
Abstract
Key message
This study found that the compact spike locus of ANK-15 is on chromosome 5D instead of 2B. We have identified a new allele of AP2L-D5 as the candidate causal polymorphism.
Abstract
Spike architecture is a key determinant of wheat yield, a crop which supports much of the human diet but whose yield gains are stagnating. Spike architecture mutants offer opportunities to identify genetic factors contributing to inflorescence development. Here, we investigate the locus underlying the compact spike phenotype of mutant line ANK-15 by conducting mRNA-sequencing and genetic mapping using ANK-15 and its non-compact spike near-isogenic line Novosibirskaya 67 (N67). Previous literature has placed the compact spike locus of ANK-15 to chromosome 2B. However, based on the single nucleotide polymorphisms (SNPs) identified using mRNA-seq data, we were unable to detect polymorphisms between N67 and ANK-15 in the putative chromosome 2B region. We performed differential expression analysis of developing rachis and found that AP2L-D5, the D homoeolog of the domestication Q gene, is upregulated in ANK-15 in comparison to N67. ANK-15 carries a SNP in the microRNA172 binding site of AP2L-D5, which is predicted to lead to higher expression of AP2L-D5 due to decreased miRNA172-mediated degradation. Furthermore, we performed genetic mapping using an ANK-15 × N67 F2 population and found a single quantitative trait locus on chromosome 5D coinciding with the position of AP2L-D5. This result suggests that AP2L-D5 is likely the underlying causal gene for the compact spike phenotype in ANK-15. We performed a field trial to investigate the effect of the AP2L-D5 allele on agronomic traits and found that the AP2L-D5 allele from ANK-15 is associated with a significant reduction in height, increased thousand grain weight (TGW), and increased grain width.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) has played a crucial role in supporting human civilization as we rely on wheat for approximately 20% of our daily caloric and protein intake (FAO 2023). However, wheat yield increases have stagnated in recent decades, especially in major wheat-growing areas (Ray et al. 2012; Schauberger et al. 2018). Spike architecture is crucial to determine yield potential. Spike length, for example, has been shown to exhibit a high prediction ability for yield (Guo et al. 2018b). Identifying novel genetic factors regulating spike architecture can therefore provide insights into spike developmental processes and identify possible targets for yield improvement.
Several major loci which determine spike length and related spike morphological traits have been characterized. These include the Q gene on chromosome 5A and the compactum (C) locus on chromosome 2D. The dominant C allele is responsible for the morphology of club wheat, T. aestivum ssp. compactum, and has recently been fine-mapped to a 11 Mbp region from 231 to 242 Mb on the Chinese Spring reference genome (Kajla et al. 2023). Q has been molecularly characterized as a member of the APETALA2-like (AP2L) family of transcription factors (Simons et al. 2006) and is also known as AP2L-A5 (the A subgenome homoeolog of AP2L5; Debernardi et al. 2020). AP2L5 is a transcriptional repressor containing two AP2 protein domains, and its function as a transcriptional repressor is mediated by an ethylene-responsive element binding factor-associated amphiphilic repression (EAR) motif (Liu et al. 2018). AP2L genes are regulated post-transcriptionally by microRNA172 (miR172), which targets AP2L mRNAs for cleavage (Debernardi et al. 2017). Mutations in the miR172 binding site have been shown to lead to increased AP2L gene expression by reducing microRNA-mediated degradation of transcripts (Greenwood et al. 2017; Xu et al. 2018). A point mutation in the miR172 target site of AP2L-A5 led to the transition of the ancestral q allele, which conferred a ‘speltoid’ spike shape with an elongated rachis, to the modern Q allele ubiquitous in common wheat, which confers a subcompact square-shaped spike (Debernardi et al. 2017; Simons et al. 2006). In addition to controlling spike morphology, AP2L5 is known for conferring the free-threshing character and pleiotropically influences plant height, flowering time, and grain morphology (Debernardi et al. 2022; Simons et al. 2006; Xie et al. 2018). Multiple miR172 binding site alleles of AP2L5 have been generated with a range of phenotypic consequences. In addition to these major loci, other quantitative trait loci (QTLs) influencing spike length have been identified on several chromosomes (Liu et al. 2022; Wolde et al. 2019; Yu et al. 2022). However, few QTLs contributing to spike morphology have ultimately been cloned, suggesting that further genetic dissection is warranted.
Spike morphological mutants are useful resources for studying the mechanisms underlying inflorescence development as they represent deviations from the normal process of spike formation. To identify novel genes controlling spike length, we used near-isogenic lines (NIL) that have contrasting spike morphology. ANK-15 is near-isogenic to Novosibirskaya 67 (N67) and carries a locus that confers compact spike morphology (Koval et al. 1988). Amagai et al. (2016) showed that the compact spike locus of ANK-15 is not allelic to the C locus on chromosome 2D or the Q gene on chromosome 5A. Instead, Amagai et al. (2016) proposed the underlying gene to be allelic to a compact spike locus designated as Cg from the Japanese landrace, Nakate Gumbai, on the long arm of chromosome 2B.
In this study, we conducted mRNA-sequencing on the developing rachis of the ANK-15 NILs in combination with genetic mapping to identify the underlying gene. To our surprise, our analysis mapped the compact spike locus onto chromosome 5D instead of the previously proposed chromosome 2B. Differential gene expression and sequence analysis support that a polymorphism on the miRNA targeted site of AP2L-D5, the D genome homoeolog of Q, is the causal mutation. Furthermore, we carried out field evaluation of the NILs to characterize the phenotypic effects of the AP2L-D5 allele on spike morphology, plant architecture, phenology, and grain morphology.
Materials and methods
Plant materials and growth conditions
We developed an F2 mapping population derived from crossing near-isogenic lines ANK-15 and N67 (n = 207). Seeds were stratified at 4 °C for 48 h, then sown in John Innes Cereal Mix (65% peat, 25% loam,10% grit, 3 kg/m3 dolomitic limestone, 1.3 kg/m3 PG mix, 3 kg/m3 Osmocote Exact) in glasshouses at the John Innes Centre, Norwich, UK. Plants were grown under long-day (16-h light: 8-h dark) conditions at 21 °C and 16 °C during the day and night, respectively. Alongside the ANK-15 F2 population, we also grew the parental varieties N67 (n = 6 plants) and ANK-15 (n = 5 plants) for phenotypic comparison with the F2 progenies.
