induced diabetic mice treated with regular insulin alone using an osmotic micropump (5 mU·g–1·day–1 for 12 days), indicating the induction of severe insulin-deficient diabetes (Fig. 6A). In this study, chronic administration of leptin alone (40 ng·g–1·h–1) via the osmotic micropump did not improve the diabetes throughout the experiment. However, co-administration of leptin (40 ng·g–1·h–1) and insulin (5 mU·g–1·day–1) normalized the hyperglycaemia in STZ-induced diabetic mice 12 days after the co-administration. Before the co-administration, plasma insulin concentrations in all the groups were <0.10 ng/ml. Plasma insulin concentrations were <0.10 ng/ml in the V and L groups throughout the experiment, whereas they were up to 0.22±0.04 and 0.19±0.04 ng/ml in the I and I + L groups, respectively, 12 days after the co-administration (n=6) (Fig. 6B). Plasma leptin concentrations were markedly increased in the L and I + L groups 12 days after the co-administration (38.1±3.0 and 38.3±7.1 ng/ml in L and I + L groups, respectively, n=6) (Fig. 6C), which were roughly comparable to those in TGM. In this study, there was no difference in cumulative food intake between the V and I groups (63.9±2.3 vs 61.9±4.6 g/24 h, n=6) (Fig. 6D). In contrast, the L and I + L groups were less hyperphagic than the V group 12 days after the co-administration. Cumulative food intake during the co-administration study was reduced in the I + L group relative to the L group (41.2±1.4 vs 50.8±3.2 g/12 days, n=6, p<0.01). At the end of the co-administration study, body weight increased, although not significantly, in the I group relative to V group (1.9±0.5 vs 0.9±0.6 g/12 days, n=6), whereas hyperglycaemia was not normalized in the I group. No difference in body weight was noted between the L and I + L groups (Fig. 6E).

Effect of co-administration of leptin and insulin in normal mice rendered diabetic by the STZ treatment. (A) Time course of blood glucose concentrations in diabetic mice during the leptin and insulin co-administration. Vehicle-treated groups (V); ○, Insulin-treated groups (I); ■, Leptin-treated groups (L); ◇, Leptin- and insulin-treated groups (I+L); ▲. **p<0.01 vs V group. (B) Plasma insulin concentrations in diabetic mice 12 days after the co-administration. (C) Plasma leptin concentrations in diabetic mice 12 days after the co-administration. (D) Cumulative food intake in diabetic mice during 12 days of the co-administration. (E) Body weight change in diabetic mice 12 days after the co-administration. V, I, L, and I+L indicate the same groups as in (A). Closed bars indicate groups treated with leptin. *p<0.05, **p<0.01
Discussion
The data of this study showed that TGM develop insulin-deficient diabetes similar to that induced in WT by the STZ treatment. These observations indicate that chronic hyperleptinaemia cannot prevent the progression of STZ-induced insulin-deficient diabetes, suggesting that insulin is required for the anti-diabetic effect of leptin. In this study, the insulin tolerance test showed an exaggerated hypoglycaemic response to exogenously administered insulin in diabetic TGM relative to diabetic WT. One possible explanation for the insulin sensitization in TGM with dramatic loss of visible adipose tissue might be the reduced levels of triglycerides and FFAs both in the circulation and in body tissue [8, 12]. These observations led us to speculate that a smaller amount of insulin is sufficient to improve the diabetes in diabetic TGM than in diabetic WT. To address this possibility, we examined the effect of insulin administration in diabetic TGM. We showed that hyperglycaemia is normalized in diabetic TGM, when treated with less than 1·10–1 of insulin of which can improve the diabetes in diabetic WT. We also examined the effect of chronic co-administration of leptin and insulin in STZ-induced diabetic mice and found that a sub-threshold dose of insulin, in conjunction with leptin, is sufficient to normalize the hyperglycaemia in these mice. These findings indicate that leptin can reduce the insulin requirement to achieve good glycaemic control. Thus, leptin could act as an insulin-sensitizing anti-diabetic agent in insulin-deficient diabetes. This is reminiscent of a previous report that the anti-diabetic effect of metformin results in a reduction of insulin requirement in the treatment of insulin-deficient diabetes [27, 28]. It was reported that leptin alone is capable of improving the diabetes in STZ-induced diabetic rats [16], in which plasma insulin concentrations were reduced (25.4 pmol/l=~0.2 ng/ml). Furthermore, central leptin infusion alone can improve glucose metabolism in STZ-induced diabetic rats [17, 18]. It is conceivable that exogenous leptin (administered either centrally or peripherally) acts as an anti-diabetic agent in concert with the residual insulin, thereby improving the hyperglycaemia in STZ-induced diabetic rats. In this regard, using graded doses of STZ, we have produced mice with varying degrees of insulin-deficient diabetes and confirmed that leptin's ability to achieve good glycaemic control is correlated with the residual amount of insulin. Collectively, we postulate that leptin could be therapeutically used as an adjunct of insulin therapy in insulin-deficient diabetes.
