
Abstract
Aims/hypothesis
Kallikrein-binding protein (KBP) is a serine proteinase inhibitor (serpin). It specifically binds to tissue kallikrein and inhibits kallikrein activity. Our study was designed to test its effects on retinal neovascularization and vascular permeability.
Methods
Endothelial cell proliferation was determined by [3H] thymidine incorporation assay and apoptosis quantified by Annexin V staining and flow cytometry. Effect on retinal neovascularization was determined by fluorescein angiography and count of pre-retinal vascular cells in an oxygen-induced retinopathy (OIR) model. Vascular permeability was assayed by the Evans blue method. Vascular endothelial growth factor (VEGF) was measured by Western blot analysis and ELISA.
Results
Kallikrein-binding protein specifically inhibited proliferation and induced apoptosis in retinal capillary endothelial cells. Intravitreal injection of KBP inhibited retinal neovascularization in an OIR model. Moreover, KBP decreased vascular leakage in the retina, iris and choroid in rats with OIR. Blockade of kinin receptors by specific antagonists showed significantly weaker inhibition of endothelial cells, when compared to that of KBP, suggesting that the anti-angiogenic activity of KBP is not through inhibiting kallikrein activity or kinin production. KBP competed with 125I-VEGF for binding to endothelial cells and down-regulated VEGF production in endothelial cells and in the retina of the OIR rat model.
Conclusion/interpretation
Kallikrein-binding protein is a multi-functional serpin, and its vascular activities are independent of its interactions with the kallikrein-kinin system. Inhibition of VEGF binding to its receptors and down-regulation of VEGF expression could represent a mechanism for the vascular activities of KBP.
Similar content being viewed by others


Angiogenesis in the retina is controlled by a delicate balance between angiogenic stimulators (e.g., vascular endothelial growth factor, VEGF) and angiogenic inhibitors (e.g., pigment epithelium-derived factor, PEDF) [1, 2, 3]. Under certain pathological conditions such as diabetic retinopathy and retinopathy of prematurity (ROP), the retinal cells increase the production of angiogenic stimulators while decreasing angiogenic inhibitors in response to local hypoxia [4, 5]. These changes break the balance in angiogenesis control and consequently, resulting in over-proliferation of capillary endothelial cells and retinal neovascularization which is a common cause of blindness [2, 3, 6]. The molecular mechanism leading to retinal neovascularization is presently not known.
The retina and vitreous fluid contain endogenous angiogenic inhibitors [7, 8, 9, 10, 11]. PEDF, a serine proteinase inhibitor (serpin), has been identified as a potent angiogenic inhibitor endogenously expressed in the retina [12]. Angiostatin has also been identified in human vitreous fluid [13]. Decreased concentrations of angiostatin and PEDF have been shown to correlate with the development of proliferative diabetic retinopathy [13, 14].
The tissue kallikrein-kinin system consists of tissue kallikrein, kallikrein-binding protein (KBP), kinins, kininogens (precursors of kinins), kininases and bradykinin receptors [15]. Tissue kallikrein is a serine proteinase which cleaves kininogens to release vasoactive kinins. Kinins interact with bradykinin receptors on the cell surface and exert a variety of biological effects. It is known that most functions of kinins such as vasodilation, regulation of local blood flow and tissue metabolic rate, production of pain and inflammatory responses, are mediated by the B2 kinin receptor [15, 16]. Kinins also have a direct mitogenic effect on endothelial cells [15, 16]. It has been shown that the angiogenic activity of kinins is mediated by the B1 kinin receptor [17, 18].
KBP was originally identified from rat serum as it binds to tissue kallikrein, forming a SDS-stable complex [19, 20]. It inhibits kallikrein activity in transgenic mouse over-expressing kallikrein. KBP has been shown to have vascular function independent of its interactions with the kallikrein-kinin system [21, 22]. KBP is predominantly produced in the liver. It has been identified in a number of other tissues including the retina and vitreous [23]. KBP shares a considerable sequence homology with other serpins such as α1-antitrypsin and α1-antichymotrypsin, suggesting that it belongs to the serpin super family [24].
The serpin super family consists of multiple proteins with widely diverse functions [25]. Some of the serpin members, such as PEDF, antithrombin and maspin, have been shown to have anti-angiogenic activity [12, 26, 27]. Previous evidence indicates that KBP is involved in blood pressure regulation, inflammatory response and animal growth [28, 29, 30]. In ocular tissues, KBP concentrations were reduced in the retina of rats with streptozotocin (STZ)-induced diabetes and in vitreous from patients with proliferative diabetic retinopathy [23, 31]. These results suggest that KBP has certain functions independent of its interactions with the kallikrein-kinin system [32].
We determined the effect of KBP on angiogenesis and vascular leakage and possible mechanism of action using primary endothelial cells and a rat model of oxygen-induced retinopathy (OIR).
Subjects and Methods
Materials
The rat Müller cell line, rMC-1, was a generous gift from Dr. V. Sarthy at the Northwestern University. Retinal capillary endothelial cells (RCEC) and pericytes were isolated from bovine eyes following a protocol described previously [33, 34]. The identity of RCEC was confirmed by a characteristic cobblestone morphology and the incorporation of acetylated low-density lipoprotein labelled with a fluorescent probe, DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) (Biomedical Technologies, Stoughton, Mass., USA). Purity of the pericyte culture was determined by immunostaining using an FITC-conjugated antibody specific to α-smooth-muscle actin (Sigma, St. Louis, Mo., USA).