We grew N67 and ANK-15 at the Dorothea de Winton field station in Bawburgh, UK (52°37′50.7"N 1°10′39.7"E) during the 2023 growing season (sown 27 March 2023) to further characterize the phenotypic effects of the allele under field conditions. Each NIL was drilled in 1.3 m2 (1 m × 1.3 m) double-row plots with a row spacing of 0.17 m in randomized complete block design with six blocks. Plant growth regulator (3C Chlormequat 750; BASF) was applied to all plots 53 days after sowing at the rate of 0.6 L/ha.
Phenotyping
To phenotype the N67 × ANK-15 F2 population, we measured the spike length (SL), spikelet density (SD), spikelet number per spike (SNS), and plant height at maturity for the primary spike. We also visually scored plants as having either a non-compact (wild-type) or compact (ANK-15-like) spike morphotype. SL is the distance between the first basal spikelet node to the tip of the spike, excluding awns. SD is calculated as \(\frac{{Spikelet\;number\;per\;spike}}{{Spike\;length}}\). Plant height was measured as the distance between the soil and the tip of the tallest culm without awns.
In our field evaluation of the AP2L-D5 NILs, heading date was the number of days at which 75% of spikes in a plot had emerged. We assigned a lodging score to each plot at the end of the growing period based on lodging incidence, in which a score of 1 indicated high lodging and 9 indicated no lodging. Five representative primary spikes were selected for phenotyping SL, SNS, and SD in each of the six plots. In addition, we recorded architectural traits including the lengths of the primary tiller (plant height), stem (distance from base of culm to base of spike), and peduncle (base of spike to last node on stem). To assess the effects of the compact spike trait on grain morphology, we used a MARVIN grain analyzer (GTA Sensorik GmbH, Germany) to record average grain morphometric traits, including grain number and grain weight per spike, grain width, length, area, and thousand grain weight (TGW), of the five subsamples per plot.
Rachis transcriptome analysis of ANK-15 and N67
We collected and froze the rachises of four ANK-15 and four N67 spikes (one rachis per one plant) at green anther stage (Guo et al. 2018b) in liquid nitrogen and homogenized individual samples with a mortar and pestle. Using the Spectrum Plant Total RNA kit (Sigma), we isolated RNA from each sample according to the manufacturer’s protocol and performed On-Column DNAse I digestion (Sigma) to remove contaminating genomic DNA. To quantify the RNA concentration of each preparation, we used a NanoDrop 1000 spectrophotometer (Thermo Fisher). The mRNA-sequencing was performed by Novogene (Cambridge, UK) using the NovaSeq 6000 platform (Illumina, United States), generating paired-end 150 bp reads. We aligned these reads to the Chinese Spring genome RefSeq v1.0 assembly (IWGSC et al. 2018) using HiSAT2 (v2.1.0; Kim et al. 2019). Using SAMtools (v1.4.1; Danecek et al. 2021), the resulting alignments were converted to BAM files, sorted, and indexed, allowing visualization in the Integrative Genomics Viewer browser (IGV; Robinson et al. 2011).
To identify differentially expressed genes (DEGs) between the NILs, we quantified the abundance of transcripts with Kallisto (v0.48.0), using 30 bootstrap samples (Bray et al. 2016). Reads were pseudoaligned to a transcriptome index constructed using the IWGSC RefSeqv1.0 assembly and v1.1 annotation. Differential analysis of the quantified transcripts was then performed using the R package Sleuth (v0.30.1; Pimentel et al. 2017). We identified DEGs that function in infloresence development by using the web-based gene function discovery platform KnetMiner with the search term “inflorescence” (Hassani‐Pak et al. 2021). Gene ontology (GO) terms of DEGs were extracted with Ensembl BioMart (Kinsella et al. 2011) and analyzed in the PANTHER GO enrichment analysis tool on the Gene Ontology Resource webpage (Mi et al. 2013).
Genotyping
To identify polymorphisms between parental lines, we called variants from RNA-seq alignments using FreeBayes (v1.3.1; Garrison and Marth 2012) with a minimum alternate allele count of 5. We retained SNPs with a quality score of at least 1000 and a read depth of at least 40. SNPs on candidate chromosomes were selected for conversion to Kompetitive Allele-Specific PCR (KASP) markers using the PolyMarker (Ramirez-Gonzalez et al. 2015) primer design pipeline. The 5′ termini of allele-specific forward primers were labeled with the FAM (5′- GAAGGTGACCAAGTTCATGCT-3′) or HEX (5′-GAAGGTCGGAGTCAACGGATT-3′) tail sequence. All primers specific to nucleotide from N67 were FAM-compatible (Supplementary Table 4).
To genotype the N67 × ANK-15 F2 population, we conducted KASP assays in a 384-well format. For each marker, we prepared a primer assay mix consisting of both allele-specific forward primers (12 μM) and the common reverse primer (30 μM) (Supplementary Table 4). Each reaction consisted of 2 μL DNA (10–50 ng), 1.944 μL PACE Genotyping Master Mix (Standard ROX; 3CR Bioscience), and 0.056 μL primer assay mix. PCR was carried out according to the following conditions: initial denaturation and hot-start enzyme activation at 94 °C for 15 min; 10 touchdown cycles of template denaturation (94 °C, 20 s) followed by annealing/extension with decrease of 0.8 °C per cycle (65–57 °C, 60 s); and 40 cycles of amplification consisting of a denaturation step (94 °C, 20 s) followed by annealing/extension (57 °C, 60 s). Fluorescence of PCR products was detected with a PheraStar plate reader (BMG Labtech, Germany). We identified genotypes manually in KlusterCaller (version 3.4.1; LGC Biosearch Technologies, UK). Samples showing ambiguous clustering were subject to additional 5-cycle amplifications (94 °C, 20 s; 57 °C, 60 s) to improve separation of clusters.
Sequencing of the candidate gene AP2L-D5
After identifying AP2L-D5 as a possible candidate gene, we amplified its sequence in ANK-15 and N67 (Supplementary Table 5). PCR was carried out according to the conditions specified in the source publication of the primer sequences (Zhao et al. 2018). PCR products were purified using the Wizard SV Gel and PCR Clean-Up System (Promega) in line with the manufacturer’s protocol. Samples were sequenced using the amplicon sequencing service provided by Plasmidsaurus which used Oxford Nanopore sequencing.
