
Abstract
Key message
Map-based cloning, subcellular localization, virus-induced-gene-silencing and transcriptomic analysis reveal HvTUB8 as a candidate gene with pleiotropic effects on barley spike and leaf development via ethylene and chlorophyll metabolism.
Abstract
Barley lateral spikelet morphology and grain shape play key roles in grain physical quality and yield. Several genes and QTLs for these traits have been cloned or fine mapped previously. Here, we report the phenotypic and genotypic analysis of a barley mutant with round lateral spikelet (rls) from cv. Edamai 934. rls had round lateral spikelet, short but round grain, shortened awn, thick glume and dark green leaves. Histocytologic and ultrastructural analysis revealed that the difference of grain shape of rls was caused by change of cell arrangement in glume, and the dark leaf color resulted from enlarged chloroplast. HvTUBULIN8 (HvTUB8) was identified as the candidate gene for rls by combination of RNA-Seq, map-based-cloning, virus-induced-gene-silencing (VIGS) and protein subcellular location. A single G-A substitution at the third exon of HvTUB8 resulted in change of Cysteine 354 to tyrosine. Furthermore, the mutant isoform Hvtub8 could be detected in both nucleus and cytoplasm, whereas the wild-type protein was only in cytoplasm and granular organelles of wheat protoplasts. Being consistent with the rare phenotype, the “A” allele of HvTUB8 was only detected in rls, but not in a worldwide barley germplasm panel with 400 accessions. VIGS confirmed that HvTUB8 was essential to maintain spike integrity. RNA-Seq results suggested that HvTUB8 may control spike morphogenesis via ethylene homeostasis and signaling, and control leaf color through chlorophyll metabolism. Collectively, our results support HvTUB8 as a candidate gene for barley spike and leaf morphology and provide insight of a novel mechanism of it in barley development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare L.) is the fourth largest cereal crop worldwide. As a staple food of ancient civilizations, domesticated from Hordeum vulgare subsp. Spontaneum ~ 10,000 years ago (Harlan et al. 1973; Zohary and Hopf 2000), cultivated barley is now used mainly for animal feed and malting. Selection of varieties with high yield potential is always the ultimate goal for barley breeding. Inflorescence architecture is one of the key factors that affect grain yield in cereal crops, such as rice, maize, barley and wheat (Wang et al. 2021a). Among them, barley and wheat usually form typical compact and branchless “spike” inflorescences, characterized by sessile spikelets produced directly from alternating, opposing nodes on the branchless main axis or rachis, which is specific to the tribe Triticeae, while inflorescences of maize and rice have highly branched tassels or panicles (Koppolu et al. 2013). Moreover, being different from its close relative wheat, barley inflorescence comprises a multi-node central stalk (rachis) with tripartite clusters of unifloretted spikelets attached alternately along its length. This distinctive triple spikelet arrangement arises when each inflorescence node forms a triple spikelet meristem (TSM) that cleaves into a large central spikelet meristem (CSM) bordered by two smaller lateral spikelet meristems (LSMs) (Zwirek et al. 2019). Relative fertility of lateral spikelets within each cluster leads to spikes with two or six row of spike, or an intermediate morphology. Two-rowed spikes bear a single grain from a fertile central spikelet flanked by two sterile lateral spikelets within each rachis node, while all three spikelets are fertile and develop into grains in six-rowed barley (Ramsay et al. 2011a). The two-rowed state is ancestral, being found in the wild progenitor of cultivated barley (Hordeum vulgare ssp. Spontaneum), where the sterile spikelets form part of the seed dispersal mechanism (Zohary and Hopf 2000).
Up to date, five major genes, namely SIX-ROWED SPIKE (VRS) genes, controlling lateral spikelet (LS) development has been identified by forward genetics, including VRS1, VRS2, VRS3 (syn. INTERMEDIUM-A), VRS4 (syn. INTERMEDIUM-E) and VRS5 (syn. INTERMEDIUM-C). VRS1 on chromosome 2H encodes a homeodomain-leucine zipper class I protein (HD-ZIP1) (He et al. 2004; Komatsuda et al. 2007). VRS2 mutant can develop supernumerary spikelets at its base and occasionally enlarged and fertile LSs toward the center of the spike, but almost two-rowed-like, sterile LSs at its tip. VRS2 is on the long arm of chromosome 5H, encoding a homologue of the Arabidopsis SHORT INTERNODES (SHI) transcriptional regulator (Youssef et al. 2017). Barley two-rowed (in the lower portion of the spike) and six-rowed (in the upper part) spikes-controlled gene VRS3 encodes a putative Jumonji C-type H3K9me2/me3 demethylase, which is on the short arm of chromosome 1H (Bull et al. 2017). Apart from lateral spikelet fertility, vrs4 mutants show indeterminate triple spikelet meristems, thereby producing additional spikelets/florets. VRS4 is on the short arm of chromosome 3H and encodes a LATERAL ORGAN BOUNDARIES (LOB) transcription factor orthologous to the maize RAMOSA2 gene, which prevents ectopic branching in maize ears and tassels (Koppolu et al. 2013). VRS5 on short arm of chromosome 4H encodes a class II TCP transcription factor whose homologues in maize, rice, wheat and Arabidopsis repress tiller bud outgrowth to promote apical dominance (Ramsay et al. 2011b). The re-sequence analysis of HvMADS-box family genes in 30 spike-contrasting barley lines revealed that HvMADS56 also played an important role in lateral spikelet development (Sayed et al. 2021).
Grain shape is another essential factor for barley grain physical quality and yield. In wheat, a single amino acid substitution in STKc_GSK3 kinase conferred semispherical grains of Triticum sphaerococcum (Cheng et al. 2020). A number of QTLs for barley grain shape determinants (grain length, grain width, grain thickness) had been mapped throughout the whole genome (Fang et al. 2020; Wang et al. 2019a, 2021b). Some major QTLs have been fine mapped, such as qGL5H (Watt et al. 2019) and qGL2H (Watt et al. 2020) for grain length locus. However, the genetic and biological basis of round grain development still remains largely unknown.
Typically, barley has long awns, but short-awn variants exist. Awn plays an important role in seed dispersal, protection as well as photosynthesis and yield (Abebe et al. 2009; Elbaum et al. 2007), whereas, awnless and short awn cultivars are likely to perform better under extreme conditions such as drought (Rebetzke et al. 2016). Given the potential impact of awn length on barley yield, a number of QTLs affect this trait have been identified on barley genome, both from modern cultivars (Chen et al. 2012; Gyenis et al. 2007) and wild accessions (Liller et al. 2017). Three awnless-specific genes have so far been cloned in barley. One was Knox3 transcription factor for the hood-shaped awn (Müller et al. 1995), and mutation of the SHI-family transcription factor lks2 produced awns that were only a half of the normal ones (Yuo et al. 2012), while vrs1 was allelic to the reduced lateral spikelet appendage (lr) locus, characteristic of having normal and long awns on the central spikelets and awnless on the lateral spikelets (Komatsuda et al. 2007). Genetic analysis further revealed that the loci/genes underling barley awn trait diversity could interact with each other (Huang et al. 2020).
Previously, a mutant library of Edamai 934 (referred to E934 in this study) was constructed by combined 60Co-γ and EMS treatment. E934 is a two-rowed feeding barley variety bred by Hubei Academy of Agricultural Sciences (Hubei, China) (Qin et al. 2021). The barley mutant Round Lateral Spikelets (rls), which was identified from the E934 mutant library, exhibits dark leaf color, round lateral spikelet, round grain, shortened awn and abnormal glume. Here, we identified the causal gene through positional cloning. A G-A nucleotide substitution in HvTUB8 gene may contribute to the pleiotropic changes on barley spike and leaf morphology in rls. The current study will not only lay a foundation for unraveling the function and mechanisms of HvTUB8 in spike and leaf development, but also be useful for barley breeding using rls and HvTUB8 as a resource.
Materials and methods
Plant materials
Rls mutant was identified from 60Co-γ and EMS induced E934 mutant library in our previous work, which was mainly characterized as a mutant with abnormal spike morphology. rls was crossed with Yan 03174, a typical two-rowed barley variety, which was bred by Yancheng Academy of Agricultural Sciences (Jiangsu, China). Genetic characterization of the gene (s) controlling the rls phenotype was analyzed based on the spike morphology of F1 hybrid, F2 population and F2:3 families. Chi-squared test was used to determine the suitability of observed data with expected segregation ratios. All of the field work was conducted in the experimental station of Hubei Academy of Agricultural Sciences (Wuhan, China). Relative chlorophyll concentration in the fully developed leaf on the main tiller of rls and E934 was measured using SPAD-502 at the booting stage (Fig. 1a). Average of five leaves for rls and E934 were measured and compared. Dried seeds of rls and E934 were subjected to micro-CT scanning (SkyScan 1275, Bruker) to evaluate differences in three dimensions.
