
Abstract
Aims/Hypothesis
Increased serum inflammatory markers have been found in obesity and insulin-resistant states, and could play a causative role in insulin resistance, atherosclerosis and cardiovascular disease. The polycystic ovary syndrome represents a human model of insulin resistance because both lean and obese polycystic ovary syndrome patients are insulin-resistant compared with non-hyperandrogenic women. We evaluated whether obesity, insulin resistance, or both, are related to the increased concentrations of inflammatory markers in pre-menopausal women.
Methods
We compared 35 patients with polycystic ovary syndrome and 28 healthy women, paired for BMI, prevalence of obesity and smoking. Measurements included serum inflammatory markers, BMI, waist-to-hip ratio, blood pressure, serum glucose, insulin, lipid and hormone concentrations, and insulin sensitivity index.
Results
The insulin sensitivity index was reduced in polycystic ovary syndrome patients compared with controls. However, no differences were observed between both groups in C-reactive protein, interleukin 6, tumour necrosis factor-α, soluble type 2 tumour necrosis factor receptor, and soluble intercellular cell adhesion molecule-1. When considering patients and controls as a whole, C-reactive protein and interleukin 6, were increased in obese subjects compared with lean women. Inverse correlations existed between insulin sensitivity index and C-reactive protein, interleukin 6, tumour necrosis factor-α, soluble type 2 tumour necrosis factor receptor, and soluble intercellular cell adhesion molecule-1. Only the weak correlation with C-reactive protein persisted after controlling for BMI.
Conclusion/interpretation
Obesity, and not insulin resistance, is the major determinant of serum inflammatory cardiovascular risk markers in pre-menopausal women.
Similar content being viewed by others

Insulin resistance is defined by the decreased ability of insulin to stimulate glucose disposal into target tissues, resulting in a compensatory increase in circulating insulin concentrations [1]. Insulin resistance is involved in the pathogenesis of several disorders, such as Type 2 diabetes, central obesity, arterial hypertension, and atherosclerosis, finally resulting in cardiovascular disease. These disorders are frequently termed the metabolic syndrome.
Chronic inflammation could also be involved in the development of the metabolic syndrome and cardiovascular disease [2], and serum markers of chronic inflammation and insulin resistance are increasingly being considered as cardiovascular risk factors. Together with impaired glucose tolerance, dyslipidaemia, hyperhomocysteinaemia, increased coagulation and impaired fibrinolysis, and increased circulating concentrations of cell adhesion molecules and markers of endothelial dysfunction [2], inflammatory markers cluster in patients with cardiovascular disease, suggesting a role in the pathogenesis of atherosclerosis.
Clustering of these markers partly explains the frequent finding of correlations between measures of insulin resistance and markers of inflammation and endothelial dysfunction. Among others, inverse correlations have been reported between indexes of insulin sensitivity and circulating concentrations of TNF-α [3], soluble type 2 tumour necrosis factor receptor (sTNFR2) [4], IL-6 [5], C-reactive protein (CRP) [5], and soluble intercellular cell adhesion molecule-1 (sICAM-1) [5].
The adipose tissue plays a central role in the relationship between cytokines and insulin resistance. The expression of TNF-α and TNFR2 in adipose tissue is increased in obesity [6, 7]. TNF-α expression correlates with indexes of insulin resistance, and decreases with weight loss in parallel with the improvement in insulin sensitivity [6]. Similar results have been reported for IL-6 [8]. Moreover, inflammatory cytokines might induce insulin resistance by direct actions on insulin-signalling postreceptor molecules [9] or by inducing central obesity through activation of the hypothalamic-pituitary-adrenal axis [10].
As obesity induces insulin resistance, and inflammatory cytokines are secreted by adipose tissue [6, 8], it is difficult to evaluate if the relationship between serum markers of inflammation and insulin resistance results from a pathophysiological link, or is merely reflecting the stronger relationship between obesity and insulin resistance.
The polycystic ovary syndrome (PCOS), as defined by the presence of hyperandrogenic oligo-ovulation [11], can be considered a human model of insulin resistance. Both lean and obese women with PCOS are insulin resistant when compared with their non-hyperandrogenic counterparts [12]. As PCOS patients are usually diagnosed before the onset of the other components of the metabolic syndrome and of atherosclerosis and cardiovascular disease—which could have acted as confounding factors in previous studies—PCOS offers a unique model to study the relationship between serum markers of inflammation, insulin resistance and obesity.