Brown Norway rats were purchased from Harlan (Indianapolis, Ind., USA). Care, use and treatment of all animals in this study were in strict agreement with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, as well as the guidelines set forth in the Care and Use of Laboratory Animals by the Medical University of South Carolina.
Expression and purification of recombinant KBP
The KBP cDNA containing a sequence coding for the full-length mature peptide was amplified from the total RNA of rat liver by reverse transcription-polymerase chain reaction (RT-PCR) as described previously [35]. The 5′ PCR primer (5′-GTCGGATCCTGATGGCATACTGGGAAG-3′) and the 3′ primer (5′-GTGGAGCTCATGGGGTTAGTGACTTTG-3′) contain a Bam HI and Sac I site, respectively. The PCR product was cloned into the pET28 vector (Novagen, Madison, Wis., USA) at the Bam HI and Sac I sites in frame with the sequence encoding the 6x His tag at its 3′ end.
The KBP/pET28 construct was introduced into E. coli strain BL-21/DE3 (Novagen, Madison, Wis., USA). The expression and purification were carried out as described previously [36]. Endotoxin concentrations were monitored using a limulus amebocyte kit (Biowhittaker, Walkersville, Md., USA).
Quantification of viable cells
Cells were plated in 12-well plates in triplicate and cultured in the growth medium until they reached 60 to 70% confluency. The culture medium was replaced with a medium containing 1% fetal bovine serum (FBS). Recombinant KBP was added to the cover medium to various concentrations and incubated with the cells for 72 h. The viable cells were quantified by the MTT assay (3-[4,5-dimethylthiazol-2-yl]-2,5-dephenyl tetrazolium bromid, Roche, Mannheim, Germany) following a protocol recommended by the manufacturer.
[3H] thymidine incorporation assay
RCEC were seeded in 24-well plates in 1:1 DMEM + F-12 nutrient mixture plus 10% FBS and cultured in a CO2 incubator to reach 60 to 70% confluency. The cells were washed three times with PBS and the growth medium replaced by a medium containing 1% FBS and different concentrations of KBP. After a 24-h culture, [3H] thymidine was added to the medium (2 µCi/well) and incubated with the cells at 37°C for 12 h. Free [3H] thymidine was removed by three washes with PBS, and a solution of 6% TCA was added to the wells. The TCA solution was removed, and the wells were washed once with PBS. The remaining material was solubilized with 200 µl of 1 mol/l NaOH [37]. Incorporated [3H] thymidine was measured with a microplate scintillation counter (Packard Instrument, Meriden, Conn.).
Quantitative analysis of apoptosis by flow cytometry
RCEC were plated at a density of 105 cells per well in 6-well plates. Two days after seeding, the cells were exposed to KBP at a different concentration for 24 h and harvested for Annexin and propidium iodide (PI) staining using the Annexin V-FITC Apoptosis Detection Kit (Sigma, St. Louis, Mo., USA) following the protocol recommended by the manufacturer. Colchicine (Sigma), which is known to induce apoptosis by disrupting microtubules and preventing its polymerization, was used as a positive control. The cells were subsequently counted by flow cytometry.
Induction of retinal neovascularization and intravitreal injection of KBP
Retinal neovascularization was induced as described in another study [38] with some modifications. Briefly, newborn pigmented Brown Norway rats at postnatal day 7 (P7) were exposed to hyperoxia (75% O2) for 5 days and then normoxia. Animals were anesthetized, and KBP was injected into the vitreous of the right eye through the pars plana using a glass capillary. The left eye received the same volume of PBS as the control. After injection, the animals were kept in normoxia for another 5 days for further analyses.
Retinal angiography with high molecular weight fluorescein and quantification of neovascularization
Retinal angiography was as described previously [38]. Briefly, rats were anesthetized and perfused with fluorescein via intra-ventricle injection of 50 mg/ml of high molecular weight (2×106) fluorescein isothiocyanate-dextran (Sigma). The animals were immediately killed and the eyes were enucleated and fixed in 4% paraformaldehyde for 10 min. The retina was dissected free of the lens and vitreous and incubated in 4% paraformaldehyde for 3 h. The retina was cut and flat-mounted on a gelatin-coated slide. The vasculature was then examined under a fluorescent microscope (Axioplan2 Imaging, Zeiss).
Retinal neovascularization was quantified by counting pre-retinal vascular cells as described previously [36]. The average number of pre-retinal vascular nuclei was compared to the PBS control group by a Student's t test.