Scanning electron microscopy
Immature spike (3 mm to 5 mm) and seedling leaves from rls and E934 were pre-fixed with 2.5% glutaraldehyde in phosphate buffer (PH = 7.0) at 4 ℃ overnight and further treated as described in our previous work (Qin et al. 2015). The specimens were observed under a Hitachi HT7700 scanning electron microscope (Hitachi, Japan).
Plant softening paraffin embedding experiment
Intact glumes of E934 and rls were picked from the middle of the spike when it extracted from flag leaf completely. Then glumes were fixed in FAA at 4 ℃ overnight. Subsequent softening, dehydration, paraffin embedding, splicing and statistical analysis were conducted by Servicebio® (Wuhan, China) according to the standard procedure.
BSA based on RNA-Seq (BSR)
Young leaves of 60 mutant type individuals and 60 wild-type individuals were picked up from 60 heterozygous F2:3 families and pooled together to construct the mutant pool and wild-type pool, respectively. RNA was extracted using TRIzol (Rio et al. 2010). RNA-Seq was carried out on an Illumina HiSeq 2500 platform following the manufacturer’s protocol. Both RNA extraction and RNA-Seq were conducted by Beijing Novogene Bioinformatics Technology Co. Ltd. Raw RNA-Seq reads were assessed for quality control by Trimmomatic v 0.32 (Bolger et al. 2014) after removing adapter sequences and low-quality bases. Adapter sequences were removed by identifying the pairing part between the two paired-end reads, followed by trimming bases that were not paired at read ends. Low-quality bases at read ends whose quality values (Phred-value) were lower than 3 were trimmed, and low-quality bases in the middle of each read were identified by a 10 bps sliding window method (mean Phred-value < 15 in the window) (Wang et al. 2017a, b). High-quality reads were aligned to the barley cv. Morex reference genomic sequence (2016 version 1) (Mascher et al. 2017). After alignments, the mapping results were filtered using an in-house Perl script. Only uniquely mapped reads with Phred-value quality larger than 40 were kept. Variant calling and subsequent identification of variants possibly linked to the target gene was carried using the Haplotype Caller module in GATK v3.2–2 as previously described in wheat (Wang et al. 2017a, b; Zhang et al. 2020). Briefly, the read count information for each allele per variant was obtained from the GATK output. Then, a Fish exact test was applied on read counts for each variant site (Indels and SNPs) between the two bulks to identify variants possibly linked to the target gene. Additionally, the allele frequency for each variant and the allele frequency difference (AFD) between the two bulks were calculated. Variants with p < 1e−10 and AFD > 0.6 were considered to be putatively target gene linked (Wang et al. 2017a, b; Hua et al. 2020).
DNA isolation
Genomic DNA of E934, rls and Yan 03174 was extracted from young leaves using TaKaRa MiniBEST Universal Genomic DNA Extraction Kit following the manufacture’s instruction. Genomic DNA of the F2, F3 and F4 plants derived from Yan 03174 and rls was extracted from young leaves using CTAB method.
Genotyping based on Penta-primer amplification refractory mutation system (PARMS)
Indel and SNP markers were developed according to the BSR analysis. These markers were firstly used to screen Yan 03174 and rls. Then polymorphic markers were used to genotype the F2, F3 and F4 plants derived from Yan 03174 and rls based on PARMS (Lu et al 2020). All PARMS analysis was conducted by Gentides Biotech Co., Ltd (Wuhan, China). Five μl PCR system contained 2.5 μl 2 × PARMS PCR mix, 150 nM each allele-specific primer, 400 nM locus-specific primer, and 1.4 μl alkaline lysis DNA template. The thermal cycler program included denaturing at 95 °C for 15 min and then 10 cycles of denaturation at 95 °C for 20 s and annealing started at 65 °C for 1 min and then decreasing 0.8 °C per cycle to the annealing temperature at 57 °C. This was followed by 32 cycles of denaturation at 95 °C for 20 s and annealing at 57 °C for 1 min. The well plate was read using a TECAN infinite M1000 plate reader; SNP calling and plots were carried out by an online software snpdecoder (http://www.snpway.com/snpdecoder/) combining manual modification. Linkage analysis was conducted using Mapmaker 3.0 and a LOD score threshold of 3.0 (Lander et al. 1987).
Analysis of linked markers and cloning of candidate genes
Sequences of flanking markers were conducted by BLASTN analysis online (https://galaxy-web.ipk-gatersleben.de/) (MorexV3 assembly) (Mascher et al. 2021) to get the updated genomic region containing the gene. High-confident genes in the target region were extracted according to MorexV3 assembly. Several gene-specific overlapped primers were designed based on the genomic sequence of these genes using DNAMAN5.0 software. Specific PCR products from genomic DNA of E934, rls and Yan 03174 were sequenced and compared with each other. cDNA from 5 to 10 mm young panicle of rls and E934 was used to clone CDS of HvTUB8. 1000bps upstream region of the candidate gene was also sequenced from E934 and rls according to the reference genomic sequence of cv. Morex. All the PCRs were conducted using TaKaRa Premix TaqTM (Japan). 20 μl PCR included 2 × TaKaRa Premix 10 μl, 10 μM Forward primer 0.4 μl, 10 μM Reverse primer 0.4 μl, DNA template 10–100 ng, PCR water several μl. PCR mixtures were subjected to Touch-down PCR program (95 °C 5 min; ten cycles of 95 °C 30 s, 62–57 °C (− 0.5 °C /cycle) 45 s, 72 °C 1–2 min; 35 cycles of 95 °C 30 s, 57 °C 45 s; 72 °C 1–2 min; 72 °C 5 min) on ABI VeritiPro PCR machine. Specific PCR products with expected size were subjected to Sanger sequencing. Secondary structure of proteins was predicted by DNAMAN5.0.
Phylogenetic analysis of HvTUB8 homologue genes
Homologue genes of HvTUB8 in other gramineae species were downloaded from NCBI according to BLASTP results. Accession numbers of other barley β-TUBULIN genes were extracted according to BLASTN search in database “Barley all CDS Morex V3.0 (Jul 2020)” (https://galaxy-web.ipk-gatersleben.de/), and then, sequences were downloaded from “BARLEX” (https://apex.ipk-gatersleben.de/apex/f?p=284:10::::::). Sequences of OsTUB proteins were also extracted from NCBI according to their accession numbers (Yoshikawa et al. 2003). Alignment and phytogenetic tree of these HvTUB8 homologue genes was constructed based on their protein sequences using DNAMAN5.0.
Spatio-temporal expression of HvTUB8 in barley
Quantitative Real-Time PCR was performed to analyze expression of HvTUB8 in different organs and tissues of E934, including leaf at three-leaf stage (seedling leaf); internode below the spike (internode); young panicle (0.5–1 cm, > 1 cm); glume, stamen, pistil, awn and flag leaf at heading stage; grain at 10 DAP (10DAP). Gene-specific primers for HvTUB8 are shown in Supplementary Table S1. Barley gene Actin 1 was used as an endogenous control.
Genetic variation of HvTUB8 in barley accessions
Allelic variation of HvTUB8 was analyzed in the 20 accessions according to pan-genome data (Jayakodi et al. 2020), including 1000 bps upstream and downstream regions as well as coding region of B1K-04-12 (FT11), HOR 21599, HOR7552, HOR 13942, OUN333, HOR 3365 (HHOR), HOR 10350, ZDM02064 (CHIBA), HOR 9043, ZDM01467 (DULIHUANG), HOR 3081, HOR 13821, Morex, HOR 8148, Akashinriki, Igri, Hockett, Barke, RGT Planet and Golden Promise (Jayakodi et al. 2020). Genotyping of the gene-specific maker was also conducted on a barley natural population consisting of 400 landraces and modern cultivars with typical two-rowed or six-rowed spikes from worldwide based on PARMS, including China, Australia, Canada, USA and so on.
