
Abstract
Aims/hypothesis
Insulin-dependent glucose influx in skeletal muscle and adipocytes is believed to rely largely on GLUT4, but this has not been confirmed directly. We assessed the relative functional contribution of GLUT4 in experimental models of skeletal muscle and adipocytes using the HIV-1 protease inhibitor indinavir.
Methods
Indinavir (up to 100 µmol/l) was added to the glucose transport solution after insulin stimulation of wild-type L6 muscle cells, L6 cells over-expressing either GLUT4myc or GLUT1myc, 3T3-L1 adipocytes, isolated mouse brown or white adipocytes, and isolated mouse muscle preparations.
Results
100 µmol/l indinavir inhibited 80% of both basal and insulin-stimulated 2-deoxyglucose uptake in L6GLUT4myc myotubes and myoblasts, but only 25% in L6GLUT1myc cells. Cell-surface density of glucose transporters was not affected. In isolated soleus and extensor digitorum longus muscles, primary white and brown adipocytes, insulin-stimulated glucose uptake was inhibited 70 to 80% by indinavir. The effect of indinavir on glucose uptake was variable in 3T3-L1 adipocytes, averaging 45% and 67% inhibition of basal and maximally insulin-stimulated glucose uptake, respectively. In this cell, fractional inhibition of glucose uptake by indinavir correlated positively with the fold-stimulation of glucose uptake by insulin, and was higher with sub-maximal insulin concentrations. The latter finding coincided with an increase only in GLUT4, but not GLUT1, in plasma membrane lawns.
Conclusion/interpretation
Indinavir is a useful tool to assess different functional contributions of GLUT4 to glucose uptake in common models of skeletal muscle and adipocytes.
Similar content being viewed by others
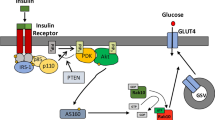


The glucose transporter GLUT4 is generally thought to be the major contributor to insulin-stimulated glucose uptake in adipocytes and skeletal muscle, based on its restricted tissue distribution and its capacity to translocate from intracellular pools to the cell surface in response to insulin. Conversely, GLUT1 is frequently viewed as the major transporter isoform through which glucose enters the cells in non-stimulated conditions, largely based on its ubiquitous expression and surface localization under these conditions [1, 2]. The contributions of GLUT1 and GLUT4 to glucose uptake in cells and tissues that express both isoforms have not been directly assessed. Commonly used measurements of the total cellular content or of the subcellular location of a given glucose transporter (GLUT) isoform provide only an indirect and incomplete assessment of their functional importance to overall glucose influx into cells.
Genetic ablation of GLUT4 has confirmed its importance for maintaining normal insulin sensitivity [3, 4, 5]. However, it is difficult to deduce the functional contribution of GLUT4 to glucose influx into normal tissues using this approach, since compensatory mechanisms in the affected tissue are induced [3, 6]. Thus, in cells expressing multiple isoforms of glucose transporters, a direct, 'functional' measurement of the glucose influx through a specific transporter isoform is required to decipher its relative contribution to the overall influx of glucose.
The direct assessment of glucose uptake through a specific isoform is mostly limited by the lack of inhibitors with sufficient selectivity for a specific GLUT. Compounds like cytochalasin B are potent inhibitors of glucose transport [7, 8], with similar binding affinities for GLUT1 and GLUT4 [9]. Forskolin binds to glucose transporters [10], with moderately increased binding affinity to GLUT4 compared with GLUT1 [9]. This finding could explain the observed ability of forskolin to inhibit more potently insulin-stimulated rather than basal glucose uptake in cells that express both GLUT1 and GLUT4 [11]. Recently, certain HIV-1 protease inhibitors were shown to inhibit GLUT4 preferentially over other isoforms in experiments using Xenopus oocytes exogenously over-expressing different GLUTs [12]. In this system, the IC50 of indinavir on glucose uptake was 50 to 100 µmol/l for cells expressing GLUT4, greater than 500 µmol/l for those expressing GLUT2, and even higher for GLUT1, GLUT3 or a GLUT8 mutant directed to the cell surface [13]. In primary adipocytes, the inhibitory effect of indinavir on glucose uptake was further shown to be reversible and non-competitive [13].
In this study, the selectivity of indinavir towards GLUT4 was tested in skeletal muscle L6 cells overexpressing either myc-tagged GLUT4 protein or myc-tagged GLUT1 protein. The compound was then used to examine the relative functional contribution of GLUT4 to basal and insulin-stimulated glucose uptake in isolated mouse skeletal muscle and adipocytes.
Materials and methods
Human insulin (Humulin R) was obtained from Eli Lilly Canada (Toronto, Ont., Canada). DNase I and phthalic acid dinonyl ester were from Sigma (St. Louis, Mo., USA). Collagenase type 1 and type 2 was purchased from Worthington (Lakewood, N.J., USA). Polyclonal anti-GLUT1 was raised in our laboratory against a C-terminal epitope [14]. Cy3-conjugated goat anti-rabbit was from Molecular Probes (Eugene, Ore., USA). Polyclonal anti-GLUT4 antibody was from Biogenesis (Kingston, N.H., USA). Indinavir was kindly provided by Merck (Rahway, N.J., USA).
Cell culture
Wild-type L6, L6 GLUT4myc and L6GLUT1myc myoblasts were differentiated into myotubes as described previously [15]. Prior to all experimental manipulations, myotubes were deprived of serum for 4 h. 3T3-L1 cells were grown in monolayer culture in DMEM supplemented with 20% (v/v) calf serum and 1% (v/v) antibiotic solution (10,000 units/ml penicillin and 10 mg/ml streptomycin) in an atmosphere of 9% CO2 at 37°C. 3T3-L1 fibroblasts were differentiated into adipocytes as described previously [16]. Cells exhibited more than95% adipocyte morphology by light microscopy and lipid accumulation by Oil-Red-O staining.