Energy prediction of miR172-target interaction
To predict the binding affinities of the wild-type and mutant AP2L-D5 alleles with miR172, we used the RNAhybrid online web tool (Rehmsmeier et al. 2004) which computes the minimum free energy (MFE) hybridization of a pair of RNA molecules. The mRNA sequences of AP2L-D5 from N67 and ANK-15 and the sequence of wheat miR172 (Debernardi et al. 2017) were uploaded to the RNAhybrid server.
Quantitative Reverse Transcription PCR of candidate genes
To verify the results from RNA-seq, we compared the expression levels of the candidate gene and its homoeologs in rachises collected at yellow anther stage between NILs grown in the field (Guo et al. 2018b). Four biological replicates, consisting of one rachis from each of the four plots, were sampled from each genotype. We extracted and quantified RNA as above and synthesized first-strand complementary DNA (cDNA) from 1 μg of total RNA per sample using the SuperScript III First-Strand Synthesis System (Invitrogen). Each reaction consisted of 5 μL LightCycler 480 SYBR Green Master mix (Roche), 0.5 μL each of the forward and reverse primers (100 μM; Supplementary Table 5), 3 μL nuclease-free water, and 1 μL of diluted cDNA (1:5) for a total volume of 10 μL. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was selected as an endogenous reference gene for expression normalization.
For qRT-PCR, we performed three technical replicates and three no template control (NTC) reactions for each biological replicate. qRT-PCR was conducted on a LightCycler 480 Instrument II (Roche) under the following conditions: initial template denaturation at 95 °C (5 min); 45 amplification cycles, each consisting of denaturation at 95 °C (10 s), annealing at 62 °C (15 s), and extension at 72 °C (30 s); generation of a dissociation curve from 60 °C to 95 °C. Upon completion of the qRT-PCR, we verified that the amplified products were target-specific through melting curve analysis. To analyze the relative expression levels of the target genes, we employed the 2−ΔΔCT method (Livak and Schmittgen 2001) and compared gene expression between N67 and ANK-15 using Student’s t-tests.
QTL mapping and statistical analysis
QTL analyses were conducted using the R/qtl package (v1.60; Broman et al. 2003) in R (v4.3.0; R Core Team, 2023). We calculated conditional genotype probabilities and marker recombination frequencies in the F2 populations, following Haldane’s mapping function. Assuming a single QTL, we conducted standard interval mapping using the scanone function with the expectation–maximization algorithm. We employed the normal model for data with a normal distribution and the non-parametric model for data that failed the Shapiro–Wilk test.
Statistical analyses of phenotypic data were performed in R. To determine whether parametric statistical tests were appropriate, we conducted Shapiro–Wilk tests to assess the normality of phenotypic data. We conducted one-way analysis of variance (ANOVA) tests to evaluate the effect of AP2L-D5 alleles on the traits measured. Tukey’s Honestly Significant Difference (HSD) test was implemented to further identify pairwise differences between the genotypes.
For the field evaluation of AP2L-D5b, we assessed the phenotypic effects of the genotype and the blocking factor (plot) by aggregating the five subsamples taken from each plot to obtain a plot mean for each phenotype and performed two-way ANOVAs including genotype and block as factors. To evaluate the effect of genotype and block when a non-parametric test was required, we conducted permutational two-way ANOVA based on 5000 permutations using the aovp() function from the R package lmPerm (v2.1.0; Wheeler and Torchiano 2016).
Results
The AP2L-D5 allele of ANK-15 is overexpressed and has a mutated microRNA172 (miR172) binding site.
ANK-15 is near-isogenic to Novosibirskaya 67 (N67), and has a compact spike locus introduced from a mutant line of wheat cultivar Skala (Fig. 1a; Amagai et al. 2016; Koval et al. 1988). Recent work suggested that the compact spike phenotype mapped to chromosome 2B (Amagai et al. 2016; Koval et al. 1988). To identify the gene underlying the ANK-15 compact spike phenotype, we conducted mRNA sequencing of the developing rachises of N67 and ANK-15 at green anther stage. We chose this stage as this is where most of the spike elongation occurs in wheat (Guo et al. 2018a). We therefore hypothesized that the causal gene for the compact spike phenotype would be differentially expressed between NILs at this stage.

AP2L-D5 of ANK-15 contains a single nucleotide polymorphism in the miR172 binding site. a Representative image of N67 and ANK-15 spikes grown in the glasshouse. b Intron–exon structure of AP2L-D5 of N67 and ANK-15. Both 5′ and 3′ untranslated regions are colored in grey, exons are depicted as black boxes, introns as black lines, AP2 protein domains are highlighted in blue, EAR binding motifs are highlighted in green and the miR172 binding site is highlighted in red. Mismatches to miR172 are indicated in red font. The putative causal SNP leading to the compact spike phenotype from ANK-15 is shown in bold red font and is proposed as the AP2L-D5b allele
To check the isogenic status between N67 and ANK-15, we identified polymorphisms by aligning the RNA-seq dataset against the Chinese Spring genome (RefSeq v1.0; IWGSC et al. 2018). On average, we obtained 119 million reads per sample with 91.9% of the reads aligned to RefSeqv1.0 (Supplementary Table 1). As expected, we detected limited polymorphisms between ANK-15 and N67 supporting that the two lines are near-isogenic (Supplementary Fig. 1). There were limited polymorphisms (68 out of 1978 detected polymorphisms; 3.4%) identified on chromosome 2B. We inspected the putative interval on chromosome 2B (487 to 685 Mbp) but did not identify any polymorphisms in that interval with the closest detectable SNP about 100 Mbp away. We also identified a large polymorphic region extending across the short arm of chromosome 5A between ANK-15 and N67 (1063 out of 1978 detected polymorphisms; 53.7%).