Barley stripe mosaic virus (BSMV) virus-induced gene silencing (VIGS)
BSMV-VIGS was used for functional characterization of HvTUB8 in E934. Specific primers for HvTUB8 (Supplementary Table S1) were designed to amplify the target fragments of 296 bps region in the first exon, which was inserted into the digested BSMV-γ genome to generate the recombinant BSMV:HvTUB8 construct. Infection with a BSMV-TaPDS control indicated infection and gene-silencing efficiency by bleaching of leaf color. Following inoculation and quantification were conducted as described before (Shang et al. 2020). Six plants of E934 were infected at two-leaf seedling stage for both BSMV:HvTUB8 and BSMV control. When bleaching was visible in BSMV-TaPDS-infected plants, Real-time PCR was conducted to analyze the expression of the target gene in the newly developed leaves after BSMV:HvTUB8 infection as mentioned above.
Subcellular localization analysis
Complete ORF of wild-type HvTUB8 in E934 and the variant in rls (designed as Hvtub8 in this study) was amplified with gene-specific primers (Supplementary Table S1), and the product was digested by Eco31I and cloned into the expression cassette of modified pCAMBIA1302 (Biorun, Wuhan, China) to generate pCAMBIA1302-35S::HvTUB8-eGFP and pCAMBIA1302-35S::Hvtub8-eGFP vectors, respectively. The construction was confirmed by enzyme digestion and sequencing. The confirmed constructs and vector alone were transformed into wheat protoplasts via co-cultured with PEG4000 (Yoo et al. 2007). The GFP signal was observed by Confocal Laser Scanning Fluorescence Microscopy after 18–24-h incubation in dark (LSFM, Carl Zeiss).
Transcriptome analysis
Immature spikes from rls and E934 at glume primordium (GP) to lemma primordium (LP) formation stage (about 0.5–1 cm) were collected and frozen in liquid nitrogen and then stored at − 80 ℃, three biological repeats for both E934 and rls. Total RNA extraction, library construction and RNA-seq were performed by Beijing Novogene Bioinformatics Technology Co. Ltd (Beijing, China) as mentioned above. The clean reads of each sample were mapped to MorexV3 assembly (Mascher et al. 2021). Transcript quantification from RNA-Seq data was analyzed using the Salmon (1.9.0) tool. Expression levels of mapped reads were quantified as Transcripts Per Kilobase of exon model per Million mapped reads (TPM) (Patro et al. 2017). The Bioconductor package DESeq2 (Love et al. 2014) was used to perform differential expression analysis. Differential expression genes (DEGs) between rls and E934 were defined as |log2 fold change (LFC)|≥ 1.5, adjusted P ≤ 0.05. GO Enrichment analysis of DEGs was carried out online (http://wheat.cau.edu.cn/TGT/) (Chen et al. 2020). GO terms with a corrected FDR of < 0.05 were considered to be significantly enriched. The percentage of DEGs for each GO term was calculated with reference to all transcripts in MorexV3 assembly. Expression of the genes in ethylene and chlorophyll pathways was illustrated by indoor perl script.
Results
Phenotypic analysis of barley rls mutant
The mutant rls was initially identified from screening of 60Co-γ and EMS-treated mutant library in the genetic background of E934. Leaf color of rls was much darker than that of E934 (Fig. 1a) during the whole life, and chlorophyll concentration reflected by SPAD was 29.03 ± 1.11 and 52.53 ± 3.25 in E934 and rls at the booting stage, respectively. Besides, rls was mainly characterized as a mutant with abnormal spike morphology (Fig. 1b). Lateral spikelets of rls were round and sterile. In addition, awn of rls was very short, glume was harder, bilateral empty glumes were also much shorter but a little bit wider than wild type (Fig. 1c–e). Grains of rls were round and shorter than wild type, but there was no significant difference on grain width between rls and E934 (Fig. 1f). Consequently, thousand grain weight of rls was only 32.26 g, being much lower than 45.86 g of E934. Moreover, outer glume section around the embryo in dried seeds of rls (166.40 μm) was much thicker than that in E934 (108.00 μm) (Fig. 1g). rls was also characterized as dwarf, plant height of which was about 60% of wild type and heading date of rls was almost 14 days later than E934.

Seedling and spike morphology of rls. a Seedling of E934 and rls; b Wild-type (MM), medium mutant (mM) type and mutant (mm) type spikes; c central spikelets; d lateral spikelet; e bilateral empty glumes; f, g: seeds of E934 and rls
To find out which stage was crucial for the differentiation of rls spike morphology, 3 mm and 5 mm young panicles of E934 and rls were scanned under electron microscope. It was found that there was no difference between 3 mm spikes of E934 and rls. However, for 5 mm spikes, rls awn was longer than E934, especially the awn at the bottom (Fig. 2a, b), though awn of rls was much shorter than E934 at maturity stage. Despite these differences, lateral spikelets clearly increased in size with enlarged lemmas enclosing differentiating floral organs in rls as compared with E934 (Fig. 2c, d). What is more, to determine whether dark green leaf color of rls was associated with development of chloroplast, ultrastructure of chloroplast of E934 and rls leaves at booting stage was also investigated under scanning electron microscope. Consistently, chloroplast size was increased in rls as compared with that in E934, almost half of the cell being occupied by chloroplasts in rls (Fig. 2e–h).

Immature spike and ultrastructure of chloroplast from E934 and rls. a, b 5-mm spike of E934 and rls; c, d lateral spikelet of E934 and rls; e, f ultrastructure of leaf cell from E934 and rls; g, h chloroplast of E934 and rls; i, j: horizontal section of lemma from E934 and rls (bar = 100 μm). SC, silicified cells; OPC, outer parenchyma cells; IPC, inner parenchyma cells
To further reveal the cellular basis of round grain in rls mutant, cross section of hull in E934 and rls was observed. Significant differences of cell morphology were observed between E934 and rls both longitudinally and horizontally. As shown in Fig. 2, a regular pattern of three cell layers including silicified cells (SC), outer parenchyma cells (OPC) and inner parenchyma cells (IPC) was observed in WT (Fig. 2i, j). There was no significant difference between SC of E934 and rls; however, a remarkable difference was observed between OPC and IPC between them. Wider empty region in the OPC layer but a relative bigger and regular IPC was observed both in the lemmas and paleas from rls as compared with that from E934, which may contribute to the thicker glumes and round grains in rls (Fig. 1g).
Genetic dissection of rls mutant underling gene
To decipher genetic feature of the causal gene, rls was crossed with Yan 03174, a typical two-rowed barley variety with long thin and sterile lateral spikelet, and also long awn, to obtain the F1 seeds. It was found that spikes of the F1 plants showed intermedium type, awns of which were long and same as wild-type plants (MM), but were tighter than wild-type awns. Lateral spikelets of F1 plants were fatter than wild type and crooked, but bilateral empty glumes of lateral spikelets were the same as rls (mm) (Fig. 1b–e). Individuals with the same phenotype as F1 plants were recorded as “mM” in this study (Fig. 1b–e). While the F2 population showed a segregation of the MM, mM and mm type spikes, which included 268, 462 and 286 plants, respectively. Chi-test showed that the number of MM, mM and mm type plants in the F2 population fitted a ratio of 1:2:1. Moreover, all the F2:3 families derived from MM type F2 individuals had homozygous wide type spikes, all the F2:3 families derived from mM type F2 individuals showed a segregation of three types spikes, while all the F2:3 families derived from mm type F2 individuals had homozygous mutant type spikes, which confirmed that this changed spike morphology in rls was controlled by a single semi-dominant gene, and was designed as HvRLS in this study.
Preliminary mapping of HvRLS candidate gene via BSA based on RNA-Seq
To map the causal gene for rls phenotype, BSA based on RNA-Seq was initially conducted using mutant pool and wild-type pool derived from 60 heterozygous F2:3 families (progenies of mM type F2 individuals). Totally 494 SNPs/Indels with p < 1e−10 and AFD > 0.6 were identified as potential linkage with the phenotype, half (246) of which could be anchored to the end of long arm of chromosome 4H (Totally 647.06Mbps), falling into the interval of 619Mbp-646Mbp (MorexV1 assembly) (Fig. 3a). However, only 23, 40, 67, 42, 32 and 40 SNPs/Indels were on chromosome 1H, 2H, 3H, 5H, 6H and 7H, respectively, indicating that HvRLS gene was on chromosome 4H.