Determination of 2-deoxy-3H-D-glucose uptake in cells in culture
2-Deoxyglucose uptake was measured as described earlier [17]. An indinavir stock solution was prepared in DMSO and added to the transport solution after insulin stimulation. Control cells were treated with the same final concentration of DMSO (reaching a maximum of 0.1% v/v).
Plasma membrane lawn formation and immunofluorescence analysis
Differentiated 3T3-L1 adipocytes, grown on glass coverslips in 6-well dishes, were treated for 20 min with different concentrations of insulin as indicated in the figure legend. Plasma membrane lawns (sheets) were prepared as described previously with slight modifications [17], as follows: After incubation, the cells were placed on ice and washed twice in ice-cold PBS. One millilitre of ice cold poly-lysine (0.55 mg/ml) was added to coverslips for 1 min. Hypotonic buffer (23 mmol/l KCl, 10 mmol/l Na-HEPES, 2 mmol/l MgCl2, 1 mmol/l EGTA, pH 7.5) was added in three quick rinses. Ten millilitres of sonication buffer (70 mmol/l KCl, 30 mmol/l Na-HEPES, 5 mmol/l MgCl2, 3 mmol/l EGTA, 1 mmol/l dithiothreitol, 0.5 mmol/l PMSF, 1 µmol/l pepstatin A, pH 7.5) were added to each well. Cells were disrupted on ice using a probe sonicator (Heat Systems, Farmingdale, N.Y., USA) set at 30% maximal output, for 10 s. The coverslips were washed three times in sonication buffer and incubated with cold 3% paraformaldehyde in PBS buffer for 10 min, followed by three washes in PBS. Excess fixative was quenched with 50 mmol/l NH4Cl/PBS for 5 min, followed by three washes with PBS. The lawns were subsequently blocked by a 1-h incubation in 5% goat serum in PBS, then incubated with rabbit anti-GLUT4 antiserum (1:150) or rabbit anti-GLUT1 antiserum (1:150) for 60 min and washed three times in PBS. Cy3-conjugated goat anti-rabbit antiserum (1:250) was added for 45 min then rinsed out with four washes with PBS and the coverslips mounted with DAKO mounting solution. Background fluorescence was assessed using the same dilution of the secondary antibody, but without an incubation with the primary antibody, and was found to be negligible compared to samples incubated with either anti-GLUT1 or GLUT4 antibodies. Images were obtained using a Leica inverted fluorescence microscope with a ×100 objective. Images were collected under identical gain settings. Quantification was made using NIH Image J software.
Animals
Mice were housed in a pathogen-free environment on 12-h light to dark cycle and fed ad libitum with standard rodent chow. All protocols were approved by the Animal Care Committee of The Hospital for Sick Children, Toronto, Ontario.
2-Deoxyglucose uptake into isolated white and brown adipocytes
Brown adipose tissue was dissected from the interscapular regions of 4- to 6-week-old C57Bl/6J mice, and white adipocytes from 3-month-old animals. White and brown adipocytes were isolated by collagenase type 2/DNAse digestion (6 and 0.2 mg/ml, respectively) in Krebs Ringer HEPES buffer (KRH) containing 1% (w/v) fatty acid-free BSA as described earlier [18]. After digestion, the reaction mixture was filtered trough a 250 µm nylon mesh. Brown adipocytes were washed three times with KRH containing 1% BSA (KRH 1%), and then twice with KRH containing 4% BSA (KRH 4%). White adipocytes were washed once with KRH 4%, and then once with KRH 1%. After the final washing, the floating brown adipocytes were resuspended in KRH 4% (1% BSA for white adipocytes), and aliquoted for all experimental conditions. Cells were incubated for 30 min with or without 100 nmol/l insulin. 2-Deoxy-3H-D-glucose uptake (final 2-deoxyglucose concentration 10 µmol/l) was measured over a 5-min period with or without the presence of 100 µmol/l indinavir. Glucose uptake was stopped by separating cells from the medium by centrifugation through an oil mixture (silicone oil DC550 with phthalic acid dinonyl ester in a ratio of 2:3) and then subjected to liquid scintillation counting. Cellular lipid content was measured by lipid extraction done as previously described [19].
2-Deoxyglucose uptake into isolated skeletal muscles
Intact soleus and EDL muscles were incubated with or without 2 U/ml insulin. 2-Deoxyglucose uptake in insulin-stimulated muscles was then measured for 20 min as described [20], with or without the presence of insulin and 100 µmol/l indinavir, respectively. Non-GLUT dependent 2-deoxyglucose uptake was measured in the presence of 10 µmol/l cytochalasin B in the transport solution. The obtained value was subtracted from the value obtained in the absence of cytochalasin B.
Data analysis
Statistical analysis was done by using either Student's t test (comparison of two variables), or analysis of variance test (ANOVA, Fisher's multiple comparisons test) for comparison of more than two variables, and Pearson correlation when indicated. All statistical analyses were done by using Prism 2C software (Graphpad, San Diego, Calif., USA). A p value of less than 0.05 was considered to be statistically significant.