In this study, we have examined the circulating concentrations of serum markers of inflammation in a well-defined group of pre-menopausal women with PCOS compared with healthy controls, focusing on their relationship to insulin resistance and obesity.
Subjects, materials and methods
Subjects
Thirty-five consecutive patients diagnosed with PCOS (BMI 31.8±9.2 kg/m2, age 24.9±7.4 years) and 28 non-hyperandrogenic women (BMI 30.6±8.0 kg/m2, age 31.7±8.0 years) were included in the study. The PCOS and control groups were matched primarily for BMI, and secondarily for prevalence of obesity and prevalence of smokers. The control group included women submitted to one of the authors for treatment of obesity, and healthy lean volunteers. None of the control subjects had signs or symptoms of hyperandrogenism, menstrual dysfunction, or a history of infertility.
The diagnosis of PCOS was based on endocrine criteria [11]. All the patients had oligo-ovulation as indicated by oligomenorrhoea or amenorrhoea, or regular menstrual cycles with serum progesterone below 12.7 nmol/l between days 22 to 24 of the menstrual cycle. Menstrual calendars and basal body temperature charts served to confirm oligo-ovulation as needed. In addition, patients had hirsutism, as defined by a modified Ferriman-Gallwey score above 7 [13], acne, alopecia and/or increased circulating total or free testosterone, androstenedione or dehydroepiandrosterone-sulfate (DHEAS) concentrations. Finally, hyperprolactinaemia, androgen secreting tumours, and congenital adrenal hyperplasia were ruled out in all the patients.
Fasting glucose concentrations were below 6.11 mmol/l and BP was below 140/90 mm Hg, in all the participants with the exception of two PCOS patients who had undiagnosed and untreated hypertension. All the subjects were Caucasian and had not taken hormonal medications, including contraceptive pills, for the last six months. Data from some of the patients and control subjects regarding the molecular genetic mechanisms leading to hyperandrogenism have been published previously [14, 15, 16].
The ethics committee of the Hospital Ramón y Cajal approved the study, and informed consent was obtained from all participants.
Protocol
Clinical and anthropometric variables, including hirsutism score, BMI, WHR and BP were evaluated by a single investigator in all the subjects.
Samples were obtained between days 5 and 10 of the menstrual cycle, or during amenorrhoea after excluding pregnancy. After a 3-day 300 g carbohydrate diet and 12-h overnight fasting, samples were obtained for the measurement of serum CRP, IL-6, TNF-α, sTNFR2, sICAM-1, total testosterone, androstenedione, DHEAS, luteinizing hormone, follicle-stimulating hormone, estradiol, sex hormone-binding globulin (SHBG), calculated free testosterone [17], total cholesterol, low-density lipoprotein cholesterol (LDL), high-density lipoprotein cholesterol (HDL) and triglycerides. Then, a 75 g OGTT was carried out, and samples were obtained for measurement of serum insulin and plasma glucose at 0, 30, 60, 90 and 120 min. Samples were immediately centrifuged, and serum was separated and frozen at −20°C until assayed.
Serum CRP concentrations were measured by a high sensitivity immunochemilumenescent assay (Immulite High Sensitivity CRP, Diagnostic Products Corporation, Los Angeles, Calif., USA) with lower limit of detection of 0.01 mg/dl, and intra- and inter-assay coefficients of variation below 10%. Serum TNF-α was assayed by immunochemilumenescent assay (Immulite TNF-α, Diagnostic Products Corporation, Los Angeles, Calif., USA) with a lower limit of detection of 1.7 pg/ml, and intra- and inter-assay coefficients of variation below 7%. Serum CRP and TNF-α measurements were carried out within single assays to avoid interassay variation. Serum IL-6 concentrations were measured by ELISA (Cytoscreen Human Interleukin-6 UltraSensitive Immunoasssay Kit, Biosource International, Camarillo, Calif., USA) with a lower limit of detection of 0.104 pg/ml, and mean intra- and inter-assay coefficients of variation of 6.4% and 7.8% respectively. Serum sTNFR2 concentrations were assayed by ELISA (Quantikine Human sTNF RII, R & D Systems, Minneapolis, Minn., USA) with a lower limit of detection of less than 1 pg/ml, and mean intra- and inter-assay coefficients of variation of 2.2% and 4.1% respectively. Serum sICAM-1 concentrations were measured by ELISA (Human sICAM-1 Kit, Biosource International, Camarillo, Calif., USA) with a lower limit of detection of less than 0.04 ng/ml, and mean intra- and inter-assay coefficients of variation of 5.2% and 7.8% respectively. The technical characteristics of the assays used for plasma glucose and serum hormone measurements have been described elsewhere [14, 15, 16]. The composite insulin sensitivity index (ISI) was calculated from the circulating glucose and insulin concentrations during the OGTT according to a known method [18].