Map-based cloning of HvRLS. a Initial mapping; b, c fine mapping; d phenotype and genotype of recombinant lines. Note: A, B and H in Fig. 3d stand for homologous wild-type allele MM, homologous mutant allele mm and heterozygous allele mM, respectively
To validate the BSR analysis and fine map the gene, totally 15 polymorphic markers (Supplementary Table S1) in the target region were developed according to the BSR-seq results. These markers were then used to screen the genotypes of rls and Yan 03174 as well as the F3 and F4 plants derived from mM type F2 and F3 individuals, respectively. Based on PARMS, totally 505 mm, 16 mM and 12 MM type plants from F3 and F4 populations were genotyped and analyzed. Finally, HvRLS gene was mapped between 4H-6375 and 4H-60210 (Fig. 3b, c), which was located at 601,621,013 bp and 602,104,747 bp on chromosome 4H according to MorexV3 assembly (Mascher et al. 2021), spanning about 480 kb. Out of the 533 F3 and F4 individuals, six and four recombinant events were identified for 4H-6375 and 4H-60210, respectively (Fig. 3d). The most interesting was that the genotypic data of 4H-60205, representing a single nucleotide polymorphism (SNP) G/A, co-segregated with MM, mM and mm phenotype in the segregate populations (Fig. 3d). The co-segregation was also confirmed in the F2 population derived from rls and Yan 03174, including 268 MM, 462 mM and 286 mm individuals.
Cloning of rls candidate gene
Further analysis showed that there were 17 high-confident genes (Table 1) in this 480 kb target region. It was very interesting that the G to A substitution represented by 4H-60205 was within a β-TUBULIN gene. Further BLAST analysis showed that this β-TUBULIN gene was the previously annotated HvTUB8. According to the updated barley genome, genomic sequence and coding region of HvTUB8 was 1589 bps and 1344 bps in length, respectively, which had three exons and encoded a polypeptide composed of 447 amino acid residues harboring the conserved “TUBULIN beta chain” domain (1–430/447), and the G-A variation was on the third exon (+ 1061) of HvTUB8 (Fig. 4a).

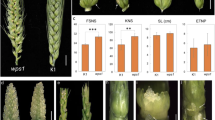
Sequence, expression and functional analysis of HvTUB8. a Gene structure of HvTUB8 (Red indicates the G-A and C-Y substitution on the third exon); b phylogenetic analysis of HvTUB8 and its homologue genes; c partial alignment of HvTUB8 and its homologue genes (red star indicates the C-Y substitution); d spatio-temporal expression of HvTUB8 in E934; e spikes of BSMV:HvTUB8-infected and non-infected E934 (WT) (color figure online)
To validate the co-segregated G-A substitution, full length coding regions of HvTUB8 were cloned from genomic DNA and cDNA of young panicles of E934 and rls subsequently. Both of the sequences from gDNA and cDNA confirmed the G to A mutation in rls, while no other variation was detected in coding region between E934 and rls. Moreover, there was no difference between the sequence of 1000bps upstream region from E934 and rls either, whereas coding sequences and their expression in immature spikes of the other 16 genes showed no difference between E934 and rls. Further study exhibited that the cysteine (C) at amino acid position 354 of HvTUB8 was replaced by a tyrosine (Y) in rls due to the G-A substitution (Fig. 4a).
Phylogenetic analysis of β-TUBULIN proteins in plant
BLASTP and BLASTN of the wide-type HvTUB8 protein was then conducted online, and homologue proteins from barley and its relatives, such as Triticum dicoccoides, Triticum aestivum, Aegilops tauschii, Oryza sativa and Zea mays, were downloaded. As shown in Fig. 4b, these proteins were highly conserved among different species, which showed 95% to 100% similarity to each other. Phylogenetic analysis indicated that HvTUB8 belonged to a mini cluster that only included members from Triticum and its ancestor (Fig. 4b). HvTUB8 protein showed 100% and 96.89% similarity to its counterparts from Triticum dicoccoides (XP_037437345.1) and Oryza sativa (OsTUB2), respectively. Though the Cys to Try substitution mentioned above located in a region that was not quite conserved as compared with other regions, the wide type Cys was highly conserved among barley and its relatives, the substitution could only be detected in rls (Fig. 4c).
Spatial and temporal expression of HvTUB8 in E934
Then spatial and temporal expression profile of HvTUB8 was analyzed in 10 tissues of E934 at different developmental stages. Result showed that expression pattern of HvTUB8 was highly tissue-specific. It mainly expressed in young panicles and leave, and a relative lower expression was also detected in stamen and internode below the spike at the heading stage (Fig. 4d). However, it was hardly detected in other tissues.
Haplotype analysis of HvTUB8 gene in barley accessions
The co-segregated maker QDD4H-60205, which represented the G to A mutation in HvTUB8, was also applied to screen a panel of 400 barley accessions collected from different countries with typical two- or six-rowed type spikes. As expected, all of these accessions had the “G” type allele as E934. To further explore the natural diversity of HvTUB8, genetic variation of the gene was analyzed in the 20 barley accessions together with E934 using the barley pan-genome data. Coding region from the start codon to stop codon was extracted from 19 accessions except for “Igri”, with nine bps deletion at the very beginning of the gene. It was also found that all these 20 accessions had the same allele (G) as E934 at + 1061. Additionally, a total of 14 other SNPs were detected in coding region of HvTUB8, which resulted in seven haplotypes (Supplementary Table S2) in the 20 accessions, while HvTUB8 in E934 was distinctive and different from all the seven haplotypes. In addition to coding region, 1000 bps upstream regions of HvTUB8 were analyzed between these 19 accessions except for HOR3365. Abundant variations were also detected in 1000 bps upstream of this gene. However, none of these haplotypes was associated with barley row type.
Functional characterization of HvTUB8 by BSMV-VIGS
To confirm the participation of HvTUB8 in barley spike development, we used BSMV-VIGS to silence the gene in wild-type E934 plants. Control infections with BSMV indicated that viral infection did not affect spike morphology of barley, and with BSMV-TaPDS indicated infection and gene-silencing efficiency, as shown by bleached lines on infected leaves. Expression analysis of the target gene in leaves of these BSMV:HvTUB8-infected plants suggested that expression of HvTUB8 was reduced about 70% as compared with in non-transformed E934. As shown in Fig. 4e, irregular spikes were consistently observed in the four survived plants infected with BSMV:HvTUB8, and extra spikes were produced along the main spike, which was different from the known barley branched spikes. However, no other difference, like grain shape, awn length, leaf color was observed in BSMV:HvTUB8-infected E934 plants as compared with the non-treated E934. Spikes of the plants infected by BSMV control showed no difference with the wide type. The results suggested that the wild-type HvTUB8 gene is essential for maintaining the integrity of barley spike.
Subcellular localization of HvTUB8 and Hvtub8 proteins
To demonstrate the subcellular localization of HvTUB8 protein, both the full-length HvTUB8 and the mutated Hvtub8 were fused with GFP reporter and expressed in wheat protoplasts. As shown in Fig. 5, in the positive control cells expressing 35S::GFP protein only, GFP signal was observed throughout the cells, but the single amino acid change of HvTUB8 caused change of subcellular localization of the protein in wheat protoplasts. In the cells expressing 35S::HvTUB8:GFP protein (Fig. 5e–h), a weak signal was detected in cytoplasm, and a strong signal was detected in a few of granular organelles. However, in the cells expressing the Hvtub8 fused GFP protein, a strong green florescence signal was detected in both the nucleus and cell membrane.

Subcellular localization of HvTUB8 protein. 35S::eGFP indicates the expression of GFP protein in wheat protoplasts as the negative control. 35S::HvTUB8-eGFP indicates the cytoplasm localization of wild-type HvTUB8 protein in wheat protoplasts. Green is GFP signal. Bright is the bright light. Merged indicates the confused with the green signal and the bright light. Bars: 10 μm
Transcriptome profiling reveals the downstream genes regulated by HvRLS
To interrogate the downstream signaling pathways regulated by HvRLS and their contributions to barley development, RNA-Seq was performed on 0.5–1 cm young panicles of rls and E934 with three biological replicates. Totally, 40,892 expressed genes were detected with 441 genes being differentially expressed (DEG) in rls relative to E934, but none of the 17 genes (including HvTUB8) within the mapping interval showed DEG between E934 and rls, and more than three quarters (340) of these differentially expressed genes (DEGs) were up-regulated by the mutation of HvRLS (Supplementary Table S3).
Then, expressions of known barley lateral spikelet development regulating genes, including VRS1, VRS2, VRS3, VRS4 and VRS5, were further analyzed, but none of them were altered in rls as compared with in wild type. Two LOB domain-containing protein genes on 3H (HORVU.MOREX.r3.3HG0233680) and 4H (HORVU.MOREX.r3.4HG0414380) were up-regulated by threefold and 7.6 fold, respectively, and a Homeobox-leucine zipper family protein (HORVU.MOREX.r3.2HG0189890) was also up-regulated by four folds in rls. However, the two LOB domain-containing genes only showed 30.35% and 33.16% similarity to VRS4/HvRA2 in the coding region, while the Homeobox-leucine zipper family protein showed low level (40.55%) of similarity to VRS1, respectively.