To identify candidate genes for the compact spike phenotype of ANK-15, we conducted differential expression analysis using the RNA-seq dataset and identified 157 differentially expressed genes (DEGs) between the NILs (q < 0.05), 93 of which were high-confidence genes (Supplementary Table 2). We performed GO enrichment analysis for the high-confidence gene set; however, we found no significant enrichment for any gene ontology (Supplementary Table 3). To identify genes associated with spike development, we used KnetMiner, which is a gene discovery tool that infers gene function based on publicly available datasets (Hassani‐Pak et al. 2021). We found that 66 out of 93 genes have functions involved in inflorescence development or are predicted to have interactions with genes of such function. Surprisingly, this includes AP2L-D5 (TraesCS5D02G486600), the D subgenome homoeolog of Q. This gene was found to be the underlying gene of RHT23 (REDUCED HEIGHT 23) and influences spike compactness and plant height (Zhao et al. 2018). This gene was upregulated in ANK-15 by 2.3-fold in the developing rachis in comparison to N67. We further performed quantitative real-time PCR (qRT-PCR) to confirm the difference in AP2L-D5 expression between N67 and ANK-15 using samples collected from the field. We found that AP2L-D5 was significantly upregulated (P < 0.05) in ANK-15 compared to N67 within the rachis (Supplementary Fig. 2). There was however no significant difference in relative expression of the B homoeolog (P = 0.37; TraesCS5B02G486900) and a borderline non-significant effect of the A homoeolog (P = 0.06; TraesCS5A02G473800). Furthermore, the AP2L-D5 allele of ANK-15 is different from N67 by a G to A transition on exon 10. We designated the wild-type AP2L-D5 allele of N67 as AP2L-D5a (same as AP2L-D5 in Chinese Spring) and the ANK-15 allele carrying the point mutation as AP2L-D5b based on the wheat gene nomenclature guidelines (Boden et al. 2023).
We aligned the sequences of AP2L-D5 in N67 and ANK-15 and found that the G/A polymorphism of ANK-15 was within the miR172 binding site (Fig. 1b). As mismatches to microRNA binding site are known to limit the efficiency of target mRNA cleavage, we assessed the effect of the SNP by comparing the predicted energy of interactions between miR172 and the mRNA sequences of AP2L-D5a and AP2L-D5b in RNAhybrid (Rehmsmeier et al. 2004). The minimum free energy (MFE) of the interaction between miR172 and AP2L-D5a was predicted to be − 38.8 kcal/mol, while for AP2L-D5b this was − 32.6 kcal/mol. The increase in the energy of interactions between miR172 and its mutated binding site on AP2L-D5b indicates a decrease in the binding affinity. We hypothesize that the G/A SNP in AP2L-D5b reduces miR172-mediated degradation of the transcript, leading to higher expression of the gene in the rachis. A different point mutation in the AP2L-D5 miR172 binding site present in the mutant line NAUH164 (we refer to this allele as AP2L-D5c; Supplementary Fig. 3) has previously been shown to increase spike compactness in addition to pleiotropically reduce plant height (Zhao et al. 2018). Given the known connection between spike compactness and polymorphisms in the miR172 binding site of Q, we hypothesize that AP2L-D5 is the underlying gene for the compact spike phenotype of ANK-15.
The compact spike locus of ANK-15 maps to chromosome 5D not chromosome 2B
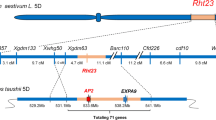
To test the hypothesis that AP2L-D5 is the candidate gene, we performed QTL analysis using a N67 × ANK-15 F2 population grown in the glasshouse. As they are near-isogenic, we only developed KASP markers for three targeted regions based on the SNP data (Supplementary Fig. 1). First, we developed KASP marker 5D:521,713,197 to distinguish AP2L-D5a and AP2L-D5b in addition to other markers spanning chromosome 5D (Supplementary Table 4). Second, ANK-15 was proposed to be allelic to a compact spike QTL interval (Cg) from a Japanese landrace, Nakate Gumbai, between 487 and 685 Mbp on the long arm of chromosome 2B (based on BLAST location of the SSR markers; Amagai et al. 2016). However, there were limited polymorphisms on chromosome 2B (Supplementary Fig. 1) so we were only able to develop three KASP markers at around 770 Mbp. Lastly, we found that ANK-15 and N67 were polymorphic on chromosome 5A (Supplementary Fig. 1). We therefore developed markers from 176 to 539 Mbp on chromosome 5A using the identified polymorphisms.
The compact spike morphology from ANK-15 has a high density of spikelets in the apical region of the spike giving it a “squarehead” morphology (Fig. 1a; Supplementary Fig. 4). Based on this, plants in the N67 × ANK-15 F2 population (n = 207 plants) could be visually distinguished into two distinct spike morphotypes of 140 compact spike to 67 non-compact spike lines (Supplementary Fig. 4). Amagai et al. (2016) considered the compact spike trait of ANK-15 to be dominant; however, a Chi-square goodness-of-fit test indicated that our observed phenotypes differed (p = 0.0144) from the expected 3:1 (155:52) ratio of compact to non-compact lines. The distributions of spike length, spikelet density, and height were normal in the F2 populations, indicating that these traits might be controlled by multiple genes in the F2 population.
We phenotyped the plants for spike length, spikelet density and spikelet number to perform QTL analysis. Consistent with our hypothesis, we found significant associations between markers on chromosome 5D with spike morphology and height (Fig. 2b; Supplementary Fig. 5). The marker that distinguishes between AP2L-D5a and AP2L-D5b (5D:521,713,197) had the highest LOD value for spike morphological traits. F2 lines homozygous with the ANK-15 allele (AP2L-D5b; 7.9 cm) at this marker were associated with shorter spikes than F2 lines homozygous for the N67 allele (AP2L-D5a; 10.2 cm). This effect was additive with heterozygous lines showing an intermediate phenotype (9.0 cm). Marker 5D:521,713,197 explained 44.86% of variance in spike length. This marker is also significantly associated with spikelet number and spikelet density (Supplementary Fig. 5). The ANK-15 allele is associated with slightly lower number of spikelets but is still associated with higher spikelet density due to the significant reduction in spike length. The allele at AP2L-D5 also co-segregated with the spike morphotypes. Among the 205 F2 plants that were successfully genotyped using marker 5D:521,713,197 (genotyping for two plants had failed), all 56 AP2L-D5a homozygous lines had non-compact spikes, while all 51 AP2L-D5b homozygous lines had compact spikes. Of the heterozygous lines, 87 had compact spikes, while 11 were classified as non-compact. Co-segregation of the 5D:521,713,197 SNP with spike morphotype supports that AP2L-D5b is the gene underlying the ANK-15 compact spike locus. We also found that there were no significant associations with spike morphology traits for markers on chromosome 2B and 5A (Fig. 2). These were the regions where the compact spike locus was proposed to be and the highly polymorphic region between N67 and ANK-15, respectively. Our genetic data thus further supports that the compact spike locus for ANK-15 is on chromosome 5D and not chromosome 2B as previously proposed, and that AP2L-D5 is the likely causal gene.