Statistical Analysis
Data are represented as means ± SD unless otherwise stated. Data were submitted to a full-factorial multivariate general linear model (GLM) to reduce the probability of a type 1 error (i.e. the probability of finding differences after repeating multiple tests simply by chance), considering the large number of variables studied here [19].
Dependent variables included hirsutism score, WHR, mean blood pressure, ISI, circulating serum inflammatory cardiovascular risk markers, serum glucose and lipid concentrations, and hormone concentrations. Independent variables were obesity, and PCOS (representing a higher degree of insulin resistance). The interaction of both independent variables was also analysed. As the controls were older compared with the PCOS patients, age was introduced as a covariate. The GLM also calculated the univariate analysis for all the dependent variables.
All the dependent variables were tested for normality using the Kolmogorov-Smirnov statistic, with a Lilliefors significance level for testing normality. Logarithmic or square root transformations were applied as needed to ensure normal distribution of the variables.
Correlation analysis and partial correlation analysis were also used as described below. Statistical analyses were done using SPSS 10 for the Macintosh (SPSS, Chicago, Ill., USA). A p value less than 0.05 was considered statitically significant.
Results
Effect of PCOS and obesity on serum markers of inflammation, and on clinical and biochemical variables
Obesity, as defined by a BMI greater than or equal to 27 kg/m2, was observed in 22 patients (62.9%) and in 17 control subjects (60.7%). Ten control subjects (36%) and 13 of the PCOS patients (37%) were smokers.
The multivariate GLM analysis detected differences in the dependent variables between PCOS patients and controls (Wilks' λ=0.282, F=6.852, p<0.0001) and between obese and lean women (Wilks' λ=0.422, F=3.682, p<0.0005). However, there was no interaction between PCOS and obesity (Wilks' λ=0.783, F=0.745, p=0.734), indicating that the influence of PCOS on the variables studied here was not different in lean women compared with obese women, and that the influence of obesity on the variables studied here was the same in the PCOS and control groups.
Due to the inclusion of age as a covariate, all analyses were corrected for the difference of age between PCOS patients and healthy controls. Specifically, age influenced the WHR (p<0.001), and circulating androstenedione (p<0.01), DHEAS (p<0.001), cholesterol (p<0.001), LDL (p<0.005), mean blood pressure (p<0.001) and serum glucose concentrations (p<0.05).
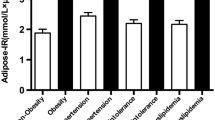
The univariate analyses regarding serum inflammatory cardiovascular risk markers were calculated in lean PCOS patients and controls, and in obese PCOS women and controls, separately (Fig. 1). However, because there was no interaction between PCOS and obesity in the GLM, pooled analysis of PCOS patients compared with control women (Table 1), and pooled analysis of obese subjects compared with lean women (Table 2) were also performed.

Influences of PCOS and obesity on the insulin sensitivity index and on serum concentrations of inflammatory cardiovascular risk markers. The multivariate GLM showed differences between PCOS patients and controls (p<0.0001) and between obese and lean women (p<0.0005). There was no interaction between PCOS status and obesity (p=0.734). Comparisons were corrected for age, because age was introduced as a covariate in the model. The box-plot includes the median (horizontal line) and the interquartile range (box), and the whiskers indicate the 5th and 95th percentiles. The figures below the x-axis indicate the number of subjects in each subgroup. * p<0.01 for the difference between obese and lean women. † p<0.01 for the difference between PCOS patients and controls
Differences between PCOS patients and controls
The ISI was reduced, and fasting and post-load serum insulin was increased, in PCOS patients compared with controls. These results confirm that PCOS can be considered a human model of insulin resistance, both in lean and obese pre-menopausal women. Therefore, in our experimental model, having PCOS is equivalent to being insulin resistant independently of the degree of obesity. The WHR showed a small but highly significant (p<0.01) increase in PCOS patients, and this increase in WHR may be related to the insulin resistance of these women.