Further GO enrichment analysis of the 441 DEGs showed that about half of the DEGs (213) could be anchored to known GO terms (Supplementary Table S4). Totally, 34, 5 and 1 GO terms were enriched for Biological Process, Molecular Function and Cellular Component, respectively. It was found that phytohormones homeostasis was extensively disturbed in rls. Notably, a 1-aminocyclopropane-1-carboxylate oxidase gene and a 1-aminocyclopropane-1-carboxylate synthase gene in “ethylene biosynthetic process”, together with 12 of the 14 ethylene-responsive transcription factors in “ethylene-activated signaling pathway” were significantly up-regulated in rls (Fig. 6). Additionally, genes responsive to other phytohormones were also enriched in DEGs, including salicylic acid, brassinosteroid and gibberellic acid, and most of them were transcription factors, such as WRKY and zinc finger proteins. Moreover, the RNA-Seq data also witnessed a large amount of other transcription factors being regulated by mutation of HvTUB8, such as NAC, F-box, MYB, CRT-binding factors, bHLH, MADS-box and so on. The majority of them were up-regulated in rls. On the other hand, expressions of 32 genes encoding various protein kinases (LRR, WAK etc.) were changed in rls, with 27 and 5 being up-regulated and down-regulated, respectively.

Expression of DEGs in ethylene and chloroplast pathways
Considering the difference of leaf color and ultrastructure of chloroplast of E934 and rls, chlorophyll biosynthesis and metabolism-related genes were analyzed subsequently. It was found that expressions of 11 genes participating in chloroplast synthesis and metabolism were changed by the mutation of HvRLS in rls, nine of which were also up-regulated (Fig. 6).
Discussion
rls is a rare and distinctive resource for barley study
Functional characterization of genes and genetic mechanisms involved in barley inflorescence performance has been well elucidated since the identification of VRS1, which was a negative regulator of barley lateral spikelet development. At least five VRSs (VRS1-5) have been identified since then. Variation at VRS1 was sufficient to control the formation of two-rowed or six-rowed barley (Ramsay et al. 2011b), while induced loss function of alleles of any of the other genes could cause complete to intermediate gains of lateral spikelet fertility (Zwirek et al. 2019). Recently, HvAP2L-H5 was identified to be required for barley inflorescence indeterminacy and spikelet determinacy (Zhong et al. 2021). Except for the vrs mutants mentioned above with partial or completed sterility of the lateral spikelet, several intermedium-spike barley mutants were also identified with significant different lateral spikelet morphology and size (Franckowiak and Lundqvist 2012; Youssef et al. 2020). For example, spike of int-f appeared six-rowed, but the lateral spikelets were small and pointed (Gustafsson and Lundqvist 1980). Most of these barley row-type-related genes have pleiotropic effects on shoot branching, grain number per spike and thousand grain weight, either background dependent or independent (Liller et al. 2015). rls in this study was mainly characterized as a mutant in spike morphology, grain shape, plant height and leaf color. Particularly, lateral spikelets of rls were completely sterile but much shorter and fatter than wild-type ones, which were different from those known vrs mutants and were described as “round” in the present study. Though the pointed lateral spikelets were also observed in some intermedium mutants, rls has pleiotropic variations of spike, grain, stem, leaf and awn, suggesting that the underling gene was different in controlling barley development. Therefore, identification and functional characterization of the causal gene will be important to fully understand the complex network in barley spike, grain and leaf development, and thus ultimately provide targets for understanding and exploiting natural or induced genetic diversity toward improving barley yield potential and grain physical quality.
HvTUB8 as a candidate gene for rls phenotype
Based on BSR and fine mapping, HvRLS was anchored within a 480 kb interval with 17 annotated genes on chromosome 4H. The gene was co-segregated with a marker 4H-60205 (Fig. 3d). Further analysis found that a G/A substitution for the marker 4H-60205 was located at the third exon of HvTUB8, a member of β-TUBULIN gene family. We further tested the G/A substitution in a natural population with 400 accessions and analyzed the gene variations in the barley pan genome panel (Jayakodi et al. 2020). The “A” allele of HvTUB8 could not be found in the natural barley population and is specific to rls, indicating that it is an artificial allele induced by chemical and physical mutation. Transcriptomic sequencing analysis further confirmed that the G/A substitution was the only variation in the coding sequences of the 17 genes within the interval between the parental line E934 and the mutant rls. As there was no detectable gene expression difference or alternative splicing between E934 and rls, any potential sequence variations in the non-coding regions within the interval are unlikely to play an important role for the observed phenotype in rls. These results indicated HvTUB8 as the candidate gene and the G/A substitution as the casual mutation, which lead to Cys354Tyr substitution in rls.
Plant TUBULIN is a dimeric protein that contributes to formation of microtubules, and it is highly conserved and composed primarily of monomeric globular polypeptides designated as α- and β-TUBULINs (Breviario et al. 2013). Microtubules are a conserved cytoplasmic structure found in all eukaryotes. They function in essential roles ranging from cellular morphogenesis to cell movement, cell division, polarity of growth, cell-wall deposition, intracellular trafficking and communications (Breviario et al. 2013; Koo et al. 2009). Cysteine residues often play essential roles in protein structure and function by conferring stability through disulfide bond formation, maintaining proper maturation and localization through protein–protein intermolecular interactions, or providing a thiol group for reactions with molecular substrates (Meitzler et al. 2013). In potato, there were 12 and 11 cysteines in α- and β-TUBULINs, respectively. These cysteine residues play vital role in accessibility of tubulin for modification by benomyl, colchicine and GTP binding (Koo et al. 2009). There were 12 cysteines in the wide type HvTUB8 protein. HvTUB8 homologue proteins from other species showed high level of similarity. Previous studies showed that the Cysteine 354 of β-TUBULIN was essential for binding of colchicine (Bai et al. 1996, 2000) and acrolein (Uemura et al. 2019), and mutation of this cysteine in yeast β-TUBULIN could result in cold-stable microtubules (Gupta et al. 2010). Our result demonstrated that the Cys354Tyr substitution resulted in changes of subcellular location of the protein. The mutant Hvtub8 protein was diffused to nuclear and membrane, while the wild-type HvTUB8 was concentrated in cytoplasm and a few of granular organelles (Fig. 5). The results indicated that mutation of the Cys354Tyr changed the protein location and interaction between HvTUB8 and its counter partner subsequently. This result is consistent with the rice paa3 protein which encoded a H+ -ATPase and controlled panicle development and seed size in rice (Yang et al. 2021). The Arg258Gln substitution caused by a G-A mutation in rice receptor-like kinase CR4 could also disturb its subcellular localization, and lead to the change of grain size and shape in mis2 mutant (Yan et al. 2020). In wheat, the grain weight controlling gene GW7 co-localized with α- and β-TUBULIN proteins in the cells, which was involved in the pathways regulating cell division and organ growth (Wang et al. 2019b). We speculated that the Cys354Tyr substitution in rls altered formation of tubulin complex and microtubules and resulted in changes of cell morphology and arrangement in spikelet hull and leaf, and thus provided further evidence to support HvTUB8 as the functional gene.
In plants, cortical microtubule arrays play a critical role in plant cell shape determination by defining the direction of cell expansion. Thus, TUBULIN genes are critical in all kinds of physiological and developmental processes, including grain shape. For example, three rice α-TUBULIN genes TubA1, TubA2 and TubA3 showed significantly different expression in flowers, roots and coleoptile segments treated with auxin (Qin et al. 1997), while members of β-TUBULIN gene may be responsible for chilling (Jeong et al. 2021) and salt tolerance (Jin et al. 2021) of rice. Rice auxin-inducible and microtubule-localized protein OsIQD14 could affect grain shape by regulating microtubule rearrangements in rice hull cells (Yang et al. 2020). What is more, members of α-TUBULIN were not only the causal gene for Small and round seed 5 (Srs5) (Segami et al. 2012), but also for Twisted dwarf 1 (Tid1) (Sunohara et al. 2009). These results are consistent with our observation that rls mutant has round and short grain and thus support HvTUB8 as the functional gene.