The Diabetes Control and Complication Trial (DCCT), a perspective, randomized study comparing the effects of intensive and standard insulin therapy in the course of long-term diabetic complications, shows that intensive insulin therapy reduces clinically distinctive progression of retinopathy and the incidence of proteinuria and neuropathy [2]. In the DCCT, rather than a standard regimen with one or two injections of insulin daily, intensive insulin therapy is often associated with an increased frequency of severe hypoglycaemic episodes and increased weight gain [29, 30, 31, 32, 33]. Weight gain might result in an increase of blood pressure and atherogenic serum lipid profile, thus increasing the risk of cardiovascular diseases. Due to the potential adverse consequences of intensive insulin therapy, efforts to prevent hypoglycaemia and unwanted weight gain associated with intensive therapy are being pursued. Here we provide evidence that leptin, when combined with insulin, can reduce the amount of insulin required to achieve good glycaemic control in insulin-deficient diabetes. In the co-administration study, we found that a combination therapy of leptin and insulin tends to reduce body weight gain relative to insulin monotherapy, which could be due to the leptin's ability to decrease food intake or the insulin requirement, or both. Thus, combination therapy of leptin and insulin might offer a novel therapeutic strategy for the treatment of insulin-deficient diabetes, with lowering the undesirable effect of insulin; such as body weight gain.
It is recognized that leptin acts directly on the hypothalamus, where it leads to the increases in anorexigenic peptides such as α-melanocyte stimulating hormone derived from POMC and reciprocal decreases in orexigenic peptides such as NPY and AGRP [34]. In this study, daily food intake is increased in diabetic WT relative to non-diabetic WT. Furthermore, hypothalamic NPY and AGRP mRNA expression is up-regulated, whereas POMC mRNA expression is down-regulated, in diabetic WT relative to non-diabetic WT [37]. These findings suggest that the altered expression of the above hypothalamic neuropeptides is involved in increased food intake in diabetic WT. It is likely that the alteration is due at least in part to hypoleptinaemia in diabetic WT, because expression of mRNAs for these neuropeptides remains unchanged in diabetic TGM. These observations support the idea that diabetic hyperphagia is a consequence of the hypoleptinaemia-induced imbalance between hypothalamic orexigenic and anorexigenic neuropeptides. Interestingly, even with chronic hyperleptinaemia, diabetic TGM became more hyperphagic than non-diabetic TGM, although less than diabetic WT. In this study, cumulative food intake is reduced in both diabetic WT and diabetic TGM relative to non-diabetic groups during the insulin administration, when plasma leptin concentrations are unchanged. These findings suggest that chronic hyperleptinaemia alone cannot prevent hyperphagia in insulin-deficient diabetes in which the leptin's effect on hypothalamus was not attenuated. A small dose of insulin that does not alter the leptin level improved hyperphagia in diabetic TGM, which is consistent with previous reports that diabetic hyperphagia is normalized by insulin [35, 36, 37]. Our data suggest that hypoinsulinaemia per se could play a role in diabetic hyperphagia through a leptin-independent mechanism.