The compact spike length locus of ANK-15 maps to chromosome 5D in the N67 × ANK-15 F2 population. a Physical maps of chromosomes 2B, 5A and 5D and the KASP markers designed for genetic mapping. The markers are named after the physical position of the polymorphisms between the parents anchored onto RefSeq v1.0 (IWGSC et al. 2018). Black circles denote approximate centromere position. Putative position of the compact spike locus Cg according to Amagai et al. 2016 is colored in purple. The KASP marker that separates AP2L-D5a and AP2L-D5b is highlighted in yellow (5D:521,713,197). b QTL analysis for spike length. Genetic positions of the markers are denoted with ticks and connected to their physical locations in a). The dashed red line denotes the significance threshold estimated based on permutations (n = 1000). Inset Spike length distribution of the F2 individuals (n = 207 plants) carrying parental or heterozygous alleles for KASP marker that separates AP2L-D5a and AP2L-D5b. Genotype groups significantly different from each other based on post-hoc Tukey test (P < 0.01) are denoted with different letters
Zhao et al. (2018) identified AP2L-D5 as the candidate gene for the REDUCED HEIGHT 23 (RHT-23) locus in NAUH164, an EMS mutant from Sumai 3. NAUH164 also carries a G to A point mutation within the miR172 binding site of AP2L-D5, although this is 10 bp upstream from the mutation that we have identified (Supplementary Fig. 3). The NAUH164 allele, which we proposed be called AP2L-D5c, led to significant reductions in plant height. We therefore hypothesize that AP2L-D5b can also influence plant height. In the F2 mapping population, we found that F2 lines with AP2L-D5b allele are associated with a significant reduction in height of 4.0 cm (Supplementary Fig. 5). This suggests that AP2L-D5b has pleiotropic effects on height in addition to spike morphology.
AP2L-D5b has pleiotropic effects on agronomic traits under field conditions
The near-isogenic nature of N67 and ANK-15 was confirmed by the low number of SNPs across most chromosomes (Supplementary Fig. 1). Therefore, we carried out a field trial to investigate the pleiotropic effects of AP2L-D5b on agronomic traits, spike and grain morphology.
Agronomic traits: Similar to what we observed in the F2 population, AP2L-D5b is associated with a decrease in plant height under field conditions (Table 1). This decrease in plant height is in part due to a significant decrease in peduncle length. This indicated that the difference in height is not solely due to the reduction in spike length and that stem elongation is also reduced by AP2L-D5b. Despite ANK-15 being 5.2 cm shorter in the field, we did not observe a difference in lodging score between the NILs. We also noticed a significant effect of AP2L-D5 alleles on heading time where AP2L-D5b is associated with a 2 day delay in heading.
Spike morphology: Similar to what was observed in the F2 population grown in the glasshouse, AP2L-D5 alleles have significant effects on spike length and spikelet density. AP2L-D5b in ANK-15 was associated with a decrease in spike length and increase in spikelet density (Table 1). Interestingly, AP2L-D5b was associated with an increase in spikelet number under field conditions but was associated with a decrease in spikelet number under our glasshouse conditions (Table 1, Supplementary Fig. 5).
Grain morphology: AP2L-D5 has a significant impact on grain width where AP2L-D5b is associated with wider grains than AP2L-D5a (Fig. 3; Table 1). This effect however was not observed for grain length (P = 0.17). The increase in grain width translated to an increase in thousand grain weight (TGW; P = 0.02; Table 1) in the AP2L-D5b NILs. However, the increase in TGW is compensated with a decrease in the number of grains per spike as measured on the primary spike. Overall, AP2L-D5 alleles, in addition to affecting spike morphology, have pleiotropic effects on agronomic traits and grain morphology.

Representative grains of N67 and ANK-15 to illustrate the effect of AP2L-D5 alleles on grain morphology
Discussion
Identifying the genes involved in spike development can reveal targets for yield improvement. In this study, we identified an allele of AP2L-D5 with a novel SNP in the miR172 binding site. Similar to the mutation in AP2L-A5 that gave rise to the Q allele, the SNP in AP2L-D5b leads to increased expression which we attribute to reduced transcript degradation due to the expected decreased binding affinity for miR172. Several natural and induced miR172-resistant mutants have been identified for Q (Debernardi et al. 2017; Greenwood et al. 2017; Liu et al. 2020; Xu et al. 2018). However, only one miR172 binding site mutant, NAUH164, has been previously reported for AP2L-D5 (Zhao et al. 2018). Our results provide greater insight into the role of miR172-mediated regulation of AP2L-D5 as well as the possible breeding utility of this novel allele.
ANK-15 carries a compact spike locus introduced from a mutant line of wheat cultivar Skala. Recent work on ANK-15 proposed that the locus is allelic to a novel compact spike locus, Cg, from the Japanese landrace Nakate Gumbai (Amagai et al. 2016). However, we did not detect any polymorphisms in the putative region between N67 and ANK-15, which are near-isogeneic lines developed using phenotypic selection (Koval et al. 1988; Supplementary Fig. 1). In addition, genetic mapping of the F2 population of N67 × ANK-15 found no significant association between the markers on chromosome 2B and spike morphology (Fig. 2b). Instead, we found significant association between the markers on chromosome 5D with spike morphology, showing that the compact spike locus of ANK-15 is on chromosome 5D. By using mRNA-seq, we found that AP2L-D5 is expressed at a higher level in ANK-15 in comparison to N67 (Supplementary Table 2; Supplementary Fig. 2). ANK-15 carries a point mutation on the miR172 binding site of AP2L-D5 which introduces a mismatch between miR172 and its binding site (Fig. 1). Study has shown that mismatch between miR172 and its binding site limit its cleave efficiency resulting in higher transcript level of its target (Debernardi et al. 2017) which we have observed in ANK-15 in both the RNA-seq and qRT-PCR (Supplementary Fig. 2). A different mutation that introduced a mismatch in the miR172 binding site of AP2L-D5 (AP2L-D5c; Supplementary Fig. 3) also led to compact spike (Zhao et al. 2018) and the same mutation as ANK-15 but on the A subgenome homoeolog (Q gene) also led to compact spike morphology (Greenwood et al. 2017). AP2L-D5b has a dosage-dependent effect on spike length and height (Supplementary Fig. 5). The AP2L-D5c allele of NAUH164 is similarly additive, as heterozygous F2 lines were intermediate for these traits (Zhao et al. 2018). Similarly, in the F2 progeny of a cross between a miR172 binding site mutant of AP2L-A5 and its wild-type cv. Sunstate, Greenwood et al. (2017) observed that heterozygotes had intermediate spike compactness and height. Based on the strong connection between AP2L5 genes and spike morphology, we propose that AP2L-D5 is likely the underlying gene of the compact spike morphology of ANK-15.