Results
Indinavir preferentially blocks glucose uptake through GLUT4 in L6 muscle cells
To assess the possible use of indinavir to dissect the contribution of different glucose transporter isoforms to overall glucose influx in mammalian cells, wild-type L6 myotubes were used first. These cells in their differentiated state of myotubes express at least three glucose transporter isoforms, GLUT1, GLUT3, and GLUT4. L6 myotubes were first incubated for 20 min with or without 100 nmol/l insulin, after which 2-deoxyglucose uptake was measured in the absence or presence of various concentrations of indinavir only in the transport solution (5-min assay). Increasing concentrations of indinavir had a minor inhibitory effect on 2-deoxyglucose uptake in unstimulated cells (Fig. 1A). In contrast, the insulin-stimulated glucose uptake activity was markedly inhibited in a dose-dependent manner. The net insulin effect above basal for each indinavir concentration is shown in Fig. 1B. At a concentration of 100 µmol/l in the transport solution, indinavir almost completely inhibited the net insulin effect, but a significant decrease (p<0.05) could be observed with as little as 20 µmol/l. This finding is consistent with the notion that basal glucose transport and the insulin-mediated stimulation of this activity are dominated by different transporter isoforms, probably GLUT1/3 and GLUT4 respectively, which are inhibited by indinavir with different potencies.

The HIV-1 protease inhibitor indinavir blocks insulin-mediated glucose uptake in wild-type L6 myotubes. Differentiated L6 myotubes were stimulated for 20 min without or with 100 nmol/l insulin. 2-deoxyglucose uptake was then assessed in the absence or presence of the indicated final concentrations of indinavir in the transport solution. Results are the means ± SE of three independent experiments, each performed in triplicate. A Shown are basal (open bars) and insulin-stimulated 2-deoxyglucose uptake values (closed bars). *p<0.05, **p<0.01, and # p<0.001 compared with the corresponding value in cells not treated with indinavir. B The net insulin effect above basal was calculated for each dose of indinavir. *p<0.05, **p<0.01, and # p<0.001 compared with net insulin effect in cells not treated with indinavir
To further verify this possibility, we used L6 muscle cells stably overexpressing either myc-tagged GLUT1 or myc-tagged GLUT4 (L6GLUT1myc and L6GLUT4myc cells, respectively). In the undifferentiated myoblast state, L6GLUT1myc cells do not express endogenous GLUT4, since GLUT4 expression is induced only upon cellular differentiation [21]. In contrast, L6GLUT4myc myoblasts express ectopically high quantities of GLUT4myc protein. The properties of L6GLUT4myc cells have been reported, and show that GLUT4myc behaves essentially as GLUT4 in terms of its intracellular traffic [22, 23, 24], regulation by various stimuli [23, 25, 26], and in its transport properties such as Km and susceptibility to cytochalasin B [27].
The two cell lines displayed a marked difference in their susceptibility to indinavir (Fig. 2A). Whereas in L6GLUT1myc cells basal and insulin-stimulated 2-deoxyglucose uptake were only mildly affected by indinavir (10% and 9%, respectively), in L6GLUT4myc myoblast glucose uptake in both conditions was highly sensitive to the drug. Twenty µmol/l indinavir only in the transport solution was sufficient to reduce significantly 2-deoxyglucose uptake (p<0.05), and 100 µmol/l indinavir inhibited basal and insulin-stimulated glucose uptake by 80.6±3.7 and 79.7±5.2%, respectively, compared to the transport activity in the absence of the drug (p<0.001). The percent inhibition was identical in unstimulated and insulin-stimulated L6LUT4myc cells, suggesting that in this cell line GLUT4myc is largely responsible for both basal and insulin-stimulated glucose uptake. We next assessed the effect of indinavir on 2-deoxyglucose uptake in the two cell lines in the myotube state (Fig. 2B). L6GLUT4myc myotubes displayed a similar sensitivity to inhibition by indinavir to that observed in the myoblasts, with identical inhibition in the basal and the insulin-stimulated states. By comparison, 2-deoxyglucose uptake in L6GLUT1myc myotubes was relatively resistant to indinavir in the basal state (26% inhibition with 100 µmol/l indinavir, p>0.05). A slightly higher sensitivity to the presence of the drug in the transport solution was observed in insulin-stimulated cells (35% inhibition with 100 µmol/l indinavir, p<0.01). These findings show that glucose uptake activity in L6GLUT1myc cells is dominated by an indinavir-resistant GLUT, in all probability the exogenously-expressed GLUT1myc. However, following insulin-stimulation of L6GLUT1myc myotubes, some contribution of endogenous GLUT4 that was induced during cellular differentiation becomes apparent.