Measurement of vascular permeability
Vascular permeability was quantified by measuring albumin leakage from blood vessels into the retina, iris and choroid using Evans blue following a documented protocol [39] with minor modifications. Evans blue dye (Sigma) was dissolved in normal saline (30 mg/ml), sonicated for 5 min and filtered through a 0.45-µm filter (Millipore, Bedford, Mass., USA). The rats were anesthetized, and Evans blue (30 mg/kg) was injected over 10 s through the femoral vein using a glass capillary under microscopic inspection. Evans blue non-covalently binds to plasma albumin in the blood stream [40]. Immediately after Evans blue infusion, the rats turned visibly blue, confirming their uptake and distribution of the dye. The rats were kept on a warm pad for 2 h to ensure the complete circulation of the dye. Then the chest cavity was opened, and the rats were perfused via the left ventricle with 1% paraformaldehyde in citrate buffer (pH=4.2) which was pre-warmed to 37°C to prevent vasoconstriction. The perfusion lasted 2 min under the physiological pressure of 120 mmHg to clear the dye from the vessel. Immediately after perfusion, the eyes were enucleated and the retina, iris and choroid were carefully dissected under an operating microscope. Evans blue dye was extracted by incubating each sample in 150 µl formamide for 18 h at 70°C. The extract was centrifuged (TL; Beckman) at 70 000 rpm (Rotor type: TLA 100.3) for 20 min at 4°C. Absorbance was measured using 100 µl of the supernatant at 620 nm. The concentration of Evans blue in the extracts was calculated from a standard curve of Evans blue in formamide and normalized by the total protein concentration in each sample. Results were expressed in micrograms of Evans blue per milligrams of total protein content.
VEGF binding assay
VEGF (PeproTech, Rocky Hill, N.J., USA) was labelled with 125I using the Chloromine T 125I Labelling Kit (ICN Pharmaceuticals, Inc. Costa Mesa, Calif., USA) following a protocol recommended by the manufacturer. For the binding assay, RCEC were seeded in 12-well plates and cultured until 80% confluency was reached. The culture medium was replaced with serum-free medium. 125I-VEGF was added to the medium, 2.5×105 CPM/well with and without different concentrations of KBP or recombinant plasminogen kringle 5 (K5) and incubated with the cells for 1 h. The medium was removed and cells washed three times with PBS. The cells were then lysed by adding 0.35 ml 10% SDS. The cell lysates were collected, and the 125I-VEGF bound to RCEC was quantified by a gamma counter.
Measurement of VEGF in the conditioned medium of RCEC by ELISA
RCEC were seeded in T75 flasks in endothelial cell growth medium and cultured in a CO2 incubator to reach 60 to 70% confluency. The cells were washed three times with PBS and the growth medium replaced by a serum-free medium containing bFGF (GIBCO-BRL, Gaithersburg, Md., USA). KBP was added to the medium to various concentrations and incubated with the cells for 24 h under normoxia or hypoxia (in a chamber that was perfused with a mixture of 95% N2 + 5% CO2). The conditioned medium was harvested for VEGF ELISA and the cells were used for Western blot analysis. The conditioned medium was centrifuged and the protein concentration in the supernatant was measured with BioRad protein assay. VEGF concentration was measured using a VEGF ELISA kit (R&D systems, Minneapolis, Minn., USA) and normalized by total protein concentration in the medium.
Western blot analysis
One hundred micrograms of total protein were used for Western blot analysis of VEGF using an ECL detection kit (Amersham International, Piscataway, N.J., USA) [5]. The same membrane was stripped and re-blotted with an antibody specific to β-actin. VEGF concentrations were normalized by β-actin.
Statistical analysis
The Student's t test was used in all statistical analyses. A p value of less than 0.05 was considered statistically significant.
Results
Expression and purification of KBP
KBP was expressed in E. coli and purified to apparent homogeneity with the His.Bind affinity column. The purified recombinant protein showed an apparent molecular weight of 45 000 Mr, matching the calculated molecular weight from the sequence (Fig. 1A). The molecular weight of the recombinant protein is different from the native KBP (60 000 Mr) due to the lack of glycosylation in E. coli [20]. The identity of the band was confirmed by Western blot analysis using an anti-His tag antibody (Fig. 1B). An average of 20 mg of purified KBP was obtained from 1 L of culture.

Expression and purification of recombinant KBP. (A) SDS-PAGE with Coomassie blue staining; (B) Western blot analysis with an antibody specific to the His-tag. Lane 1, crude cell extract before the IPTG induction; 2, crude extract after the IPTG induction and 3, affinity-purified KBP
Specific inhibition of endothelial cell proliferation by recombinant KBP
RCEC were treated with recombinant KBP at concentrations of 5,10, 20, 40, 80 and 160 nmol/l for 72 h. Viable cells were quantified by MTT assay. At a concentration as low as 5 nmol/l, KBP treatment resulted in fewer viable cells than the control cells (p<0.05, n=3). This effect appeared to be KBP concentration-dependent, with an apparent IC50 of 50 nmol/l (Fig. 2) which is similar to that of K5, a known angiogenic inhibitor [36]. At the same concentrations, KBP did not result in any inhibition of pericytes from the same origin as the RCEC or of the Müller cell line (p>0.05, n=4), suggesting that KBP inhibition is specific to endothelial cells (Fig. 2).

Effect of KBP on cell viability. Primary RCEC, pericytes and the Müller cell line were treated with recombinant KBP at concentrations as indicated for 72 h. The viable cells were quantified using the MTT assay. Values represent absorbance as percentages of respective controls (means ± SD, n=3), and * indicates the values statistically different from the control (p<0.05)
The effect of KBP on cell proliferation rate was measured by [3H]-thymidine incorporation assay. As shown in Fig. 3, KBP inhibited thymidine incorporation in RCEC in a concentration-dependent manner from 5 to 160 nmol/l.