Despite the reduced insulin sensitivity in PCOS patients, no differences were observed in this group in circulating CRP, IL-6, sICAM-1, TNF-α, and sTNFR2 compared with healthy women.
PCOS patients had increased hirsutism scores, mean blood pressure, total testosterone, free testosterone, and androstenedione concentrations, whereas SHBG and estradiol concentrations were reduced. No differences were observed between PCOS patients and controls in DHEAS, luteinizing hormone and follicle-stimulating hormone concentrations, and in serum lipid concentrations (Table 1, Fig. 1).
Differences between obese and lean women
The effect of obesity on the individual variables was also evaluated by the GLM analysis. The BMI was 36.5±6.5 kg/m2 in the obese subgroup (n=39), and 22.7±2.9 kg/m2 in the lean subgroup (n=24). Obese women were older than lean subjects (29.5±8.6 yr vs. 25.3±7.2, p=0.045) but the comparisons between the obese and lean groups were corrected for the difference in age because of the inclusion of age as a covariate in the GLM.
The ISI was reduced, and fasting glucose and insulin concentrations were higher, in obese women compared with lean women. Circulating CRP and IL-6 concentrations were higher, and sTNFR2 concentrations tended (p<0.1) to be increased, in obese women. On the contrary, no differences were observed between obese and lean women in serum TNF-α and sICAM-1 concentrations (Table 2, Fig. 1).
Obese women had increased free testosterone concentrations, whereas hirsutism scores, and SHBG and HDL concentrations were reduced, compared with lean women. No differences were observed in WHR, mean blood pressure, total testosterone, androstenedione, DHEAS, luteinizing hormone, follicle-stimulating hormone and estradiol concentrations, cholesterol, LDL and triglycerides.
A previous study by others suggested that smoking might influence the relationships between CRP, IL-6 and insulin sensitivity [21]. However, in our series, the differences described above, between PCOS and controls and between obese and lean women, remained essentially unchanged after introducing smoking as a covariate in the GLM (data not shown).
Inclusion of serum testosterone and estradiol concentrations as covariates
Besides insulin resistance and age, PCOS patients and controls had different serum androgen and estradiol concentrations. When repeating the GLM including serum markers of inflammations as dependent variables, PCOS and obesity as independent variables, and age, serum total testosterone and estradiol concentrations as covariates, no differences in serum markers of inflammation were observed between PCOS patients and controls, whereas CRP and IL-6 concentrations were increased in obese women compared with lean women (data not shown). Therefore, the different androgen and estradiol concentrations between PCOS patients and controls did not influence the results.
Relationships between ISI, BMI, and serum inflammatory markers
As there were no differences in serum markers of inflammation between PCOS patients and control women, both groups were considered as a whole for these analyses. Logarithmic transformations ensured normal distribution of the variables.
The log[ISI] showed inverse correlations with log[CRP], log[IL-6], log[sTNFR2], log[sICAM-1] and TNF-α (Fig. 2). However, the log[ISI] also correlated inversely with BMI (r=−0.55, p<0.0001), and the BMI showed stronger direct correlations with log[CRP] (r=0.75, p<0.0001), log[IL-6] (r=0.47, p<0.0001), log[sTNFR2] (r=0.48, p<0.0001) and log[sICAM-1] (r=0.27, p<0.05).

Correlations between the insulin sensitivity index and serum markers of inflammation. Logarithmic transformation was applied before analysis to ensure normality of the variables as needed. Corr = correlation analysis, PCorr = Partial correlation analysis controlling for BMI. * p<0.05, † p<0.01, ‡ p<0.001
Therefore, we suspected that the inverse correlations between the log[ISI] and the markers of inflammation were spurious, resulting from the stronger correlation of these markers with BMI, which also correlated with the log[ISI]. When submitted to partial correlation analysis controlling for BMI, most of the inverse correlations between the log[ISI] and serum markers of inflammation did not maintain statistical significance and only the weak inverse correlation with log[CRP] concentrations retained significance (Fig. 2).