Inhibition of basal and insulin-stimulated glucose uptake by indinavir and forskolin in L6GLUT1myc and L6GLUT4myc cells. A L6GLUT4myc myoblasts (triangles) or L6GLUT1myc myoblasts (squares) were treated without (open symbols) or with (filled symbols) 100 nmol/l insulin for 20 min. 2-deoxyglucose uptake was measured in the presence of the indicated final concentrations of indinavir in the transport solution. *p<0.05 as compared to L6GLUT1myc cells under the same conditions. B Same as A, but using myotubes instead of myoblasts. Results are the fractional inhibition of glucose uptake by indinavir, and are the means ± SE of at least three independent experiments, each carried out in triplicate. *p<0.05 as compared to L6GLUT1myc cells under the same conditions. C Same analysis as above in insulin-stimulated cells, with forskolin added to the transport solution. Results are the means ± SD (if two values were available) or means ± SE (when three or more values were available) of values from at least two independent experiments, each carried out in triplicate. Basal and insulin-stimulated glucose uptake values were (in pmol·mg protein−1·min−1): for L6GLUT4myc myoblasts 23.2±1.9 and 31.2±4.9, respectively; for L6GLUT4myc myotubes 14.2±2.8 and 24.1±5.3, respectively; for L6GLUT1myc myoblasts 17.8±4.5 and 22.5±5.5, respectively; for L6GLUT1myc myotubes 13.3±2.3 and 21.6±3.2, respectively
To exclude the possibility that indinavir inhibited glucose uptake by decreasing surface GLUT4, we assessed surface myc-epitope exposure. Since the myc-epitope is inserted in the first exofacial loop of the GLUT4 protein, the exposure of the GLUT4myc transporters at the cell surface can be measured in non-permeabilized cells using anti-myc primary and HRP-linked secondary antibodies [15]. Insulin stimulation resulted in a 2.0±0.2-fold increase in cell surface GLUT4myc. After insulin stimulation, addition of 50 µmol/l indinavir for 5 min did not affect the gain in surface GLUT4myc exposure (basal + indinavir: 1.1±0.1-fold, insulin-stimulated + indinavir: 1.9±0.1-fold, relative to basal surface GLUT4myc in the absence of indinavir). Similarly, indinavir did not alter surface GLUT1myc densities in either basal (1.2±0.2-fold of myc signal in the absence of indinavir), or insulin-stimulated conditions (1.5±0.1 and 1.4±0.2-fold of basal in the absence or presence of indinavir, respectively). These findings exclude the possibility that a rapid occlusion of glucose transporters is induced by indinavir. Furthermore, insulin stimulated Akt phosphorylation was unaltered by the 5-min exposure to indinavir after insulin stimulation (not shown). Collectively, the data suggest that the activity of surface GLUT4myc is much more sensitive to direct inhibition by indinavir than that of GLUT1myc, consistent with the reported selective capacity of indinavir to inhibit the untagged, wild-type GLUT4 versus GLUT1 reported in Xenopus oocytes [12].
The selective effect of indinavir on L6GLUT4myc cells contrasted with that of forskolin, an agent previously reported to inhibit glucose transporters [9, 10, 11]. Forskolin added to the transport solution exerted some selective inhibition of GLUT4myc at concentrations of 0.1 to 5 µmol/l (Fig. 2C). Yet, such selectivity was relatively minor compared to that displayed by indinavir, and did not persist when the forskolin concentration was increased above 10 µmol/l.
Indinavir inhibition of glucose uptake in 3T3-L1 adipocytes
We used indinavir to assess the contribution of GLUT4 and GLUT1 to glucose uptake in 3T3-L1 adipocytes. These cells constitute the most insulin-responsive immortalized cell line available, and are therefore extensively used to study insulin action and GLUT4 biology. However, the response of glucose uptake to insulin varies considerably in various reports, potentially reflecting batch-to-batch and sub-clone variability. To address this biological variability, we incorporated in our analysis results from different batches of 3T3-L1 adipocytes from two separate laboratories (in Toronto, Canada and in Beer-Sheva, Israel). Basal glucose transport tended to be decreased by indinavir, although this effect did not reach ANOVA statistical significance (p>0.05, Fig. 3A). This behaviour was in contrast to that of L6 muscle cells expressing primarily GLUT1myc or those expressing primarily GLUT4myc (Fig. 2), as indinavir did not inhibit glucose uptake in the former and inhibited profoundly glucose transport in the latter. In 3T3-L1 adipocytes, insulin stimulation resulted in a 9.8±1.2-fold increase in 2-deoxyglucose uptake activity, with a range between individual batches of 5- to 20-fold. On average, 50 and 100 µmol/l indinavir inhibited the net insulin effect in these cells by 49.1±1.3% and 67.2±3.0%, respectively (Fig. 3B, p<0.001). Since this mean fractional inhibition by indinavir seemed lower than that observed with the L6 cells (particularly by 50 µmol/l indinavir), we addressed the possibility that in 3T3-L1 adipocytes indinavir required a longer time to interfere with 2-deoxyglucose uptake. Pre-incubation of the cells for 5 min before and during the 2-deoxyglucose transport assay had an identical inhibitory effect to that observed in the absence of a pre-incubation period. In addition, indinavir in the transport solution blocked the uptake of 3-O-methylglucose by similar degree with respect to its effect on 2-deoxyglucose, despite the shorter duration of the assay (up to 1 min). These results show that the inhibitory effect of indinavir on hexose transport occurs very rapidly, and probably represents interaction of the drug with GLUT4 rather than with hexokinase. The latter is also supported by the findings shown in Fig. 2A,B, of a differential effect of indinavir on cells overexpressing GLUT4myc or GLUT1myc, where hexokinase amounts are deemed similar.