Inhibition of [3H] thymidine incorporation by KBP in endothelial cells. RCEC were treated with KBP, and the effect on proliferation rate was determined by [3H] thymidine incorporation assay. Bars represent [3H] incorporated into the chromosome (mean ± SD, n=4) and values statistically different from the control are indicated by * (p<0.05)
Induction of apoptosis by KBP
To determine whether KBP might induce cell death, RCEC were incubated with different concentrations of KBP or 10 µmol/l cholchicine for 24 h, and the apoptotic cells were quantified using the Annexin V-flow cytometry method. Phosphatidylserine externalizationis a characteristic of cells undergoing apoptosis. Annexin V-FITC kit allows for fluorescent detection of Annexin V bound to apoptotic cells and quantitative determination by flow cytometry. The Annexin V-FITC kit uses Annexin V conjugated with fluorescein isothiocyante (FITC) to label phosphatidylserine sites on the membrane surface. The kit includes propidium iodide (PI) to label the cellular DNA in necrotic cells where the cell membrane has been totally compromised. This combination allows the differentiation among early apoptotic cells (Annexin V positive, PI negative), necrotic cells (Annexin V positive, PI positive), and viable cells (Annexin V negative, PI negative) which can be located in the lower right, upper right, and lower left quadrants of the cytograms, respectively (Fig. 4A). As only cells that are Annexin V-positive and PI-negative are truly apoptotic cells, the percentage of this cell population was quantified. The results showed that KBP increases apoptosis in RCEC in a dose-dependent manner (Fig. 4).

Quantitative analysis of apoptosis induced by KBP in endothelial cells. RCEC were treated with different concentrations of KBP for 24 h and stained with Annexin V and PI. Apoptotic cells were quantified by flow cytometry. (A) cytograms from flow cytometric analysis. Intact cells, early apoptotic cells, and late apoptotic and necrotic cells are located in the lower left, lower right, and upper right quadrants of the cytograms, respectively. (B) percentages of early apoptotic cells (means ± SD, n=4). 1 control RCEC; 2 RCEC treated with colchicine as positive control; 3, 4, 5 and 6 RCEC treated with 40, 160, 320 and 640 nmol/l of KBP, respectively. Values significantly higher than control (p<0.05) are indicated by *
Anti-angiogenic effect of KBP in an OIR model
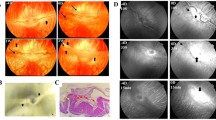
Retinal neovascularization was induced in Brown Norway rats by exposure of newborn rats to hyperoxia as described previously [5]. KBP was injected intravitreally at P12, and the control eyes received the same volume of PBS. Rats were kept under normoxia for another 5 days and retinal neovascularization examined by fluorescein angiography (P17). The control eyes (PBS-injected) developed typical retinal neovascularization, including neovascular tufts, microaneurysms, enlarged non-perfusion regions and vascular leakage (Fig. 5A). KBP injection showed an apparent improvement in retinal vasculature (Fig. 5B). KBP injection did not result in any apparent difference in retinal vasculature of normal rats (Fig. 5C, D).

Inhibition of ischaemia-induced retinal neovascularization by intravitreal injection of KBP. Retinal neovascularization was induced in newborn Brown Norway rats. Retinal vasculature was examined by angiography at 5 days after the injection of KBP or PBS (control). (A) retina from OIR rat after PBS injection; (B) retina from OIR rats after KBP injection; (C) retina from age-matched normal rats after PBS injection and (D) retina from normal rats after KBP injection. Each image is a representative from four animals of each group. (E) Pre-retinal vascular cells were counted on saggital sections from eight animals. Bars represent cell average numbers per section (means ± SD, n=8). The number in each KBP-treated group was compared with the control by Student's t test and * indicates the group with statistical difference from the control (p<0.05)
Quantification of pre-retinal neovascular cells showed that injection of 12.5 and 25 µg of KBP per eye both decreased pre-retinal vascular cells (p<0.01, n=8) (Fig. 5E). This result suggests that a single KBP injection inhibits retinal neovascularization under ischaemic conditions.
No apparent histological evidence of retinal toxicity was observed in any analysed retinal sections after the KBP injection, suggesting that KBP, at the concentrations used, does not cause any detectable toxicity to the retina or to the normal vasculature.
Effect of KBP on vascular permeability
At P14, two days after the rats were returned to normoxia, the OIR rats received an intravitreal injection of 3 µl of KBP of concentrations of 2.4, 4.8 and 9.6 mg/ml in the right eye (four animals per dose group), and the same volume of PBS in the left. Two days after the KBP injection (P16), vascular permeability was measured by the Evans blue leakage method. The rats exposed to hyperoxia showed increased vascular permeability in the retina, iris and choroid when compared to the age-matched normal rats (Fig. 6). KBP injection decreased vascular permeability in a dose-dependent manner in the retina, iris and choroid of the hyperoxia-treated rats (Fig. 6). At the high dose (9.6 mg/ml), KBP injection resulted in a decrease in permeability in all three tissues (p<0.01 in the retina and iris, and p<0.05 in the choroid, n=4). At the concentration of 4.8 mg/ml, KBP decreased vascular leakage in the retina and iris (p<0.05) but not in the choroid. At 2.4 mg/ml, KBP did not show any statistically significant effect in all three tissues (Fig. 6).