To further confirm HvTUB8 as the functional gene, we performed virus-induced gene (VIG)-silencing experiment. Silencing of HvTUB8 resulted in alteration of barley spike morphology (Fig. 4e), which confirmed that HvTUB8 was essential for maintaining barley spike integrity. However, the resulting spike phenotype was not exactly same as the rls mutant. This could be due to the following two facts: (1) the gene-silencing inoculation was conducted at the two-leaf stage, such early gene silence was effective to interfere the inflorescence development, but may not impact for the grain development at later stage; thus, we could only see abnormal spike but not the round grain; (2) VIG only partially inhibited the gene expression, but the rls resulted in cellular location change of the protein due to loss of the Cys; thus, the VIG result was in-consistent with the mutant. More research is required to confirm HvTUB8 as the functional gene through precision gene editing, e.g., prime-editing.
HvRLS participates in barley lateral spikelet development independent of known VRS gene
Being consistent with the hypothesis of gain-of-function of HvRLS, the majority (77%) of DEGs between E934 and rls were up-regulated in rls. Among those known VRS genes, VRS1 acts as a downstream integrator of VRS3, VRS4 and VRS5 to repress lateral spikelet fertility and has been considered as the switch between two-rowed and six-rowed, while VRS3 functions as an epigenetic modifier of row-type gene function (Zwirek et al. 2019). However, none of these reported lateral spikelets governing genes was differentially expressed in rls mutant, suggesting that HvRLS regulates spike development in a VRS-independent manner. As a novel gene in shaping barley spike morphology, mutation of HvRLS likely induced the expression of two LOB domain-containing protein genes and a Homeobox-leucine zipper family protein gene, which show low level sequence similarities with VRS4/HvRA2 and VRS1, respectively. Moreover, three MADS-box genes but not the HvMADS56 were also identified to be differentially expressed between E934 and rls, two of which were also up-regulated in the mutant. These results suggested that HvRLS regulate barley spike development through a different pathway but share similar regulating mechanisms with other known lateral spikelet determining genes.
HvRLS participates in barley development via ethylene signaling and chlorophyll biosynthesis
Distribution and concentration of different hormones play vital roles in barley inflorescence morphology. Loss of function of VRS2 is associated with auxin and cytokinin imbalances along the spike (Youssef et al. 2016). As one of the most important phytohormones, ethylene plays a critical role not only throughout the plant life cycle (Larsen 2015), but also in their response to biotic and abiotic threatens (Dubois et al. 2018). Arabidopsis thaliana mutants in ethylene signaling pathway presents notable alterations in seed shape (Robert et al. 2008). It has recently been proved that ethylene biosynthesis also plays a key role in maize ear elongation and kernel yield recently (Ning et al. 2021). Rice U-box E3 ubiquitin ligase gene positively regulates seed cell division and elongation through multiple hormone pathways, including ethylene (Wang et al. 2017a, 2017b). In this study, some key genes involving in ethylene biosynthesis and signaling, such as ACC synthase, ACC oxidase, ethylene-responsive transcription factors, were distinctively up-regulated upon the mutation of HvRLS in rls, suggesting potential regulatory roles of these genes in barley spike development. Actually, it has been proved previously that ethylene signaling pathways can initiated transfer cells morphology and control cell shape and vesicle transport of barley (Thiel et al. 2012).
Except for spike morphology, leaf color of rls was darker than that in E934. Accordingly, number and size of chloroplasts were increased in rls, which was also consistent with the higher chlorophyll concentration detected in rls seedling leaves when compared with that in E934. A previous study has confirmed that functional attenuations in chlorophyll biosynthesis-related genes, including HrHEMA, HrPOR and HrCAO, induce leaf color and chloroplast structure changes in Hosta (Zhang et al. 2021). Though transcriptomic analysis was conducted on young panicles of rls and E934 in this study, genes being responsible for chlorophyll biosynthesis and metabolism were indeed up-regulated in rls, such as Glutamine synthetase and decarboxylase genes, chlorophyll a-b binding protein coding genes, aminotransferase genes, protochlorophyllide reductase genes. This indicated that HvRLS may control leaf color and chloroplast structure through chlorophyll biosynthesis and metabolisms.
Data availability
All data supporting the findings of this study are available within the paper and within its supplementary materials published online.
Change history
23 March 2023
A Correction to this paper has been published: https://doi.org/10.1007/s00122-023-04323-z
References
Abebe T, Wise RP, Skadsen RW (2009) Comparative transcriptional profiling established the awn as the major photosynthetic organ of the barley spike while the lemma and the palea primarily protect the seed. Plant Genome 2:247–259
Bai R, Pei XF, Boyé O, Getahun Z, Hamel E (1996) Identification of cysteine 354 of -tubulin as part of the binding site for the a ring of colchicine. J Biol Chem 271:12639–12645
Bai R, Covell DG, Pei XF, Ewell JB, Hamel E (2000) Mapping the binding site of colchicinoids on β-tubulin. J Biol Chem 275:40443–40452
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible read trimming tool for illumina NGS data. Bioinformatics 30:2114–2120
Breviario D, Gianì S, Morello L (2013) Multiple tubulins: evolutionary aspects and biological implications. Plant J 75:202–218. https://doi.org/10.1111/tpj.12243
Bull H, Casao MC, Zwirek M, Flavell AJ, Thomas WTB, Guo W, Zhang R, Rapazote-Flores P, Kyriakidis S, Russell J, Druka A, McKim SM, Waugh R (2017) Barley SIX-ROWED SPIKE3 encodes a putative Jumonji C-type H3K9me2/me3 demethylase that represses lateral spikelet fertility. Nat Commun 8:936–936. https://doi.org/10.1038/s41467-017-00940-7
Chen G, Li HB, Zheng Z, Wei YM, Zheng YL, McIntyre CL, Zhou MX, Liu CJ (2012) Characterization of a QTL affecting spike morphology on the long arm of chromosome 3H in barley (Hordeum vulgare L.) based on near isogenic lines and a NIL-derived population. Theor Appl Genet 125:1385–1392
Chen Y, Song W, Xie X, Wang Z, Guan P, Peng H, Jiao Y, Ni Z, Sun Q, Guo W (2020) A collinearity-incorporating homology inference strategy for connecting emerging assemblies in the triticeae tribe as a pilot practice in the plant pangenomic era. Mol Plants Engl Edn 13:1694–1708
Cheng X, Xin M, Xu R, Chen Z, Cai W, Chai L, Xu H, Jia L, Feng Z, Wang Z, Peng H, Yao Y, Hu Z, Guo W, Ni Z, Sun Q (2020) A single amino acid substitution in STKc_GSK3 kinase conferring semispherical grains and its implications for the origin of triticumsphaerococcum. Plant Cell 32:923–934
Dubois M, Van den Broeck L, Inzé D (2018) The Pivotal role of ethylene in plant growth. Trends Plant Sci 23:311–323. https://doi.org/10.1016/j.tplants.2018.01.003
Elbaum R, Zaltzman L, Burgert I, Fratzl P (2007) The role of wheat awns in the seed dispersal unit. Science 316:884–886
Fang Y, Zhang X, Zhang X, Tong T, Zhang Z, Wu G, Hou L, Zheng J, Niu C, Li J, Wang W, Wang H, Xue D (2020) A high-density genetic linkage map of SLAFs and QTL analysis of grain size and weight in Barley (Hordeum vulgare L.). Front Plant Sci 11:620922
Franckowiak JD, Lundqvist U (2012) Descriptions of barley genetic stocks for 2012. Barley Genet Newsl 42:36–792
Gupta ML, Bode CJ, Dougherty CA, Marquez RT, Himes RH (2010) Mutagenesis of beta-tubulin cysteine residues in Saccharomyces cerevisiae: mutation of cysteine 354 results in cold-stable microtubules. Cell Motil Cytoskel 49:67–77
Gustafsson Å, Lundqvist U (1980) Hexastichon and intermedium mutants in barley. Hereditas 92:229–236
Gyenis L, Yun SJ, Smith KP, Steffenson BJ, Bossolini E, Sanguineti MC, Muehlbauer GJ (2007) Genetic architecture of quantitative trait loci associated with morphological and agronomic trait differences in a wild by cultivated barley cross. Genome 50:714–723
Harlan JR, de Wet JMJ, Price EG (1973) Comparative evolution of cereals. Evolution 27:311–325. https://doi.org/10.1111/j.1558-5646.1973.tb00676.x.