We previously pursued the therapeutic potential of leptin as an anti-diabetic agent by use of transgenic skinny mice and would like to propose that leptin could be therapeutically used in the treatment of diabetes of different etiologies. We have reported that transgenic overexpression of leptin can delay the onset of insulin resistance and diabetes in KKA y mice at younger ages, when they are of normal weight [11]. These findings suggest that leptin alone is effective in non-insulin-dependent diabetes or Type 2 diabetes, without obesity-induced leptin resistance. However, chronic hyperleptinaemia does not prevent the progression of diabetes in KKA y mice at older ages, when they develop full-blown obesity, suggesting that leptin alone might not be effective in Type 2 diabetes, with obesity-induced resistance to leptin and insulin. In this case, leptin could be used as an adjunct of caloric restriction at achieving good glycaemic control, because transgenic overexpression of leptin can accelerate the recovery from the insulin resistance and diabetes in calorically-restricted KKA y mice [11], which are able to respond to leptin [38]. We also found that transgenic overexpresion of leptin can rescue the insulin resistance and diabetes in a mouse model of lipoatrophic diabetes [12], showing that leptin alone should be effective in the treatment of lipoatrophic diabetes. In this study, we have added evidence that a combination therapy of leptin and insulin might be useful for the treatment of insulin-deficient diabetes or Type 1 diabetes. It is likely that the anti-diabetic effect of leptin could depend on how much insulin action and leptin sensitivity exist.
In conclusion, our study suggests that co-administration of leptin and insulin could be therapeutically used in the treatment of insulin-deficient diabetes. This study provides a further insight into the therapeutic potential of leptin as an anti-diabetic agent.
Abbreviations
- STZ:
-
streptozotocin
- TGM:
-
transgenic skinny mice overexpressing leptin
- WT:
-
non-transgenic littermates
- NPY:
-
neuropeptide Y
- AGRP:
-
agouti-related protein
- POMC:
-
proopiomelanocortin
References
Rosenzweig JL (1994) Principles of insulin therapy. In: Kahn CR, Weir GC (eds) Joslin's diabetes, 13th edn. Lea & Febiger, Tokyo, pp 460–488
The DCCT Research Group (1987) Diabetes Control and Complication Trial (DCCT): results of feasibility study. Diabetes Care 10:1–19
Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 372:425–432
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1:1311–1314
Maffei M, Halaas J, Ravussin E et al. (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1:1155–1161
Considine RV, Sinha MK, Heiman ML et al. (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334:292–295
Ogawa Y, Masuzaki H, Hosoda K et al. (1999) Increased glucose metabolism and insulin sensitivity in transgenic skinny mice overexpressing leptin. Diabetes 48:1822–1829
Aizawa-Abe M, Ogawa Y, Masuzaki H et al. (2000) Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest 105:1243–1252
Yura S, Ogawa Y, Sagawa N et al. (2000) Accelerated puberty and late-onset hypothalamic hypogonadism in female transgenic skinny mice overexpressing leptin. J Clin Invest 105:749–755
Masuzaki H, Ogawa Y, Aizawa-Abe M et al. (1999) Glucose metabolism and insulin sensitivity in trangenic mice overexpressing leptin with lethal yellow agouti mutation: usefulness of leptin for the treatment of obesity-associated diabetes. Diabetes 48:1615–1622
Ebihara K, Ogawa Y, Masuzaki H et al. (2001) Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 50:1440–1448
Chavez M, Seeley RJ, Havel PJ et al. (1998) Effect of a high-fat diet on food intake and hypothalamic neuropeptide gene expression in streptozotocin diabetes. J Clin Invest 102:340–346
Havel PJ, Uriu-Hare JY, Liu T et al. (1998) Marked and rapid decreases of circulating leptin in streptozotocin diabetic rats: reversal by insulin. Am J Physiol 274:R1482–R1491
Sindelar DK, Havel PJ, Seeley RJ, Wilkinson CW, Woods SC, Schwartz MW (1999) Low plasma leptin levels contribute to diabetic hyperphagia in rats. Diabetes 48:1275–1280