KBP Dose-dependent reduction of vascular leakage. Rats with OIR received an intravitreal injection of 3 µl of KBP at P14. Permeability was measured at P16. Evans blue-albumin leakage was normalized by total protein concentration and expressed as microgram of Evans blue per milligram total protein (means ± SD, n=4). (A) retina; (B) iris; (C) chorid. 1 age-matched normal rats injected with PBS; 2 OIR rats with PBS injection; 3, 4 and 5 OIR rats injected with 2.4, 4.8 and 9.6 mg/ml KBP, respectively. Values with statistical difference from the PBS-injected OIR control are indicated by *
Effects of B1 and B2 kinin receptor antagonists on RCEC
To test whether the anti-angiogenic activity of KBP is via reducing kinin production by inhibiting the kallikrein activity, RCEC were treated with 5 µmol/l des-Arg9-[Leu8]-bradykinin, a specific antagonist of the B1 kinin receptor or Hoe-140, a specific B2 kinin receptor antagonist in the presence or absence of 40 nmol/l KBP for 48 h, and viable cells were quantified by MTT assay. KBP treatment resulted in viable cell numbers of approximate 50% of the control (p<0.01, n=4), while the B1 antagonist treatment resulted in viable cells of 85% of the control (p<0.05, n=4) (Fig. 7). The B2 antagonist showed no statistically significant inhibition to RCEC at a high concentration (5 µmol/l) (p>0.05, n=4) (Fig. 7). The complete blockade of the B1 receptor showed weaker inhibition of RCEC, compared to KBP alone (p<0.01, n=4), suggesting that the KBP-induced inhibition of RCEC cannot be through reducing kinin production.

Effects of the B1 and B2 receptor antagonists on RCEC proliferation. RCEC were separately treated with KBP, the B1 receptor antagonist and B2 receptor antagonist for 48 h and viable cells quantified by MTT assay. Viable cell numbers are expressed as percentages of the control (means ± SD, n=4). 1 control cells treated with PBS; 2 40 nmol/l KBP alone; 3 5 µmol/l of the B1 antagonist and 4 5 µmol/l of the B2 antagonist
Inhibition of VEGF binding to RCEC by KBP
Incubation of 125I-VEGF with RCEC for 1 h resulted in binding of VEGF to RCEC. To determine the competition between KBP and VEGF in RCEC binding, 125I-VEGF was added to RCEC together with 0.5, 5 and 50 µg of unlabelled KBP to result in VEGF:KBP molar ratios of 1:5, 1:50 and 1:500, respectively. In the presence of excess amounts of KBP, VEGF bound to RCEC was decreased in a KBP concentration-dependent manner (Fig. 8). In contrast, K5 did not inhibit VEGF binding with RCEC in the same concentration range, suggesting different mechanisms of action between KBP and K5 (Fig. 8), although they both specifically inhibit endothelial cells.

Inhibition of VEGF binding to RCEC by KBP. 125I-VEGF was incubated with RCEC in the absence and presence of excess amounts of KBP or K5 as indicated. The binding of VEGF on RCEC was measured. Bars represent the bound VEGF (CPM) per well (means ± SD, n=3) and * indicates the values statistically different from the control (VEGF alone) (p<0.05)
Down-regulation of VEGF expression by KBP
As increased VEGF expression in the retina and vitreous play a key role in the development of retinal neovascularization, we have determined the effect of KBP on the expression of VEGF in cultured RCEC. VEGF secreted into the conditioned medium was measured by VEGF ELISA and normalized by total protein concentration in the medium. The result showed that KBP treatment resulted in reduced VEGF in the medium and the effect appeared to be dependent on the concentration of KBP (Fig. 9A). Western blot analysis showed that KBP also reduced VEGF concentrations in the cell lysate of RCEC in a concentration-dependent manner (Fig. 9B).

Down-regulation of VEGF expression by KBP in RCEC and in the retina. RCEC were treated with various concentrations of KBP under hypoxia for 24 h. The conditioned medium and cells were separately harvested for VEGF measurements. (A) KBP decreased VEGF concentrations in the conditioned medium. VEGF concentrations in the conditioned medium were measured by ELISA, normalized by total protein concentrations in the medium and expressed as picogram of VEGF per milligram of total protein (mean ± SD, n=4). 1 medium from normoxic culture; 2 medium from hypoxic culture; 3, 4, 5, 6 and 7 medium from hypoxic culture treated with 5, 10, 20, 40 and 80 nmol/l KBP, respectively. (B) KBP decreased cellular VEGF concentrations in RCEC. VEGF concentrations in cell lysates were measured by Western blot analysis, semi-quantified by densitometry and normalized by β-actin concentration. The relative VEGF concentrations were expressed as percentages of that in the control cultured under normoxia (mean ± SD, n=3). Lane 1 control cells under normoxia; 2 cells under hypoxia; 3, 4 and 5 cells treated with 40, 160 and 640 nmol/l KBP, respectively, under hypoxia. (C) Intravitreal injection of 25 µg KBP decreased retinal VEGF concentrations. Rats with retinal neovascularization were injected with KBP or the same volume of PBS (control). Retinal VEGF concentrations were measured by Western blot analysis, semi-quantified by densitometry, normalized by β-actin and expressed as percentages of the control (mean ± SD, n=3). 1 OIR retina with PBS injection and 2 OIR retina with KBP injection
The effect of KBP on VEGF expression was also examined in vivo. After the intravitreal injection of 25 µg KBP, VEGF concentrations were measured in the retina with OIR. Consistent with the results in cultured RCEC, KBP injection decreased retinal VEGF concentrations to approximate 35% of the control (p<0.01, n=3) (Fig. 9C), suggesting that the vascular activities of KBP in this animal model could be through down-regulation of VEGF expression in the retina.