Conversely, and in agreement with the major influence of BMI on serum markers of inflammation, after controlling for log[ISI] by partial correlation analysis, the correlations of BMI with log[CRP] (r=0.63, p<0.001), log[IL-6] (r=0.34, p<0.01) and log[sTNFR2] (r=0.36, p<0.005) persisted. These results further support that body fat, but not insulin resistance, is the major determinant of the serum concentrations of inflammatory cardiovascular risk markers in pre-menopausal women.
Finally, we studied the correlation between log[CRP] and WHR, to evaluate whether or not visceral adiposity contributed to the increase in CRP concentrations observed in obese women. log[CRP] correlated with the WHR (r=0.393, p=0.001), but this correlation weakened after controlling for the influence of BMI on both variables (r=0.249, p=0.051), further supporting the dominant influence of the degree of adiposity, represented by the BMI, on serum CRP concentrations.
Discussion
Insulin resistance and serum markers of inflammation, such as cytokines and adhesion molecules, are increasingly being considered as predictors of cardiovascular disease [22]. Atherogenesis is associated with chronic local inflammation, and the inflammatory process could result in the release of cytokines into the circulation such as TNF-α or IL-6, and of adhesion molecules such as ICAM-1 [10, 22, 23]. Moreover, the low-grade chronic inflammatory process associated with atherogenesis might be related to relatively small increases in CRP, only measurable using high sensitivity assays [10, 22, 23]. Increased CRP concentrations are characteristic of obesity [10, 22, 23], suggesting the existence of low-grade chronic inflammation also in this condition.
Recently, low-grade inflammation has been proposed as an integral part of insulin resistance, based on the results of a large elderly population of non-diabetic people [5]. However, in this study the associations between serum CRP, IL-6, sICAM-1 and post-load insulin weakened after adjustment for measures of obesity, smoking and cardiovascular disease [5].
Considering that obesity is intimately related to insulin resistance, and that some serum markers of inflammation are secreted at least in part by adipose tissue [8, 24, 25], or influence adipose tissue physiology [26], the possibility exists that instead of being related to insulin resistance, the concentrations of serum markers of inflammation actually depend on obesity [27, 28]. This hypothesis is fully supported by our results, at least for pre-menopausal women.
Due to the homogeneity of the population studied here in terms of race, ethnicity, gender, BMI and smoking habits, our experimental design was adequate to delimitate the role of obesity and insulin resistance on the increase in serum markers of inflammation. Also, all the women were young adults, had normal fasting glucose concentrations, and did not have any clinical evidence of atherosclerosis and cardiovascular disease. The use of a strict and versatile multivariate GLM analysis allowed us to delineate the relative contribution of obesity, PCOS (representing a higher degree of insulin resistance), and the interaction of both factors, to the concentrations of serum markers of inflammation. Of note, multivariate GLM analysis adjust for the possible confounding influence of obesity on the differences between PCOS and controls, and for the possible confounding influence of PCOS on the differences between lean and obese women. In other words, the lack of interaction between PCOS and obesity indicates that the influence of PCOS on the variables studied here was not different in lean and in obese women, and that the influence of obesity on the variables studied here was the same in the PCOS and control groups. Moreover, the use of a multivariate GLM avoids type 1 errors and also permits the inclusion of age as a covariate, correcting the analysis for the difference in age between PCOS patients and control subjects, and for the difference in age between the obese and lean groups.
Although in our series PCOS patients had decreased insulin sensitivity, no differences were observed between PCOS patients and controls in any of the serum markers of inflammation studied here, whereas obesity increased serum CRP and IL-6 concentrations, and tended to increase sTNFR2 concentrations. As stated above, the influence of obesity on these markers was not different in PCOS patients and in controls suggesting that this influence was independent on the degree of insulin resistance. Moreover, these results were confirmed when repeating the GLM including serum testosterone and estradiol concentrations, and smoking, as covariates, ruling out any important influence of these factors on our results.