Chinookoswong N, Wang JL, Shi ZQ (1999) Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes 48:1487–1492
Hidaka S, Yoshimatsu H, Kondou S et al. (2002) Chronic leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. FASEB J 16:509–518
Lin CY, Higginbotham DA, Judd RL, White BD (2002) Central leptin increases insulin sensitivity in streptozotocin-induced diabetic rats. Am J Physiol 282:E1084–E1091
Kiess W, Anil M, Blum WF et al. (1998) Serum leptin levels in children and adolescents with insulin-dependent diabetes mellitus in relation to metabolic control and body mass index. Eur J Endocrinol 138:501–509
Hathout EH, Sharkey J, Racine M, Ahn D, Mace JW, Saad MF (1999) Changes in plasma leptin during the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab 84:4545–4548
Ebihara K, Ogawa Y, Katsuura G et al. (1999) Involvement of agouti-related protein, an endogenous antagonist of hypothalmic melanocortin receptor, in leptin action. Diabetes 48:2028–2033
Shintani M, Ogawa Y, Ebihara K et al. (2001) Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes 50:227–232
Kamohara S, Burcellin R, Hallas JL, Friedman JM, Charron MJ (1997) Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 389:374–377
Liu L, Karakanias GB, Morales JC et al. (1998) Intrecerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem 273:31160–31167
Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL (1999) Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401:73–76
Arioglu E, Simha V, Ruiz E et al. (2002) Leptin-replacement therapy for lipodystrophy. N Engl J Med 346:570–578
Gin H, Messerchmitt C, Brottier E, Aubertin J (1985) Metformin improved insulin resistance in type I, insulin-dependent diabetic patients. Metabolism 34:923–925
Bailey CJ, Mynett KJ (1994) Insulin requirement for the antihyperglycaemic effect of metformin. Br J Pharmacol 111:793–796
White NH, Skor DA, Cryer PE, Levandoski LA, Bier DM, Santiago JV (1983) Identification of type I diabetic patient at increased risk for hypoglycemia during intensive therapy. N Engl J Med 308:485–491
The DCCT Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
The DCCT Research Group (1995) Adverse events and their association with treatment regimens in the Diabetes Control and Complications Trial. Diabetes Care 18:1415–1427
The DCCT Research Group (1997) Hypoglycemia in the Diabetes Control and Complications Trials. Diabetes 46:271–286
The DCCT Research Group (1988) Weight gain associated with intensive therapy in the diabetes control and complication trial. Diabetes Care 11:567–573
Elmquist JK, Maratos-Flier E, Saper CB, Flier JS (1998) Unraveling the central nervous system pathway underlying responses to leptin. Nat Neurosci 1:445–450
Sipols AJ, Baskin DG, Schwartz MW (1995) Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes 44:147–155
Kim EM, Grace MK, Welch CC, Billigton CJ, Levine AS (1999) STZ-induced diabetes decreases and insulin normalizes POMC mRNA in arcuate nucleus and pituitary in rats. Am J Physiol 276:R1320–R1326
Havel PJ, Hahn TM, Sindelar DK et al. (2000) Effects of streptozotocin-induced diabetes and insulin treatment on the hypothalamic melanocortin system and muscle uncoupling protein 3 expression in rats. Diabetes 49:244–252
Marsh DJ, Hollopeter G, Huszar D et al. (1999) Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21:119–122
Acknowledgements
We thank M. Aizawa-Abe for discussions. The authors also acknowledge I. Kitahata and M. Nagamoto for secretarial and technical assistance. This work was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Yamanouchi Foundation for Research on Metabolic Disorders, the Inamori Foundation, Yamaguchi Endocrine Research Association, and the ONO Medical Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miyanaga, F., Ogawa, Y., Ebihara, K. et al. Leptin as an adjunct of insulin therapy in insulin-deficient diabetes. Diabetologia 46, 1329–1337 (2003). https://doi.org/10.1007/s00125-003-1193-6
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1193-6