Discussion
KBP is a member of the serpin super family that specifically binds to tissue kallikrein, forming a covalent complex [20]. We have shown that KBP inhibited the development of retinal neovascularization and decreased vascular leakage in the retina, iris and choroid in a rat model of OIR. Our results also showed that KBP blocks VEGF binding to its receptors and down-regulates VEGF expression, which could represent a mechanism responsible for its anti-angiogenic activity.
KBP is known to form a covalent complex with tissue kallikrein [20]. Delivery of the KBP gene into the transgenic mouse over-expressing kallikrein reverses the effect of kallikrein on blood pressure regulation, which provides in vivo evidence that KBP inhibits the activity of tissue kallikrein, and this inhibition could contribute to the regulation of vasodilation and local blood flow [29]. KBP is present in the retina and vitreous at high concentrations, suggesting that it could have physiological functions in the ocular tissues [23, 31]. The vitreous KBP concentrations were decreased in patients with proliferative diabetic retinopathy, suggesting its possible role in diabetic retinopathy [31]. Previous studies have shown that KBP is a potent angiogenic inhibitor [22]. Our results confirmed its anti-angiogenic activity in a retinal neovascularization model. Moreover, the present study has shown a new activity of this serpin, i.e. decreasing vascular permeability and vascular leakage.
As KBP can inhibit the releases of bioactive kinins from kininogen [41], and kinin promotes angiogenesis through the B1 receptor [17, 18], a natural question is whether the anti-angiogenic activity of KBP is through its inhibition of kallikrein activity and consequent reduction of kinin production. We used selective B1 and B2 kinin receptor antagonists to treat endothelial cells and compare their inhibitory effects with that of KBP alone. It has been shown previously that at 1 µmol/l, the B1 receptor antagonist des-Arg9-[Leu8]-bradykinin is able to completely block the bradykinin-induced endothelial cell proliferation [42]. Here, we used a high concentration (5 µmol/l) of des-Arg9-[Leu8]-bradykinin to ensure a complete blockade of the B1 receptor. The result showed that the inhibitory effect on RCEC by complete blockade of the B1 receptor was weaker than that of KBP alone (p<0.01), while blocking the B2 receptor had no inhibition. These results suggest that the anti-angiogenic activity of KBP cannot be ascribed to the inhibition of kinin production. This observation is consistent with previous findings [21, 22]. It is possible that KBP is a multi-functional protein which has several independent activities, i.e, binding with tissue kallikrein, inhibiting angiogenesis and decreasing vascular permeability. We hypothesize that these functions involve distinct structural domains in KBP. The multi-functional feature has also been documented in other serpins. Antithrombin III is known to inhibit thrombin and also has anti-angiogenic activity [26]. PEDF, a non-inhibitory serpin, possesses both neurotrophic and anti-angiogenic activities [12, 43].
In the past few years, a number of endogenous angiogenic inhibitors have been identified. Most of these inhibitors can be classified into two major groups: serpins including PEDF, maspin and anti-thrombin III [12, 26, 27], and peptide fragments of extracellular proteins including endostatin, angiostatin, K5 and tumstatin [44, 45, 46, 47, 48]. Recently, it has been shown that several fragments of extracellular proteins, e.g. angiostatin, endostatin and tumstatin bind to integrins, and their anti-angiogenic activities have been suggested to be through interfering with the integrin signalling [48, 49, 50]. However, the molecular mechanisms of the anti-angiogenic serpins are still unknown. Our results showed that KBP inhibits VEGF binding to its receptors on endothelial cells. Efficient binding of VEGF to its receptors is known to depend on heparin binding [51, 52]. As KBP is also a heparin-binding protein [20], the inhibition of VEGF binding to its receptors by KBP could be through competing on heparin binding. Our results also show that KBP down-regulates VEGF expression under hypoxia. The mechanism responsible for the KBP-mediated down-regulation of VEGF is presently not known. VEGF is a potent endothelial cell growth factor, and increased VEGF expression is a major cause of pathological angiogenesis and vascular leakage as found in diabetic retinopathy [53]. Inhibition of VEGF binding to its receptors and down-regulation of endogenous VEGF could represent a mechanism underlying the anti-angiogenic activity of KBP and its effect on vascular leakage.