As the wheat spike is determinate, spikelet number per spike (SNS) is limited early in development by the number of axillary meristems formed before the terminal spikelet stage. Delaying the terminal spikelet stage may therefore lead to higher spikelet numbers, albeit often with an associated delay in flowering time. Greenwood et al. (2017) found an increase in rachis node number (which are the nodes that individual spikelets are attached to) with increased AP2L-A5 expression. Conversely, SNS decreased in ap2l5 null mutants generated by Debernardi et al. (2020) and in a Q truncation mutant identified by Zhang et al. (2022). These results indicate that AP2L5 can delays formation of the terminal spikelet leading to higher SNS. Consistent with these results, our field-grown ANK-15 plants with increase AP2L-D5 expression exhibited greater number of spikelets in comparison with N67 (Table 1). However, AP2L-D5b was associated with a decrease in SNS in F2 lines grown in the glasshouse. These discrepancies may be explained by differences between glasshouse and field conditions. Uniform temperatures and photoperiod in the glasshouse may have promoted greater uniformity and faster floral development across the F2 genotypes.
A delay in terminal spikelet formation is one component of altered phenology caused by increased AP2L5 expression. The ap2l5 null mutants developed by Debernardi et al. (2020) as well as transgenic miR172 overexpression lines (Debernardi et al. 2017) flowered earlier than their wild-type lines, which is reflected in our field evaluation by the delayed heading exhibited by ANK-15 (Table 1). Through differential expression analysis, we found that TERMINAL FLOWER1 (TFL1; TRAESCS5A02G128600), a floral repressor, was upregulated in ANK-15 during spike development, suggesting it may be regulated (directly or indirectly) by AP2L-D5 (Supplementary Table 2). TFL1, a phosphatidylethanolamine-binding protein, regulates inflorescence meristem identity and is a known negative regulator of flowering time in Arabidopsis (Simon et al. 1996as). Yant et al. (2010) demonstrated that TFL1 was downregulated in ap2 mutants. The transcription factor AP1, a target of AP2, directly represses TFL1 activity (Kaufmann et al. 2010), which may underlie the upregulation of TFL1 in AP2 family overexpression mutants such as ANK-15. However, the association between TFL1 and AP2L5 expression in wheat has not been characterized to date. Our results suggests that it would be of interest to further explore if AP2L5 regulates meristem transition through TFL1.
Although an increase in spikelet number due to delayed terminal spikelet formation may be expected to increase grain number per spike, our grain morphometric analysis revealed that ANK-15 spikes had a significantly lower average grain number per spike compared to N67 (Table 1). These results suggest that AP2L-D5 might influence the distribution of yield components, as the reduction in grain number corresponded to an increase in TGW and grain width (Table 1; Fig. 3). Our ANOVA results also suggest that AP2L-D5b may positively influence grain area. Xie et al. (2018) demonstrated that the transformation from q to Q caused by a miR172 binding site SNP, led to increased grain width and TGW. Larger grain size is beneficial not only from a yield perspective but is also associated with increased early vigor (Zhao et al. 2019). In addition, milling yield is positively correlated with grain size (Marshall et al. 1986). Hence, the enhancement of grain width represents a potential way in which this novel allele may contribute to variety improvement. Interestingly, the miR172-resistant AP2L-D5 allele of NAUH164 conferred a reduction in both TGW and grain number per spike (Chen et al. 2015; Zhao et al. 2018), suggesting that different polymorphisms in the miR172 binding site may differ in their pleiotropic effects on yield and quality-related traits. The difference between the studies can also be attributed to the difference in genetic background and environment in which these studies were conducted.
Beyond inflorescence development and grain morphology, AP2L-D5b also has pleiotropic effects on plant height (Table 1). The mechanism by which AP2L5 genes affect height has not been fully characterized. Patil et al. (2019) proposed that miR172-mediated HvAP2 degradation in barley ultimately facilitates gibberellin (GA)-promoted stem elongation, finding that a miR172-resistant mutant showed inhibited growth responses to GA. Several semi-dwarfing loci in use since the Green Revolution, such as Rht-B1b and Rht-D1b, encode truncated DELLA proteins that constitutively repress GA responses (Thomas 2017; Van De Velde et al. 2021). The use of these GA-insensitive semi-dwarfing alleles leads to reduction in internode elongation, but the disruption of the GA signal transduction pathway also confers undesirable pleiotropic effects on seedling emergence and growth due to coleoptile shortening (Allan 1980; Tang et al. 2009). Therefore, alternative semi-dwarfing genes are currently sought after (Borrill et al. 2022). While exogenous application of GA to the miR172-resistant NAUH164 does not lead to the restoration of wild-type height, an alpha amylase assay demonstrated that the GA signaling pathway was not disrupted in the mutant at the seed stage (Chen et al. 2015). This may indicate that GA insensitivity caused by AP2L5 overexpression could be constrained to the stem elongation process. Although further insight into the relationship between AP2 genes and the GA pathway is needed, miR172-resistant alleles such as AP2L-D5b may present alternative sources of height reduction that avoid the limitations of the Rht1 alleles caused by constitutive GA insensitivity. The effect of AP2L5 alleles on spikelet density might be a concern for mechanical harvest but production of club wheat with similar spike structure, to the best of our knowledge, has not been linked with harvest issues.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author.