He C, Sayed-Tabatabaei BE, Komatsuda T (2004) AFLP targeting of the 1-cM region conferring the vrs1 gene for six-rowed spike in barley, Hordeum vulgare L. Genome 47:1122–1129
Huang B, Huang D, Hong Z, Owie SO (2020) Genetic analysis reveals four interacting loci underlying awn trait diversity in barley (Hordeum vulgare). Sci Rep 10:12535
Hua W, Tan C, Xie J, Zhu J, Shang Y, Yang J, Zhang XQ, Wu X, Wang J, Li C (2020) Alternative splicing of a barley gene results in an excess-tillering and semi-dwarf mutant. Theor Appl Genet 133:163–177. https://doi.org/10.1007/s00122-019-03448-4
Jayakodi M, Padmarasu S, Haberer G, Bonthala VS, Gundlach H, Monat C, Lux T, Kamal N, Lang D, Himmelbach A, Ens J, Zhang X-Q, Angessa TT, Zhou G, Tan C, Hill C, Wang P, Schreiber M, Boston LB, Plott C, Jenkins J, Guo Y, Fiebig A, Budak H, Xu D, Zhang J, Wang C, Grimwood J, Schmutz J, Guo G, Zhang G, Mochida K, Hirayama T, Sato K, Chalmers KJ, Langridge P, Waugh R, Pozniak CJ, Scholz U, Mayer KFX, Spannagl M, Li C, Mascher M, Stein N (2020) The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588:284–289. https://doi.org/10.1038/s41586-020-2947-8
Jeong B, Lee Y, Kwon Y, Kim JH, Ham TH, Kwon SW, Lee J (2021) Genome-wide association study reveals the genetic basis of chilling tolerance in rice at the reproductive stage. Plants 10:1722
Jin CH, Dongwon B, Jun JB, Min CH, Suk PM, Hyeon LS, Hyeon LL, Jin CY, DongWon B, Tae KS, DaeJin Y, Chul KM (2021) Microtubule dynamics plays a vital role in plant adaptation and tolerance to salt stress. Int J Mol Sci 22:5957–5957
Komatsuda T, Pourkheirandish M, He C, Azhaguvel P, Kanamori H, Perovic D, Stein N, Graner A, Wicker T, Tagiri A, Lundqvist U, Fujimura T, Matsuoka M, Matsumoto T, Yano M (2007) Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc Natl Acad Sci USA 104:1424–1429. https://doi.org/10.1073/pnas.0608580104
Koo B, Kalme S, Yeo SH, Lee SJ, Yoon MY (2009) Molecular cloning and biochemical characterization of a- and b-tubulin from potato plants (Solanum tuberosum L.). Plant Physiol Biochem Paris 47:761–768
Koppolu R, Anwar N, Sakuma S, Tagiri A, Lundqvist U, Pourkheirandish M, Rutten T, Seiler C, Himmelbach A, Ariyadasa R, Youssef HM, Stein N, Sreenivasulu N, Komatsuda T, Schnurbusch T (2013) Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc Natl Acad Sci USA 110:13198–13203. https://doi.org/10.1073/pnas.1221950110
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newherg LA (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181. https://doi.org/10.1016/0888-7543(87)90010-3
Larsen PB (2015) Mechanisms of ethylene biosynthesis and response in plants. Essays Biochem 58:61–70. https://doi.org/10.1042/bse0580061
Liller CB, Neuhaus R, von Korff M, Koornneef M, van Esse W (2015) Mutations in Barley row type genes have pleiotropic effects on shoot branching. PLoS ONE 10:e0140246–e0140246. https://doi.org/10.1371/journal.pone.0140246
Liller CB, Walla A, Boer MP, Hedley P, Macaulay M, Effgen S, von Korff M, van Esse GW, Koornneef M (2017) Fine mapping of a major QTL for awn length in barley using a multiparent mapping population. Theor Appl Genet 130:269–281
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Lu J, Hou J, Ouyang Y, Luo H, Zhao J, Mao C, Han M, Wang L, Xiao J, Yang Y, Li X (2020) A direct PCR-based SNP marker-assisted selection systhem (D-MAS) for different crops. Mol Breed 40:9. https://doi.org/10.1007/s11032-019-1091-3
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang XQ, Zhang Q, Barrero RA, Li L, Taudien S, Groth M, Felder M, Hastie A, Šimková H, Staňková H, Vrána J, Chan S, Muñoz-Amatriaín M, Ounit R, Wanamaker S, Bolser D, Colmsee C, Schmutzer T, Aliyeva-Schnorr L, Grasso S, Tanskanen J, Chailyan A, Sampath D, Heavens D, Clissold L, Cao S, Chapman B, Dai F, Han Y, Li H, Li X, Lin C, McCooke JK, Tan C, Wang P, Wang S, Yin S, Zhou G, Poland JA, Bellgard MI, Borisjuk L, Houben A, Doležel J, Ayling S, Lonardi S, Kersey P, Langridge P, Muehlbauer GJ, Clark MD, Caccamo M, Schulman AH, Mayer KFX, Platzer M, Close TJ, Scholz U, Hansson M, Zhang G, Braumann I, Spannagl M, Li C, Waugh R, Stein N (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433. https://doi.org/10.1038/nature22043
Mascher M, Wicker T, Jenkins J, Plott C, Lux T, Koh CS, Ens J, Gundlach H, Boston LB, Tulpová, Holden S, Hernández-Pinzón I, Scolz U, Mayer KFX, Spannagl M, Pozniak C, Sharpe AG, Šimková H, Moscou MJ, Grimwood J, Schmutz J, Stein N (2021) Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33:1888–1906. https://doi.org/10.1093/plcell/koab077
Müller KJ, Romano N, Gerstner O, Garcia-Marotot F, Pozzi C, Salamini F, Rohde W (1995) The barley Hooded mutation caused by a duplication in a homeobox gene intron. Nature 374:727–730. https://doi.org/10.1038/374727a0
Meitzler JL, Hinde S, Bánfi B, Nauseef WM, Ortiz de Montellano PR (2013) Conserved cysteine residues provide a protein-protein interaction surface in dual oxidase (DUOX) proteins. J Biol Chem 288:7147–7157. https://doi.org/10.1074/jbc.M112.414797
Ning Q, Jian Y, Du Y, Li Y, Shen X, Jia H, Zhao R, Zhan J, Yang F, Jackson D, Liu L, Zhang Z (2021) An ethylene biosynthesis enzyme controls quantitative variation in maize ear length and kernel yield. Nat Commun 12:5832–5832. https://doi.org/10.1038/s41467-021-26123-z
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419. https://doi.org/10.1038/nmeth.4197
Qin D, Xu F, Xu Q, Peng Y, Ge S, Dong J, Jiao C (2021) Construction of a mutant library in Barley and preliminary cytological analysis of leaf width mutants. J Nucl Agric Sci 35:9
Qin D, Dong J, Xu F, Guo G, Ge S, Xu Q, Xu Y, Li M (2015) Characterization and fine mapping of a novel barley stage green-revertible albino gene (HvSGRA) by bulked segregant analysis based on SSR assay and specific length amplified fragment sequencing. BMC Genom 16:838
Qin X, Giani S, Breviario D (1997) Molecular cloning of three rice alpha-tubulin isotypes: differential expression in tissues and during flower development. Biochimica Biophysica Acta (BBA) Gene Struct Exp 1354:19–23
Ramsay L, Comadran J, Druka A, Marshall DF, Waugh R (2011a) INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat Genet 43:169–172
Ramsay L, Comadran J, Druka A, Marshall DF, Thomas WTB, Macaulay M, MacKenzie K, Simpson C, Fuller J, Bonar N, Hayes PM, Lundqvist U, Franckowiak JD, Close TJ, Muehlbauer GJ, Waugh R (2011b) INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat Genet 43:169–172. https://doi.org/10.1038/ng.745
Rebetzke GJ, Bonnett DG, Reynolds MP (2016) Awns reduce grain number to increase grain size and harvestable yield in irrigated and rainfed spring wheat. J Exp Bot 67:2573–2586. https://doi.org/10.1093/jxb/erw081
Rio DC, Ares M Jr, Hannon GJ, Nilsen TW (2010) Purification of RNA using TRIzol (TRI reagent). Cold Spring Harbor Protocol 2010:pdb.prot5439. https://doi.org/10.1101/pdb.prot5439.