Anti-angiogenic proteins or peptide fragments can offset increased angiogenic stimulators under hypoxia and thus, are believed to have therapeutic potential. Moreover, reduction of vascular leakage by KBP can be a beneficial effect in the treatment of macular oedema in diabetic retinopathy. KBP can be produced with a high yield in E. coli as a soluble protein with kallikrein-binding activity and inhibitory effects on angiogenesis and vascular leakage [54]. It is relatively stable and has low cytotoxicity to other cell types including pericytes and Müller cells. Intravitreal injection of KBP does not cause any detectable inflammatory response or toxicity to retinal tissues and normal vasculature. Moreover, KBP is endogenously expressed in multiple tissues including the retina and vitreous. These features suggest that KBP is a promising candidate for effective anti-angiogenic reagents in the treatment of neovascular disorders and vascular leakage such as proliferative diabetic retinopathy and solid tumor.
Abbreviations
- bFGF:
-
Basic fibroblast growth factor
- FBS:
-
fetal bovine serum
- IPTG:
-
isopropylthio-β-galactoside
- KBP:
-
kallikrein-binding protein
- OIR:
-
oxygen-induced retinopathy
- PEDF:
-
pigment epithelium-derived factor
- PI:
-
propidium iodide
- ROP:
-
retinopathy of prematurity
- RCEC:
-
retinal capillary endothelial cell
- STZ:
-
streptozotocin
- VEGF:
-
vascular endothelial growth factor
References
Jimenez B, Volpert OV (2001) Mechanistic insights on the inhibition of tumor angiogenesis. J Mol Med 78:663–672
Miller JW (1997) Vascular endothelial growth factor and ocular neovascularization. Am J Pathol 151:13–23
Bussolino F, Mantovani A, Persico G (1997) Molecular mechanisms of blood vessel formation. Trends Biochem Sci 22:251–256
Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE (1995) Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA 92:905–909
Gao GQ, Li Y, Zhang DC, Gee S, Crosson C, Ma J-X (2001) Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett 489:270–276
Blom ML, Green WR, Schachat AP (1994) Diabetic retinopathy: a review. Del Med J 66:379–388
Preis I, Langer R, Brem H, Folkman J (1977) Inhibition of neovascularization by an extract derived from vitreous. Am J Ophthalmol 84:323–328
Lutty GA, Thompson DC, Gallup JY, Mello RJ, Patz A, Fenselau A (1983) Vitreous: an inhibitor of retinal extract-induced neovascularization. Invest Ophthalmol Vis Sci 24:52–56
Lutty GA, Mello RJ, Chandler C, Fait C, Bennett A, Patz A (1985) Regulation of cell growth by vitreous humour. J Cell Sci 76:53–65
Jacobson B, Basu PK, Hasany SM (1984) Vascular endothelial cell growth inhibitor of normal and pathologic human vitreous. Arch Ophthalmol 102:1543–1545
Raymond L, Jacobson B (1982) Isolation and identification of stimulatory and inhibitory cell growth factors in bovine vitreous. Exp Eye Res 34:267–286
Dawson DW, Volpert OV, Gillis P et al. (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285:245–248
Spranger J, Hammes HP, Preissner KT, Schatz H, Pfeiffer AF (2000) Release of the angiogenesis inhibitor angiostatin in patients with proliferative diabetic retinopathy: association with retinal photocoagulation. Diabetologia 43:1404–1407
Spranger J, Osterhoff M, Reimann M et al. (2001) Loss of the antiangiogenic pigment epithelium-derived factor in patients with angiogenic eye disease. Diabetes 50:2641–2645
Bhoola KD, Figueroa CD, Worthy K (1992) Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 44:1-80
Schachter M (1983) Kallikreins and kinins, an overview: some thoughts old and new. Adv Exp Med Biol 156:13–27
Hu DE, Fan TP (1993) [Leu8]des-Arg9-bradykinin inhibits the angiogenic effect of bradykinin and interleukin-1 in rats. Br J Pharmacol 109:14–17
Emanueli C, Bonaria Salis M, Stacca T et al. (2002) Targeting kinin B(1) receptor for therapeutic neovascularization. Circulation 105:360–366
Chao J, Tillman DM, Wang MY, Margolius HS, Chao L (1986) Identification of a new tissue-kallikrein-binding protein. Biochem J 239:325–331
Chao J, Chai KX, Chen LM et al. (1990) Tissue kallikrein-binding protein is a serpin. I. Purification, characterization, and distribution in normotensive and spontaneously hypertensive rats. J Biol Chem 265:16394–16401
Chao J, Miao RQ, Chen V, Chen LM, Chao L (2001) Novel roles of kallistatin, a specific tissue kallikrein inhibitor, in vascular remodeling. Biol Chem 382:15–21
Miao RQ, Agata J, Chao L, Chao J (2002) Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood 100:3245–3252
Hatcher HC, Ma J-X, Chao J, Chao L, Ottlecz A (1997) Kallikrein-binding protein levels are reduced in the retinas of streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci 38:658–664
Chai KX, Ma J-X, Murray SR, Chao J, Chao L (1991) Molecular cloning and analysis of the rat kallikrein-binding protein gene. J Biol Chem 266:16029–16036
Silverman GA, Bird PI, Carrell RW et al. (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 276:33293–33296