References
Allan RE (1980) Influence of semidwarfism and genetic background on stand establishment of wheat1. Crop Sci 20:634–638. https://doi.org/10.2135/cropsci1980.0011183X002000050022x
Amagai Y, Kuboyama T, Watanabe N (2016) Microsatellite mapping of the gene conferring compact spike in Japanese “Gumbai” landraces of common wheat (Triticum aestivum L.). Euphytica 209:709–714. https://doi.org/10.1007/s10681-016-1661-y
Boden SA, McIntosh RA, Uauy C, Krattinger SG, Dubcovsky J, Rogers WJ, Xia XC, Badaeva ED, Bentley AR, Brown-Guedira G et al (2023) Updated guidelines for gene nomenclature in wheat. Theor Appl Genet 136(4):72. https://doi.org/10.1007/s00122-023-04253-w
Borrill P, Mago R, Xu T, Ford B, Williams SJ, Derkx A, Bovill WD, Hyles J, Bhatt D, Xia X, Macmillan C, White R, Buss W, Molnár I, Walkowiak S, Olsen O-A, Doležel J, Pozniak CJ, Spielmeyer W (2022) An autoactive NB-LRR gene causes Rht13 dwarfism in wheat. Proc National Acad Sci 119(48):e2209875119. https://doi.org/10.1073/pnas.2209875119
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. https://doi.org/10.1038/nbt.3519
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890. https://doi.org/10.1093/bioinformatics/btg112
Chen S, Gao R, Wang H, Wen M, Xiao J, Bian N, Zhang R, Hu W, Cheng S, Bie T, Wang X (2015) Characterization of a novel reduced height gene (Rht23) regulating panicle morphology and plant architecture in bread wheat. Euphytica 203:583–594. https://doi.org/10.1007/s10681-014-1275-1
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H (2021) Twelve years of SAMtools and BCFtools. GigaScience 10(2):giab008. https://doi.org/10.1093/gigascience/giab008
Debernardi JM, Lin HQ, Chuck G, Faris JD, Dubcovsky J (2017) microRNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 144:1966–1975. https://doi.org/10.1242/dev.146399
Debernardi JM, Greenwood JR, Jean Finnegan E, Jernstedt J, Dubcovsky J (2020) APETALA 2-like genes AP2L2 and Q specify lemma identity and axillary floral meristem development in wheat. Plant J 101:171–187. https://doi.org/10.1111/tpj.14528
Debernardi JM, Woods DP, Li K, Li C, Dubcovsky J (2022) MiR172-APETALA2-like genes integrate vernalization and plant age to control flowering time in wheat. PLoS Genet 18:e1010157. https://doi.org/10.1371/journal.pgen.1010157
FAO (2023) Food Balances (2010-). In: FAO (ed), Rome
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv. https://doi.org/10.48550/arXiv.1207.3907
Greenwood JR, Finnegan EJ, Watanabe N, Trevaskis B, Swain SM (2017) New alleles of the wheat domestication gene Q reveal multiple roles in growth and reproductive development. Development 144:1959–1965. https://doi.org/10.1242/dev.146407
Guo Z, Zhao Y, Röder MS, Reif JC, Ganal MW, Chen D, Schnurbusch T (2018a) Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat. Sci Rep-Uk 8(1):14435. https://doi.org/10.1038/s41598-018-31977-3
Guo ZF, Chen DJ, Schnurbusch T (2018b) Plant and floret growth at distinct developmental stages during the stem elongation phase in wheat. Front Plant Sci 9:330. https://doi.org/10.3389/fpls.2018.00330
Hassani-Pak K, Singh A, Brandizi M, Hearnshaw J, Parsons JD, Amberkar S, Phillips AL, Doonan JH, Rawlings C (2021) KnetMiner: a comprehensive approach for supporting evidence-based gene discovery and complex trait analysis across species. Plant Biotechnol J 19:1670–1678. https://doi.org/10.1111/pbi.13583
IWGSC, Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ, Stein N, Choulet F, et al. (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 361(6403), eaar7191. https://doi.org/10.1126/science.aar7191
Kajla A, Schoen A, Paulson C, Yadav IS, Neelam K, Riera-Lizarazu O, Leonard J, Gill BS, Venglat P, Datla R, Poland J, Coleman G, Rawat N, Tiwari V (2023) Physical mapping of the wheat genes in low-recombination regions: radiation hybrid mapping of the C-locus. Theor Appl Genet 136(7):159. https://doi.org/10.1007/s00122-023-04403-0
Kaufmann K, Wellmer F, Muiño JM, Ferrier T, Wuest SE, Kumar V, Serrano-Mislata A, Madueño F, Krajewski P, Meyerowitz EM, Angenent GC, Riechmann JL (2010) Orchestration of floral initiation by APETALA1. Science 328:85–89. https://doi.org/10.1126/science.1185244
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. https://doi.org/10.1038/s41587-019-0201-4
Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, Kersey P, Flicek P (2011) Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database-Oxford. https://doi.org/10.1093/database/bar030
Koval SF, Metakovsky EV, Sozinov AA (1988) A series of near-isogenic spring bread wheat lines on the basis of the variety Novosibirskaya 67. Cereal Research Communications 16:183–187
Liu P, Liu J, Dong H, Sun J (2018) Functional regulation of Q by microRNA172 and transcriptional co-repressor TOPLESS in controlling bread wheat spikelet density. Plant Biotechnol J 16:495–506. https://doi.org/10.1111/pbi.12790
Liu H, Wang K, Tang H, Gong Q, Du L, Pei X, Ye X (2020) CRISPR/Cas9 editing of wheat TaQ genes alters spike morphogenesis and grain threshability. J Genet Genomics 47:563–575. https://doi.org/10.1016/j.jgg.2020.08.004
Liu H, Shi Z, Ma F, Xu Y, Han G, Zhang J, Liu D, An D (2022) Identification and validation of plant height, spike length and spike compactness loci in common wheat (Triticum aestivum L.). Bmc Plant Biol 22:568. https://doi.org/10.1186/s12870-022-03968-0
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Marshall DR, Mares DJ, Moss HJ, Ellison FW (1986) Effects of grain shape and size on milling yields in wheat. 2. Exp Stud Aust J Agr Res 37:331–342. https://doi.org/10.1071/Ar9860331
Mi H, Muruganujan A, Casagrande JT, Thomas PD (2013) Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 8:1551–1566. https://doi.org/10.1038/nprot.2013.092