Our finding of increased CRP and IL-6 concentrations in obese subjects is not surprising considering that adipose tissue IL-6 concentrations correlate with serum CRP concentrations [29], and that the synthesis of CRP by the liver is largely regulated by IL-6 [30]. Also, IL-6 can be considered as an endocrine cytokine [31], in contrast to other cytokines such as TNF-α that exert their metabolic influences predominantly through autocrine and paracrine mechanisms [32]. The latter could explain the absence of differences in serum TNF-α and s-ICAM concentrations between obese and lean subjects in our series, whereas the tendency towards increased sTNFR2 concentrations in obese women might indicate increased shedding of TNFR2 after increased TNF-α local activity [7].
The correlation and partial correlation analyses, although partly limited by the strong correlation between BMI and ISI, also support our conclusion that serum markers of inflammation and adhesion molecules are mostly related to obesity, and cast doubt upon their usefulness as surrogate markers of insulin resistance in pre-menopausal women. After controlling for the influence of BMI, only CRP maintained the inverse correlation with the ISI, but this correlation was too weak (r=−0.29) to represent any biologically plausible relationship [19].
In agreement with our results, a previous study has shown that serum IL-6 concentrations do not correlate with indexes of insulin resistance in women [21]. However, the same study suggests that the relationships between CRP, IL-6 and insulin resistance are affected by sex, because such relationships are observed in men [21]. Due to the use of PCOS as a model of insulin resistance, our results only apply to pre-menopausal women, precluding any extrapolation to other populations, such as men or postmenopausal women.
Moreover, our results do not argue against a role for inflammatory cytokines in the pathogenesis of insulin resistance, but rather suggest that serum markers of inflammation do not reflect accurately the local tissue mechanisms mediating the induction of insulin resistance by inflammatory cytokines. Moreover, we only measured these serum markers at one time-point, and therefore we cannot rule out that integrated cytokine concentrations measured over time might yield different results. There is substantial evidence indicating that inflammatory cytokines act through paracrine and autocrine mechanisms to induce insulin resistance [33, 34]. Nevertheless, there are conflicting reports on the usefulness of their circulating concentrations to detect this process, and several studies have failed to identify relationships between serum markers of inflammation and measures of insulin resistance [35], especially when the correlations were controlled for the influence of obesity [36, 37, 38].
Among the serum markers of inflammation studied here, only CRP showed a weak correlation with insulin resistance in our series, in agreement with a very recent report in Pima Indians [39]. Therefore, CRP might translate to some extent the chronic local inflammatory process associated with insulin resistance, by reflecting the action of inflammatory mediators in the liver.
However, our experimental design had some limitations. Possibly the most important was the relatively small sample size and the use of a strict multivariate GLM analysis. The latter decreased the probability of type 1 errors, yet at the expense of decreasing the statistical power of our experimental design. Therefore we cannot exclude completely that the non-significant differences in serum inflammatory markers between PCOS and controls might reach statistical significance if the sample size was increased. This consideration is especially important regarding CRP concentrations, which was the inflammatory marker showing the larger increase in PCOS patients compared with controls, and it is possible that this increase might have reached statistical significance if a larger series was studied. Nevertheless, the non-significant increases of CRP, TNF-α and sICAM-1 in PCOS patients were small, and the mean values of IL-6 and sTNFR2 even tended to be lower in PCOS controls compared with normal controls. Even assuming that these differences would reach statistical significance by increasing sample size, we could conclude that not all serum inflammatory markers were increased in insulin-resistant PCOS subjects. Secondly, the OGTT-based method to assess insulin sensitivity used here is relatively inaccurate compared to euglycaemic clamp techniques, and we cannot exclude the possibility that the use of clamp techniques would uncover a stronger relation between insulin resistance and inflammatory markers. Other surrogate markers of insulin resistance such as the increase in WHR and in fasting and post-load insulin concentrations also supported the existence of insulin resistance in both lean and obese PCOS patients, compared with their non-hyperandrogenic counterparts. Therefore, we do not believe that our overall conclusions are invalidated by the use of OGTT to evaluate insulin sensitivity. Finally, there is the possibility that the mechanisms underlying insulin resistance in PCOS are different than those involved in the insulin resistance associated with obesity, and obesity-related insulin resistance might trigger inflammatory cytokine production while that associated with PCOS does not. Nevertheless, to our knowledge there is no experimental data to support this hypothesis.