Robert C, Noriega A, Tocino Á, Cervantes E (2008) Morphological analysis of seed shape in Arabidopsis thaliana reveals altered polarity in mutants of the ethylene signaling pathway. J Plant Physiol 165:911–919
Sayed MA, Allam M, Heck QK, Urbanavičiūtė I, Rutten T, Stuart D, Zakhrabekova S, Börner A, Pillen K, Hansson M, Youssef HM (2021) Analyses of MADS-box genes suggest HvMADS56 to regulate lateral spikelet development in Barley. Plants (basel) 10:2825
Segami S, Kono I, Ando T, Yano M, Kitano H, Miura K, Iwasaki Y (2012) Small and round seed 5 gene encodes alpha-tubulin regulating seed cell elongation in rice. Rice (NY) 5:4–4. https://doi.org/10.1186/1939-8433-5-4
Shang Y, Yuan L, Di Z, Jia Y, Zhang Z, Li S, Xing L, Qi Z, Wang X, Zhu J, Hua W, Wu X, Zhu M, Li G, Li C (2020) A CYC/TB1 type TCP transcription factor controls spikelet meristem identity in barley (Hordeum vulgare L.). J Exp Bot 71:7118–7131
Sunohara H, Kawai T, Shimizu-Sato S, Sato Y, Sato K (2009) A dominant mutation of TWISTED DWARF 1 encoding an alpha-tubulin protein causes severe dwarfism and right helical growth in rice. Genes Genet Syst 84:209–218
Thiel J, Riewe D, Rutten T, Melzer M, Friedel S, Bollenbeck F, Weschke W, Weber H (2012) Differentiation of endosperm transfer cells of barley: a comprehensive analysis at the micro-scale. Plant J 71:639–655
Uemura T, Suzuki T, Ko K, Watanabe K, Igarashi K (2019) Inhibition of dendritic spine extension through acrolein conjugation with α-, β-tubulin proteins. Int J Biochem Cell Biol 113:58–66
Wang C, Yang X, Li G (2021a) Molecular Insights into Inflorescence Meristem Specification for Yield Potential in Cereal Crops. Int J Mol Sci 22:3508
Wang J, Wu X, Yue W, Zhao C, Yang J (2021b) Identification of QTL for barley grain size. PeerJ 9:e11287
Wang N, Xing Y, Lou Q, Feng P, Liu S, Zhu M, Yin W, Fang S, Lin Y, Zhang T (2017a) Dwarf and short grain 1, encoding a putative U-box protein regulates cell division and elongation in rice. J Plant Physiol 209:84–94
Wang Q, Sun G, Ren X, Du B, Cheng Y, Wang Y, Li C, Sun D (2019a) Dissecting the genetic basis of grain size and weight in Barley (Hordeum vulgare L.) by QTL and comparative genetic analyses. Front Plant Sci 10:469
Wang W, Pan Q, Tian B, He F, Chen Y, Bai G, Akhunova A, Trick HN, Akhunov E (2019b) Gene editing of the wheat homologs of TONNEAU1-recruiting motif encoding gene affects grain shape and weight in wheat. Plant J 100:251–264
Wang Y, Xie J, Zhang H, Guo B, Ning S, Chen Y, Lu P, Wu Q, Li M, Zhang D, Guo G, Zhang Y, Liu D, Zou S, Tang J, Zhao H, Wang X, Li J, Yang W, Cao T, Yin G, Liu Z (2017b) Mapping strip rust resistance gene YrZH22 in Chinese wheat cultivar Zhoumai 22 by bulked segregant RNA-Seq (BSR-Seq) and comparative genomicis analyses. Theor Appl Genet 130:2191–2201. https://doi.org/10.1007/s00122-017-2950-0
Watt C, Zhou G, McFawn L-A, Li C (2020) Fine mapping qGL2H, a major locus controlling grain length in barley (Hordeum vulgare L.). Theor Appl Genet 133:2095–2103. https://doi.org/10.1007/s00122-020-03579-z
Watt C, Zhou G, McFawn L-A, Chalmers KJ, Li C (2019) Fine mapping of qGL5H, a major grain length locus in barley (Hordeum vulgare L.). Theor Appl Genet 132:883–893. https://doi.org/10.1007/s00122-018-3243-y
Yan C, Fang J, Zafar SA, Shang J, Li X (2020) MINI SEED 2 ( MIS2) encodes a receptor-like kinase that controls grain size and shape in rice. Rice 13:7
Yang BJ, Wendrich JR, De Rybel B, Weijers D, Xue HW (2020) Rice microtubule-associated protein IQ67-DOMAIN14 regulates grain shape by modulating microtubule cytoskeleton dynamics. Plant Biotechnol J 18:1141–1152
Yang F, Xiong M, Huang M, Li Z, Li Y (2021) Panicle apical abortion 3 controls panicle development and seed size in rice. Rice 14:68–68
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Yoshikawa M, Yang G, Kawaguchi K, Komatsu S (2003) Expression analyses of β-tubulin isotype genes in rice. Plant Cell Physiol 44:1202–1207. https://doi.org/10.1093/pcp/pcg150
Youssef HM, Allam M, Boussora F, Himmelbach A, Milner SG, Mascher M, Schnurbusch T (2020) Dissecting the genetic basis of lateral and central spikelet development and grain traits in intermedium-spike Barley (Hordeum vulgare Convar. Intermedium). Plants 9:1655. https://doi.org/10.3390/plants9121655
Youssef HM, Kai E, Koppolu R, Alqudah AM, Schnurbusch T (2016) VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat Genet 49:157
Youssef HM, Eggert K, Koppolu R, Alqudah AM, Poursarebani N, Fazeli A, Sakuma S, Tagiri A, Rutten T, Govind G, Lundqvist U, Graner A, Komatsuda T, Sreenivasulu N, Schnurbusch T (2017) VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat Genet 49:157–161. https://doi.org/10.1038/ng.3717
Yuo T, Yamashita Y, Kanamori H, Matsumoto T, Lundqvist U, Sato K, Ichii M, Jobling SA, Taketa S (2012) A SHORT INTERNODES (SHI) family transcription factor gene regulates awn elongation and pistil morphology in barley. J Exp Bot 63:5223–5232
Zhang J, Sui C, Liu H, Chen J, Han Z, Yan Q, Liu S, Liu H (2021) Effect of chlorophyll biosynthesis-related genes on the leaf color in Hosta (Hosta plantaginea Aschers) and tobacco (Nicotiana tabacum L.). BMC Plant Biol 21:45–45. https://doi.org/10.1186/s12870-020-02805-6
Zhang P, Guo G, Wu Q, Chen Y, Xie J, Lu P, Li B, Dong L, Li M, Wang R, Yuan C, Zhang H, Zhu K, Li W, Liu Z (2020) Identification and fine mapping of spot blotch (Bipolaris sorokiniana) resistance gene Sb4 in wheat. Theor Appl Genet 133:2451–2459. https://doi.org/10.1007/s00122-020-03610-3
Zhong J, Esse GW, Bi X, Lan T, Walla A, Sang Q, Franzen R, Korff M (2021) INTERMEDIUM-M encodes an HvAP2L-H5 ortholog and is required for inflorescence indeterminacy and spikelet determinacy in barley. Proc Natl Acad Sci USA 118:2011779118. https://doi.org/10.1073/pnas.2011779118
Zohary D, Hopf M (2000) Domestication of plants in the old world: the origin of spread of cultivated plants in West Asia, Europe, and the Nile Valley (Domestication of Plants in the Old World: The Origin of Spread of Cultivated Plants in West Asia, Europe, and the Nile Valley).
Zwirek M, Waugh R, Mckim SM (2019) Interaction between row-type genes in barley controls meristem determinacy and reveals novel routes to improved grain. New Phytol 221:1950–1965
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2018YFD1000706, 2018YFD1000700), National Natural Science Foundation of China (31771774), Visiting scholar program of China Scholarship Council (201808420082), Hubei Hongshan Laboratory and China Agriculture Research System of MOF and MARA (CARS-05).
Funding
This work was financially supported by the National Key Research and Development Program of China (2018YFD1000706, 2018YFD1000700), National Natural Science Foundation of China (31771774), Visiting scholar program of China Scholarship Council (201808420082), Hubei Hongshan Laboratory and China Agriculture Research System of MOF and MARA (CARS-05).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. DQ performed all the experiments and drafted the manuscript; RL performed gene cloning and expression analysis; GL, CW and YL analyzed the bioinformatics data; FX, QX, YP and SG took part in the field work; GD analyzed gene sequence; GG, JD and CL conceived the experiment and revised the manuscript. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Andreas Graner.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, D., Liu, G., Liu, R. et al. Positional cloning identified HvTUBULIN8 as the candidate gene for round lateral spikelet (RLS) in barley (Hordeum vulgare L.). Theor Appl Genet 136, 7 (2023). https://doi.org/10.1007/s00122-023-04272-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04272-7