Patil V, McDermott HI, McAllister T, Cummins M, Silva JC, Mollison E, Meikle R, Morris J, Hedley PE, Waugh R, Dockter C, Hansson M, McKim SM (2019) APETALA2 control of barley internode elongation. Development 146(11):dev170373. https://doi.org/10.1242/dev.170373
Pimentel H, Bray NL, Puente S, Melsted P, Pachter L (2017) Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14:687–690. https://doi.org/10.1038/nmeth.4324
Ramirez-Gonzalez RH, Uauy C, Caccamo M (2015) PolyMarker: a fast polyploid primer design pipeline. Bioinformatics 31:2038–2039. https://doi.org/10.1093/bioinformatics/btv069
Ray DK, Ramankutty N, Mueller ND, West PC, Foley JA (2012) Recent patterns of crop yield growth and stagnation. Nat Commun 3:1293. https://doi.org/10.1038/ncomms2296
Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R (2004) Fast and effective prediction of microRNA/target duplexes. RNA 10:1507–1517. https://doi.org/10.1261/rna.5248604
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26. https://doi.org/10.1038/nbt.1754
Schauberger B, Ben-Ari T, Makowski D, Kato T, Kato H, Ciais P (2018) Yield trends, variability and stagnation analysis of major crops in France over more than a century. Sci Rep-Uk 8:16865. https://doi.org/10.1038/s41598-018-35351-1
Simon R, Igeño MI, Coupland G (1996) Activation of floral meristem identity genes in Arabidopsis. Nature 384:59–62. https://doi.org/10.1038/384059a0
Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai Y-S, Gill BS, Faris JD (2006) Molecular characterization of the major wheat domestication gene Q. Genetics 172:547–555. https://doi.org/10.1534/genetics.105.044727
Tang N, Jiang Y, He B-r, Hu Y-g (2009) The effects of dwarfing genes (Rht-B1b, Rht-D1b, and Rht8) with different sensitivity to GA3 on the coleoptile length and plant height of wheat. Agri Sci China 8:1028–1038. https://doi.org/10.1016/S1671-2927(08)60310-7
Thomas SG (2017) Novel Rht-1 dwarfing genes: tools for wheat breeding and dissecting the function of DELLA proteins. J Exp Bot 68:354–358. https://doi.org/10.1093/jxb/erw509
Van De Velde K, Thomas SG, Heyse F, Kaspar R, Van Der Straeten D, Rohde A (2021) N-terminal truncated RHT-1 proteins generated by translational reinitiation cause semi-dwarfing of wheat green revolution alleles. Mol Plant 14:679–687. https://doi.org/10.1016/j.molp.2021.01.002
Wheeler RE, Torchiano M (2016) Permutation tests for linear models in R. R package version 2
Wolde GM, Trautewig C, Mascher M, Schnurbusch T (2019) Genetic insights into morphometric inflorescence traits of wheat. Theor Appl Genet 132:1661–1676. https://doi.org/10.1007/s00122-019-03305-4
Xie Q, Li N, Yang Y, Lv Y, Yao H, Wei R, Sparkes DL, Ma Z (2018) Pleiotropic effects of the wheat domestication gene Q on yield and grain morphology. Planta 247:1089–1098. https://doi.org/10.1007/s00425-018-2847-4
Xu B-J, Chen Q, Zheng T, Jiang Y-F, Qiao Y-Y, Guo Z-R, Cao Y-L, Wang Y, Zhang Y-Z, Zong L-J, Zhu J, Liu C-H, Jiang Q-T, Lan X-J, Ma J, Wang J-R, Zheng Y-L, Wei Y-M, Qi P-F (2018) An overexpressed Q allele leads to increased spike density and improved processing quality in common wheat (Triticum aestivum). G3: Genes Genomes, Genetics 8(3):771–778. https://doi.org/10.1534/g3.117.300562
Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, Chen X, Schmid M (2010) Orchestration of the floral transition and floral development in arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 22:2156–2170. https://doi.org/10.1105/tpc.110.075606
Yu Q, Feng B, Xu Z, Fan X, Zhou Q, Ji G, Liao S, Gao P, Wang T (2022) Genetic dissection of three major quantitative trait loci for spike compactness and length in bread wheat (Triticum aestivum L.). Front Plant Sci 13:882655. https://doi.org/10.3389/fpls.2022.882655
Zhang J, Xiong H, Guo H, Li Y, Xie X, Xie Y, Zhao L, Gu J, Zhao S, Ding Y, Liu L (2022) Identification of the Q gene playing a role in spike morphology variation in wheat mutants and its regulatory network. Front Plant Sci 12:807731. https://doi.org/10.3389/fpls.2021.807731
Zhao K, Xiao J, Liu Y, Chen S, Yuan C, Cao A, You FM, Yang D, An S, Wang H, Wang X (2018) Rht23 (5Dq′) likely encodes a Q homeologue with pleiotropic effects on plant height and spike compactness. Theor Appl Genet 131:1825–1834. https://doi.org/10.1007/s00122-018-3115-5
Zhao Z, Rebetzke GJ, Zheng B, Chapman SC, Wang E (2019) Modelling impact of early vigour on wheat yield in dryland regions. J Exp Bot 70:2535–2548. https://doi.org/10.1093/jxb/erz069
Acknowledgements
We thank the JIC Field Experimentation and Horticultural Services teams for their support in glasshouse and field experiments. We thank Dr. Clare Lister on her guidance on lodging testing in the field. We thank Professor Nobuyoshi Watanabe for supplying the germplasm.
Funding
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC) through grant BB/S016945/1 and the Institute Strategic Programmes Delivering Sustainable Wheat (DSW) (BB/X011003/1) and Building Robustness in Crops (BRiC) (BB/X01102X/1). Additional funding was provided by the European Research Council (866328). YC was supported by the John Innes Foundation and Natural Sciences and Engineering Research Council of Canada (NSERC).
Author information
Authors and Affiliations
Contributions
Conceptualization: YC; Data curation: VZ, YC; Formal analysis: VZ, YC; Funding acquisition: CU; Investigation: VZ, YC; Methodology: VZ, YC; Project administration: YC, CU; Supervision: YC, CU; Visualization: VZ, YC; Writing-original draft: VZ, YC; Writing-review & editing: VZ, YC, CU.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Xianchun Xia.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, V., Uauy, C. & Chen, Y. Identification of a novel SNP in the miR172 binding site of Q homoeolog AP2L-D5 is associated with spike compactness and agronomic traits in wheat (Triticum aestivum L.). Theor Appl Genet 137, 13 (2024). https://doi.org/10.1007/s00122-023-04514-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04514-8