Focusing on the group of PCOS patients, our results are largely confirmatory with regards to the presence of hyperandrogenism, reduction in SHBG concentrations, and increased insulin resistance compared with non-hyperandrogenic women. On the contrary, we have not found the increased CRP concentrations recently reported in a smaller series of PCOS patients and healthy controls from Scotland [40]. Nevertheless, we have shown that hyperandrogenism is influenced by certain TNF-α, TNFR2 and IL-6 genotypes [14, 15, 16], supporting that low grade chronic inflammation might influence the pathogenesis of PCOS. However, our results suggest that serum markers of inflammation do not reflect accurately the local tissue mechanisms mediating this process.
In conclusion, the circulating concentrations of serum inflammatory cardiovascular risk markers in pre-menopausal women are determined mainly by obesity and, for a similar BMI, do not change as a function of insulin resistance.
Abbreviations
- CRP:
-
C-reactive protein
- GLM:
-
multivariate general linear model
- ISI:
-
insulin sensitivity index
- PCOS:
-
polycystic ovary syndrome
- sICAM-1:
-
soluble intercellular adhesion molecule-1
- sTNFR2:
-
soluble type 2 TNF receptor
References
Reaven GM (1988) Role of insulin resistance in human disease. Diabetes 37:1595–1607
Frishman WH (1998) Biologic markers as predictors of cardiovascular disease. Am J Med 104:18S–27S
Zinman B, Hanley AJ, Harris SB, Kwan J, Fantus IG (1999) Circulating tumor necrosis factor-alpha concentrations in a native Canadian population with high rates of type 2 diabetes mellitus. J Clin Endocrinol Metab 84:272–278
Fernandez-Real JM, Broch M, Ricart W et al. (1998) Plasma levels of the soluble fraction of tumor necrosis factor receptor 2 and insulin resistance. Diabetes 47:1757–1762
Hak AE, Pols HA, Stehouwer CD et al. (2001) Markers of inflammation and cellular adhesion molecules in relation to insulin resistance in nondiabetic elderly: the Rotterdam study. J Clin Endocrinol Metab 86:4398–4405
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415
Hotamisligil GS, Arner P, Atkinson RL, Spiegelman BM (1997) Differential regulation of the p80 tumor necrosis factor receptor in human obesity and insulin resistance. Diabetes 46:451–455
Bastard JP, Jardel C, Bruckert E et al. (2000) Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab 85:3338–3342
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271:665–668
Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V (2000) Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 148:209–214
Zawadzki JK, Dunaif A (1992) Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine FP, Merriam GR (eds) Polycystic ovary syndrome. Blackwell, Boston, pp 377–384
Dunaif A (1997) Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 18:774–800
Hatch R, Rosenfield RL, Kim MH, Tredway D (1981) Hirsutism: implications, etiology, and management. Am J Obstet Gynecol 140:815–830
Escobar-Morreale HF, Calvo RM, Sancho J, San Millán JL (2001) TNF-α and hyperandrogenism: a clinical, biochemical and molecular genetic study. J Clin Endocrinol Metab 86:3761–3767
Villuendas G, San Millan JL, Sancho J, Escobar-Morreale HF (2002) The -597 GA and -174 GC polymorphisms in the promoter of the interleukin 6 gene (IL6) are associated with hyperandrogenism. J Clin Endocrinol Metab 87:1134–1141
Peral B, San Millan JL, Castello R, Moghetti P, Escobar-Morreale HF (2002) The methionine 196 arginine polymorphism in exon 6 of the TNF receptor 2 gene (TNFRSF1B) is associated with the polycystic ovary syndrome and hyperandrogenism. J Clin Endocrinol Metab 87:3977–3983
Vermeulen A, Verdonck L, Kaufman JM (1999) A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab 84:3666–3672
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22:1462–1470
Dawson B, Trapp RG (2001) Basic & clinical biostatistics, 3rd edn. Lange Medical Books/McGraw-Hill, New York
Legro RS, Kunselman AR, Dunaif A (2001) Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med 111:607–613
Fernandez-Real JM, Vayreda M, Richart C et al. (2001) Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J Clin Endocrinol Metab 86:1154–1159