Indinavir-mediated inhibition of glucose uptake in 3T3-L1 adipocytes and its relation to GLUT4 and GLUT1 translocation. 3T3-L1 adipocytes were treated as described above for L6 cells. A Basal (open bars) and insulin-stimulated glucose uptake (100 nmol/l, full bars) were measured in the absence or presence of 50 or 100 µmol/l indinavir in the transport solution. Results are the means ± SE of five independent experiments, each carried out in duplicate. # p<0.001 compared to insulin-stimulated glucose uptake in the absence of indinavir. B The net insulin effect above basal was calculated for each dose of indinavir. # p<0.001 compared to insulin-stimulated glucose uptake in the absence of indinavir. C After treatment of 3T3-L1 adipocytes with the indicated concentrations of insulin, glucose uptake was measured, and expressed as the % inhibition by indinavir of the net insulin response. Results are means ± SE of four independent experiments, carried out in duplicate. D Plasma membrane lawns were prepared as stated in Methods and immunostained with either anti-GLUT1 or anti-GLUT4 antibodies. Values were expressed as the percent of the maximal fluorescent signal observed with each GLUT. The maximum fold-stimulation over basal was 3.91±1.10 and 2.21±0.19-fold for GLUT4 (full bars) and GLUT1 (open bars), respectively. Results are the means ± SE of values from four independent experiments. *p<0.05 and **p<0.01
Given the large range of the fold response to insulin observed among different batches of 3T3-L1 adipocytes, we addressed the possibility that this variability reflects different contributions of GLUT4 to glucose transport. Pearson correlation between the fold response to insulin and the percent inhibition of the net insulin effect by indinavir showed coefficients of correlation of r=0.61 (p=0.03) and r=0.93 (p=0.02) for 50 and 100 µmol/l indinavir, respectively. A statistically significant correlation was not observed between the fold insulin response and the inhibition of basal glucose uptake by indinavir. Thus, significant variability between batches of 3T3-L1 adipocytes occurs with different contributions of GLUT4 to insulin-stimulated glucose uptake. Higher fold stimulation of glucose uptake by maximal insulin concentrations correlate with higher fractional contribution of GLUT4, which can be unraveled by indinavir.
The variable contribution of GLUT4 to glucose uptake in maximally insulin-stimulated 3T3-L1 adipocytes (100 nmol/l insulin) revealed by indinavir, prompted us to assess the contribution of GLUT4 to overall glucose influx following sub-maximal insulin concentrations. The indinavir-inhibitable fraction of glucose uptake was measured after stimulation with different insulin doses, and was compared to the translocation of GLUT4 or GLUT1 to the plasma membrane. In this set of experiments 50 µmol/l indinavir inhibited approximately 20% of basal glucose uptake, consistent with a small contribution of GLUT4 under this condition. After insulin stimulation, indinavir inhibited 80% of the net effect induced by 0.01 nmol/l insulin (1.6-fold stimulation of basal), 65% of the effect by 0.1 nmol/l (3–4-fold stimulation of basal), but only 50% of the net increase in glucose uptake induced by 10 and 100 nmol/l insulin (8–9-fold stimulation in this sub-set of experiments, Fig. 3C). These results suggest that GLUT4 contributes to a larger extent to the stimulation of glucose uptake caused by lower than by higher doses of insulin. To examine whether this difference arises form different gains in GLUT4 and GLUT1, translocation of each transporter using corresponding C-terminal antibodies was assessed. Only GLUT4 was recruited to the plasma membrane at 0.01 and 0.1 nmol/l insulin (Fig. 3D), coinciding with the high susceptibility of glucose uptake to indinavir (Fig. 3B). With increasing insulin concentrations, GLUT1 was recruited in addition to GLUT4, coinciding with a smaller fractional inhibition of glucose uptake by the drug.
GLUT4-mediated glucose uptake in 3T3-L1 adipocytes and primary murine white and brown adipocytes
Since the mouse-derived 3T3-L1 adipocyte cell line is frequently used as a model of adipose tissue, we analysed the capacity of indinavir to inhibit glucose uptake in primary murine white and brown adipocytes. Table 1 shows the fractional inhibition of basal and maximally insulin-stimulated glucose uptake by indinavir added to the transport solution. Basal state glucose uptake was less sensitive to indinavir in brown adipocytes than in white adipocytes, and similar to that observed in 3T3-L1 adipocytes, suggesting a relatively higher contribution of GLUT4 to basal glucose uptake in WAT. After insulin stimulation, 100 µmol/l indinavir inhibited glucose uptake by more than 75% in both primary cell types. These results suggest that GLUT4 could have a greater role in glucose uptake in unstimulated white adipocytes compared to brown adipocytes or 3T3-L1 adipocytes. Moreover, both cell types depend greatly on GLUT4 in the insulin-stimulated state. This feature largely exceeds the contribution of GLUT4 in 3T3-L1 adipocytes.
GLUT4 contributes to basal and insulin-stimulated glucose uptake in isolated mouse soleus and EDL skeletal muscles.
The results observed in Table 1 prompted us to investigate whether a discrepancy in indinavir sensitivity also exists between ex-vivo skeletal muscle preparations and L6GLUT4myc myotubes. Table 2 presents the fractional inhibition by indinavir of basal and insulin-stimulated glucose uptake in L6GLUT4myc myotubes, isolated mouse soleus and extensor digitorum longus (EDL) muscles. Both basal and insulin-stimulated glucose uptake were markedly inhibited by indinavir in soleus or EDL muscle. In the basal state, glucose uptake was inhibited between 60 and 70% in both muscle types. In the soleus muscle the fractional inhibition of glucose uptake by indinavir was similar in the insulin-stimulated and basal states. In the EDL, insulin-stimulated glucose uptake was inhibited by indinavir more than in the basal state, but this difference did not reach statistical significance (Table 2). Moreover, the fractional inhibition was similar to that observed in primary white adipocytes (Table 1), and higher than in 3T3-L1 adipocytes.