O'Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J (1999) Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 285:1926–1928
Zhang M, Volpert O, Shi YH, Bouck N (2000) Maspin is an angiogenesis inhibitor. Nature Med 6:196–199
Yoon JB, Towle HC, Seelig S (1987) Growth hormone induces two mRNA species of the serine protease inhibitor gene family in rat liver. J Biol Chem 262:4284–4289
Ma J-X, Yang Z, Chao J, Chao L (1995) Intramuscular delivery of rat kallikrein-binding protein gene reverses hypotension in transgenic mice expressing human tissue kallikrein. J Biol Chem 270:451–455
Hatcher HC, Wright NM, Chao J, Chao L, Ma J-X (1999) Kallikrein-binding protein is induced by growth hormone in the dwarf rat. FASEB J 13:1839–1844
Ma J-X, King L, Yang Z, Crouch RK, Chao L, Chao J (1996) Quantitative comparison of kallistatin in non-diabetic and diabetic vitreous fluids. Curr Eye Res 15:1117–1123
Chen LM, Ma J-X, Liang YM, Chao L, Chao J (1996) Tissue kallikrein-binding protein reduces blood pressure in transgenic mice. J Biol Chem 271:27590–27594
Grant MB, Guay C (1991) Plasminogen activator production by human retinal endothelial cells of nondiabetic and diabetic origin. Invest Ophthalmol Vis Sci 32:53–64
Gitlin JD, D'Amore PA (1983) Culture of retinal capillary cells using selective growth media. Microvasc Res 26:74–80
Ma J-X, Zhang D, Laser M et al. (1999) Identification of RPE65 in transformed kidney cells. FEBS Lett 452:199–204
Zhang D, Kaufman PL, Gao G, Saunders RA, Ma J-X (2001) Intravitreal injection of plasminogen kringle 5, an endogenous angiogenic inhibitor, arrests retinal neovascularization in rats. Diabetologia 44:757–765
Smith LE, Shen W, Perruzzi C et al. (1999) Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat Med 5:1390–1395
Smith LE, Wesolowski E, McLellan A (1994) Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 35:101–111
Xu Q, Qaum T, Adamis AP (2001) Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest Ophthalmol Vis Sci 42:789–794
Radius RL, Anderson DR (1980) Distribution of albumin in the normal monkey eye as revealed by Evans blue fluorescence microscopy. Invest Ophthalmol Vis Sci 19:238–243
Zhou GX, Chao L, Chao J (1992) Kallistatin: a novel human tissue kallikrein inhibitor. Purification, characterization, and reactive center sequence. J Biol Chem 267:25873–25880
Morbidelli L, Parenti A, Giovannelli L, Granger HJ, Ledda F, Ziche M (1998) B1 receptor involvement in the effect of bradykinin on venular endothelial cell proliferation and potentiation of FGF-2 effects. Br J Pharmacol 124:1286–1292
Becerra SP, Sagasti A, Spinella P, Notario V (1995) Pigment epithelium-derived factor behaves like a noninhibitory serpin. Neurotrophic activity does not require the serpin reactive loop. J Biol Chem 270:25992–25999
O'Reilly MS, Boehm T, Shing Y (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88:277–285
O'Reilly MS, Holmgren L, Shing Y (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315–328
Cao Y, Chen A, An SSA (1997) Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J Biol Chem 272:22924–22928
Cao Y, Ji RW, Davidson D (1996) Kringle domains of human angiostatin. Characterization of the anti-proliferative activity on endothelial cells. J Biol Chem 271:29461–29467
Maeshima Y, Sudhakar A, Lively JC (2002) Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science 295:140–143
Tarui T, Miles LA, Takada Y (2001) Specific interaction of angiostatin with integrin alpha(v)beta(3) in endothelial cells. J Biol Chem 276:39562–39568
Rehn M, Veikkola T, Kukk-Valdre E et al. (2001) Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA 98:1024–1029
Tessler S, Rockwell P, Hicklin D (1994) Heparin modulates the interaction of VEGF165 with soluble and cell associated flk-1 receptors. J Biol Chem 269:12456–12461
Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G (1992) The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem 267:6093–6098
Robinson GS, Aiello LP (1998) Angiogenic factors in diabetic ocular disease: mechanisms of today, therapies for tomorrow. Int Ophthalmol Clin 38:89–102
Ma J-X, Chao L, Zhou G, Chao J (1993) Expression and characterization of rat kallikrein-binding protein in Escherichia coli. Biochem J 292:825–832
Acknowledgements
This work was supported by NIH grant EY12600, Research Awards from Juvenile Diabetes Foundation and American Diabetes Association, South Carolina Lions Club and an unrestricted grant to the Department of Ophthalmology, MUSC from Research to Prevent Blindness.
Author information
Authors and Affiliations
Corresponding author
Additional information
*These authors have contributed equally to this study
Rights and permissions
About this article
Cite this article
Gao*, G., Shao*, C., Zhang, S.X. et al. Kallikrein-binding protein inhibits retinal neovascularization and decreases vascular leakage. Diabetologia 46, 689–698 (2003). https://doi.org/10.1007/s00125-003-1085-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1085-9