Van Lente F (2000) Markers of inflammation as predictors in cardiovascular disease. Clin Chim Acta 293:31–52
Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW (1999) C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19:972–978
Fried SK, Bunkin DA, Greenberg AS (1998) Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83:847–850
Mohamed-Ali V, Goodrick S, Bulmer K, Holly JM, Yudkin JS, Coppack SW (1999) Production of soluble tumor necrosis factor receptors by human subcutaneous adipose tissue in vivo. Am J Physiol 277:E971–975
Dong ZM, Gutierrez-Ramos JC, Coxon A, Mayadas TN, Wagner DD (1997) A new class of obesity genes encodes leukocyte adhesion receptors. Proc Natl Acad Sci USA 94:7526–7530
Festa A, D'Agostino R Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM (2000) Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 102:42–47
Festa A, D'Agostino R Jr, Williams K et al. (2001) The relation of body fat mass and distribution to markers of chronic inflammation. Int J Obes Relat Metab Disord 25:1407–1415
Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F (1999) Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation 99:2221–2222
Heinrich PC, Castell JV, Andus T (1990) Interleukin-6 and the acute phase response. Biochem J 265:621–636
Papanicolaou DA, Vgontzas AN (2000) Interleukin-6: the endocrine cytokine. J Clin Endocrinol Metab 85:1331–1333
Lofgren P, Harmelen V van, Reynisdottir S et al. (2000) Secretion of tumor necrosis factor-alpha shows a strong relationship to insulin-stimulated glucose transport in human adipose tissue. Diabetes 49:688–692
Moller DE (2000) Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11:212–217
Dandona P, Aljada A, Mohanty P (2002) The anti-inflammatory and potential anti-atherogenic effect of insulin: a new paradigm. Diabetologia 45:924–930
Ronnemaa T, Pulkki K, Kaprio J (2000) Serum soluble tumor necrosis factor-alpha receptor 2 is elevated in obesity but is not related to insulin sensitivity: a study in identical twins discordant for obesity. J Clin Endocrinol Metab 85:2728–2732
Targher G, Bonadonna RC, Alberiche M, Zenere MB, Muggeo M, Bonora E (2001) Relation between soluble adhesion molecules and insulin sensitivity in type 2 diabetic individuals: role of adipose tissue. Diabetes Care 24:1961–1966
Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE (2001) Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res 9:414–417
Forouhi NG, Sattar N, McKeigue PM (2001) Relation of C-reactive protein to body fat distribution and features of the metabolic syndrome in Europeans and South Asians. Int J Obes Relat Metab Disord 25:1327–1331
Weyer C, Yudkin JS, Stehouwer CD, Schalkwijk CG, Pratley RE, Tataranni PA (2002) Humoral markers of inflammation and endothelial dysfunction in relation to adiposity and in vivo insulin action in Pima Indians. Atherosclerosis 161:233–242
Kelly CCJ, Lyall H, Petrie JR, Gould GW, Connell JMC, Sattar N (2001) Low grade chronic inflammation in women with polycystic ovarian syndrome. J Clin Endocrinol Metab 86:2453–2455
Acknowledgements
The authors would like to thank Ms. G. González for excellent technical help, and V. Abraira, PhD, Biostatistics Unit, Hospital Ramón y Cajal, for his help with statistical analysis.
This study was supported by Grants 08.6/0022/1998 and 08.6/0024.2/2000 from the Consejería de Educación, Comunidad de Madrid, and by Grant FIS 00/0414 from the Fondo de Investigación Sanitaria, Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Spain.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part at the 84th Endocrine Society's Annual Meeting, 19–23 June 2002, San Francisco, California, and at the 18th Annual Meeting of the European Society for Human Reproduction and Embryology, 30 June–3 July 2002, Vienna, Austria
Rights and permissions
About this article
Cite this article
Escobar-Morreale, H.F., Villuendas, G., Botella-Carretero, J.I. et al. Obesity, and not insulin resistance, is the major determinant of serum inflammatory cardiovascular risk markers in pre-menopausal women. Diabetologia 46, 625–633 (2003). https://doi.org/10.1007/s00125-003-1090-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1090-z