Discussion
It is widely accepted that the main glucose transporter responsible for insulin-stimulated glucose uptake is GLUT4. Yet, this common assumption, based on expression levels and changes in localization upon insulin stimulation, has not been 'functionally' confirmed in normal tissues and cells. Furthermore, the contribution of other glucose transporter(s) such as GLUT1 under both physiological and pathological conditions (like insulin resistance) is not known. In fact, whether GLUT1 is actually expressed in skeletal myocytes has been the subject of debate. In mouse adipocytes, GLUT4 and not GLUT1 was estimated to be responsible for as much as 90% of insulin-stimulated glucose uptake [28]. However, since GLUT4 expression is decreased in adipocytes of insulin resistant subjects [29, 30], the relative contribution of GLUT1 and other GLUTs to overall glucose influx could be more pronounced in this condition. Likewise, slow-twitch fibers of skeletal muscle are specifically depleted of GLUT4 in diabetes [31], raising again the possibility that other GLUTs could functionally contribute to glucose uptake. The importance of other glucose transporters is highlighted in animal models with genetic ablation of GLUT4, where glucose entry into skeletal muscle is not ablated, thus preventing the expected development of diabetes [3, 6]. All these findings beg the question of what is the relative functional contribution of GLUT4 to the overall glucose influx into normal skeletal muscle or adipocytes.
The HIV-1 protease inhibitor indinavir is a potent anti-retroviral agent commonly used in the treatment of HIV infection, frequently in combination with other HPIs, nucleoside analogues and non-nucleoside analogues [32]. This therapy, while improving the prognosis of patients infected by HIV, has been associated with the development of a cluster of metabolic side effects including peripheral insulin resistance, resembling the "metabolic syndrome" [33, 34, 35]. Affected patients are at risk for developing new-onset Type 2 diabetes and cardiovascular morbidity [36, 37, 38]. Although nucleoside analogues and the HIV infection itself cause metabolic alterations, recent data support a causative role for HIV protease inhibitors in the development of the metabolic side-effects [39, 40, 41, 42]. Different cellular mechanisms have been suggested to explain insulin resistance induced by HIV protease inhibitors [12, 43, 44]. In addition, recent studies offer convincing evidence that indinavir and other HIV protease inhibitors directly interfere with the activity of glucose transporters, with selectivity towards GLUT4 over GLUT2, GLUT3, GLUT1, or GLUT8 [12, 13]. Glucose uptake into Xenopus oocytes expressing GLUT4 was inhibited by indinavir (IC50 50–100 µmol/l) added to the transport solution. Indinavir caused a decrease in affinity photolabelling of GLUT4 in skeletal muscle, raising the question of whether indinavir prevented GLUT4 translocation or activity in skeletal muscle [45]. We report that surface exposure of the myc epitope of GLUT4myc in mammalian cells is not altered by indinavir present after insulin stimulation only in the transport solution, excluding the possibility that indinavir causes acute occlusion of GLUT4 within the plasma membrane. Thus it seems probable that upon short exposure indinavir directly interacts with unique epitopes of GLUT4 transporters present at the cell surface, which are required for its capacity to function as a glucose transporter.
Indinavir is a rather selective inhibitor of glucose transport through GLUT4 in mammalian cells
We have shown that indinavir inhibits basal and insulin-stimulated glucose uptake much more efficiently in L6GLUT4myc cells than in GLUT1myc cells (~80% vs ~25%, respectively), with an IC50 value of less than 20 µmol/l in L6GLUT4myc (myoblasts or myotubes). These findings confirm significant selectivity of indinavir to inhibit GLUT4-mediated transport [12], which is superior to that of forskolin (Fig. 2C). The molecular basis for such selectivity is not known. It could arise from unique structural elements in GLUT4, which are defined by the primary sequence or by post-translational modifications of the protein, such as glycosylation. The fact that indinavir behaved similarly in the basal and insulin-stimulated states, suggests that in these cells the over-expressed GLUT, be it GLUT4myc or GLUT1myc, becomes the predominant transporter isoform responsible for glucose uptake. This is in contrast to wild-type L6 cells, in which basal glucose uptake was inhibited by 25% whereas the net response to insulin was largely reduced by indinavir (Fig. 1A,B). Therefore, in wild-type L6 myotubes, GLUT4 contributes little to basal glucose uptake, but is the glucose transporter largely responsible to the net response to insulin.
In primary adipocytes and in isolated skeletal muscle preparations, insulin-stimulated glucose uptake was highly sensitive to indinavir, suggesting a large contribution of GLUT4. In addition, GLUT4 had a major role in basal glucose influx in white adipocytes and skeletal muscle tissues isolated from mice. Since this feature was also seen in L6 cells over-expressing GLUT4myc, this cell line offers a good in-vitro cellular model of insulin responsive tissues for analysis of glucose uptake through the GLUT4 system.
Indinavir unravels a variable contribution of GLUT4 to insulin-stimulated glucose uptake in 3T3-L1 adipocytes compared to primary adipocytes
3T3-L1 adipocytes are commonly used for studies related to the regulation of glucose uptake and GLUT4 traffic. It is therefore essential to assess the functional contribution of GLUT4 to glucose uptake in this cell model. Here we have reported that the well-known batch-to-batch variability in the responsiveness to insulin could reflect different functional contribution of GLUT4 to glucose uptake, which can be documented by indinavir. In batches with a 6- to 8-fold response in glucose uptake to insulin, GLUT1 could contribute up to 50% of the total response. Such a contribution is not observed in either primary mouse adipocytes, skeletal muscle preparations, or L6GLUT4myc cells, in which indinavir inhibits a larger portion of the insulin-stimulated response.
Three conclusions arise from these studies in 3T3-L1 adipocytes: (i) 3T3-L1 adipocyte clones with exquisite insulin responsiveness (>10–12-fold) rely more heavily on GLUT4 than clones with lower responsiveness; (ii) because such high responsiveness can be difficult to maintain over multiple cell passages it becomes important to document the fractional contribution of GLUT4 in each case. Our data suggest that indinavir can be used for this purpose; (iii) GLUT4 contributes to a larger extent to glucose uptake at submaximal than at maximal insulin concentrations. Typically, 100 pmol/l insulin can induce a 3- to 4-fold increase in 2-deoxyglucose uptake, 75% of which is indinavir inhibitable. Consistent with a major contribution of GLUT4 at low insulin doses is the finding that under these conditions primarily GLUT4 is mobilized to the plasma membrane. Therefore we propose that the GLUT4 system is more sensitive to insulin than GLUT1 in 3T3-L1 adipocytes.
The newly identified difference in the sensitivity of GLUT4 and GLUT1 translocation to insulin stimulation, raises intriguing questions regarding the potential role that these two transporters could play in states of impaired insulin sensitivity and/or responsiveness. Indinavir could prove to be a valuable tool for the direct analysis of the contribution of GLUT4 to overall glucose uptake in the context of insulin resistance [46].
In conclusion, in L6 GLUT4myc cells, primary adipocytes and skeletal muscle preparations, but not in wild-type L6 cells or 3T3-L1 adipocytes, GLUT4(myc) is the predominant transporter responsible for both basal and insulin-stimulated glucose uptake. GLUT4 contributes more to glucose influx into 3T3-L1 adipocytes stimulated with picomolar rather than higher concentrations of insulin. Indinavir could be utilized to analyse the 'functional relevance' of known changes in GLUT4 expression and/or activity associated with insulin resistance.
Abbreviations
- EDL:
-
extensor digitorum longus
- HIV:
-
human immunodeficiency virus
References
Kraegen EW, Sowden JA, Halstead MB et al. (1993) Glucose transporters and in vivo glucose uptake in skeletal and cardiac muscle: fasting, insulin stimulation and immunoisolation studies of GLUT1 and GLUT4. Biochem J 295:287–293
Buse MG, Robinson KA, Marshall BA, Hresko RC, Mueckler MM (2002) Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am J Physiol Endocrinol Metab 283:E241–250
Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ (1995) Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature 377:151–155
Abel ED, Peroni O, Kim JK et al. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409:729–733
Zisman A, Peroni OD, Abel ED et al. (2000) Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 6:924–928
Ryder JW, Kawano Y, Galuska D et al. (1999) Postexercise glucose uptake and glycogen synthesis in skeletal muscle from GLUT4-deficient mice. FASEB J 13:2246–2256
Ebstensen RD, Plagemann PG (1972) Cytochalasin B: inhibition of glucose and glucosamine transport. Proc Natl Acad Sci USA 69:1430–1434
Klip A, Walker D (1983) The glucose transport system of muscle plasma membranes: characterization by means of [3H]cytochalasin B binding. Arch Biochem Biophys 221:175–187
Hellwig B, Joost HG (1991) Differentiation of erythrocyte-(GLUT1), liver-(GLUT2), and adipocyte-type (GLUT4) glucose transporters by binding of the inhibitory ligands cytochalasin B, forskolin, dipyridamole, and isobutylmethylxanthine. Mol Pharmacol 40:383–389
Joost HG, Steinfelder HJ (1987) Forskolin inhibits insulin-stimulated glucose transport in rat adipose cells by a direct interaction with the glucose transporter. Mol Pharmacol 31:279–283
Klip A, Ramlal T, Douen AG, Bilan PJ, Skorecki KL (1988) Inhibition by forskolin of insulin-stimulated glucose transport in L6 muscle cells. Biochem J 255:1023–1029
Murata H, Hruz PW, Mueckler M (2000) The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem 275:20251–20254
Murata H, Hruz PW, Mueckler M (2002) Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS 16:859–863
Romanek R, Sargeant R, Paquet MR, Gluck S, Klip A, Grinstein S (1993) Chloroquine inhibits glucose-transporter recruitment induced by insulin in rat adipocytes independently of its action on endomembrane pH. Biochem J 296:321–327
Wang Q, Khayat Z, Kishi K, Ebina Y, Klip A (1998) GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett 427:193–197
Volchuk A, Wang Q, Ewart HS et al. (1996) Syntaxin 4 in 3T3-L1 adipocytes: regulation by insulin and participation in insulin-dependent glucose transport. Mol Biol Cell 7:1075–1082
Sweeney G, Somwar R, Ramlal T, Volchuk A, Ueyama A, Klip A (1999) An inhibitor of p38 mitogen-activated protein kinase prevents insulin- stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J Biol Chem 274:10071–10078
Omatsu-Kanbe M, Zarnowski MJ, Cushman SW (1996) Hormonal regulation of glucose transport in a brown adipose cell preparation isolated from rats that shows a large response to insulin. Biochem J 315:25–31
Konrad D, Bilan PJ, Nawaz Z et al. (2002) Need for GLUT4 activation to reach maximum effect of insulin-mediated glucose uptake in brown adipocytes isolated from GLUT4myc-expressing mice. Diabetes 51:2719–2726
Roy D, Perreault M, Marette A (1998) Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Physiol 274:E692–699
Mitsumoto Y, Klip A (1992) Development regulation of the subcellular distribution and glycosylation of GLUT1 and GLUT4 glucose transporters during myogenesis of L6 muscle cells. J Biol Chem 267:4957–4962
Ueyama A, Yaworsky KL, Wang Q, Ebina Y, Klip A (1999) GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am J Physiol 277:E572–578
Li D, Randhawa VK, Patel N, Hayashi M, Klip A (2001) Hyperosmolarity reduces GLUT4 endocytosis and increases its exocytosis from a VAMP2-independent pool in l6 muscle cells. J Biol Chem 276:22883–22891
Foster LJ, Li D, Randhawa VK, Klip A (2001) Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem 276:44212–44221
Khayat ZA, Tsakiridis T, Ueyama A, Somwar R, Ebina Y, Klip A (1998) Rapid stimulation of glucose transport by mitochondrial uncoupling depends in part on cytosolic Ca2+ and cPKC. Am J Physiol 275:C1487–1497
Patel N, Khayat ZA, Ruderman NB, Klip A (2001) Dissociation of 5' AMP-activated protein kinase activation and glucose uptake stimulation by mitochondrial uncoupling and hyperosmolar stress: differential sensitivities to intracellular Ca2+ and protein kinase C inhibition. Biochem Biophys Res Commun 285:1066–1070
Somwar R, Kim DY, Sweeney G et al. (2001) GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen- activated protein kinase. Biochem J 359:639–649
Pilch PF, Wilkinson W, Garvey WT, Ciaraldi TP, Hueckstaedt TP, Olefsky JM (1993) Insulin-responsive human adipocytes express two glucose transporter isoforms and target them to different vesicles. J Clin Endocrinol Metab 77:286–289
Garvey WT (1992) Glucose transport and NIDDM. Diabetes Care 15:396–417
Garvey WT, Maianu L, Zhu JH, Hancock JA, Golichowski AM (1993) Multiple defects in the adipocyte glucose transport system cause cellular insulin resistance in gestational diabetes. Heterogeneity in the number and a novel abnormality in subcellular localization of GLUT4 glucose transporters. Diabetes 42:1773–1785
Gaster M, Staehr P, Beck-Nielsen H, Schroder HD, Handberg A (2001) GLUT4 is reduced in slow muscle fibers of type 2 diabetic patients: is insulin resistance in type 2 diabetes a slow, type 1 fiber disease? Diabetes 50:1324–1329
Flexner C (1998) HIV-protease inhibitors. N Engl J Med 338:1281–1292
Carr A, Samaras K, Burton S et al. (1998) A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 12:F51–58
Carr A, Samaras K, Thorisdottir A, Kaufmann GR, Chisholm DJ, Cooper DA (1999) Diagnosis, prediction, and natural course of HIV-1 protease-inhibitor- associated lipodystrophy, hyperlipidaemia, and diabetes mellitus: a cohort study. Lancet 353:2093–2099
Vigouroux C, Gharakhanian S, Salhi Y et al. (1999) Adverse metabolic disorders during highly active antiretroviral treatments (HAART) of HIV disease. Diabetes Metab 25:383–392
Wanke CA (1999) Epidemiological and clinical aspects of the metabolic complications of HIV infection the fat redistribution syndrome. AIDS 13:1287–1293
Yarasheski KE, Tebas P, Sigmund C et al. (1999) Insulin resistance in HIV protease inhibitor-associated diabetes. J Acquir Immune Defic Syndr 21:209–216
Hadigan C, Corcoran C, Stanley T, Piecuch S, Klibanski A, Grinspoon S (2000) Fasting hyperinsulinemia in human immunodeficiency virus-infected men: relationship to body composition, gonadal function, and protease inhibitor use. J Clin Endocrinol Metab 85:35–41
Carr A, Samaras K, Chisholm DJ, Cooper DA (1998) Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 351:1881–1883
Walli R, Herfort O, Michl GM et al. (1998) Treatment with protease inhibitors associated with peripheral insulin resistance and impaired oral glucose tolerance in HIV-1-infected patients. AIDS 12:F167–173
Purnell JQ, Zambon A, Knopp RH et al. (2000) Effect of ritonavir on lipids and post-heparin lipase activities in normal subjects. AIDS 14:51–57
Noor MA, Lo JC, Mulligan K et al. (2001) Metabolic effects of indinavir in healthy HIV-seronegative men. AIDS 15:F11–18
Caron M, Auclair M, Vigouroux C, Glorian M, Forest C, Capeau J (2001) The HIV protease inhibitor indinavir impairs sterol regulatory element- binding protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistance. Diabetes 50:1378–1388
Rudich A, Vanounou S, Riesenberg K et al. (2001) The HIV protease inhibitor nelfinavir induces insulin resistance and increases basal lipolysis in 3T3-L1 adipocytes. Diabetes 50:1425–1431
Nolte LA, Yarasheski KE, Kawanaka K, Fisher J, Le N, Holloszy JO (2001) The HIV protease inhibitor indinavir decreases insulin- and contraction-stimulated glucose transport in skeletal muscle. Diabetes 50:1397–1401
Huang C, Somwar R, Patel N, Niu W, Torok D, Klip A (2002) Sustained exposure of L6 myotubes to high glucose and insulin decreases insulin-stimulated GLUT4 translocation but upregulates GLUT4 activity. Diabetes 51:2090–2098
Acknowledgements
We thank Dr. D. McClain (University of Utah, Salt Lake City) for help in establishing the assay of glucose uptake in white murine adipocytes. This work was supported by a grant to A. Klip from the Canadian Institutes of Health Research (MT 12601). A. Rudich was the recipient of the Albert Renold Career Development Award of the European Foundation for the Study of Diabetes. D. Konrad was supported by a fellowship from the Swiss National Science Foundation (grant No. 81ZH-57433), the Research Institute of The Hospital for Sick Children (Clinician Scientist Award), and the Zürcher Diabetes-Gesellschaft.
Author information
Authors and Affiliations
Corresponding author
Additional information
A.R. and D.K. contributed equally to this work
Rights and permissions
About this article
Cite this article
Rudich, A., Konrad, D., Török, D. et al. Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia 46, 649–658 (2003). https://doi.org/10.1007/s00125-003-1080-1
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1080-1