
Abstract
Key message
SYL3-k allele increases the outcrossing rate of male sterile line and the yield of hybrid F1 seeds via enhancement of endogenous GA4 content in Oryza sativa L. pistils. The change in style length might be an adaptation of rice cultivation from south to north in the northern hemisphere.
Abstract
The style length (SYL) in rice is one of the major factors influencing the stigma exertion, which affects the outcross rate of male sterile line and the yield of hybrid F1 seeds. However, the biological mechanisms underlying SYL elongation remain elusive. Here, we report a map-based cloning and characterisation of the allele qSYL3-k. The qSYL3-k allele encodes a MADS-box family transcription factor, and it is expressed in various rice organs. The qSYL3-k allele increases SYL via the elongation of cell length in the style, which is associated with a higher GA4 content in the pistil. The expression level of OsGA3ox2 in pistils with qSYL3-k alleles is significantly higher than that in pistils with qSYL3-n allele on the same genome background of Nipponbare. The yield of F1 seeds harvested from plants with 7001SSYL3−k alleles was 16% higher than that from plants with 7001SSYL3−n allele. The sequence data at the qSYL3 locus in 136 accessions showed that alleles containing the haplotypes qSYL3AA, qSYL3AG, and qSYL3GA increased SYL, whereas those containing the haplotype qSYL3GG decreased it. The frequency of the haplotype qSYL3GG increases gradually from the south to north in the northern hemisphere. These findings will facilitate improvement in SYL and yield of F1 seeds henceforward.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is one of the important cereal crops and is consumed by 3.5 billion people worldwide. With an increase in the human population and the decrease in the effective area for food production, it is imperative to raise the yield of rice per unit area to ensure food security for millions of people globally in the near future. Cultivating hybrid varieties of rice is an effective and feasible strategy to enhance the rice grain yield. Hybrid rice cultivars out yield conventional rice cultivars by approximately 20% (Lu & Hong 1999; Cheng et al. 2004). However, the annual production of fresh F1 hybrid seeds is required for large-scale hybrid rice cultivation. The area and yield of hybrid F1 seeds in previous seasons determine the acreage of hybrid rice planting for the next season. For a given amount of spikelets per unit area, the yield of F1 hybrid rice seeds is largely determined by the outcrossing seed setting rate, which is mainly affected by the stigma exertion. Increasing the percentage of exerted stigmas (PES) of male sterile lines (used as the female parent in F1 seed production in the field) could increase the chance of receiving pollen after flowering, which compensates for the poor pollination rate caused by parental differences in the flowering time (Wang et al. 2008). Additionally, there was a remarkable positive correlation between the PES and F1 hybrid seed yield in a study, with an increase of 1% in the PES of male sterile lines, 0.74–0.92% in the seed setting rate, and 47–68 kg/hectare in the seed yield (Yang 1997). The PES is largely determined by the stigma length (STL), style length (SYL), and the sum of the stigma and style length (TSSL).
In rice, the stigma characteristics, a complex quantitative trait, are controlled by multiple genes with minor phenotypic effects (Uga et al. 2003, 2010; Yu et al. 2003). To the best of our knowledge, 37 quantitative trait loci (QTLs) have been detected for the STL and they are distributed on all the 12 chromosomes (Uga et al. 2003, 2010; Yan et al. 2009; Li et al. 2010; Marathi et al. 2015; Dang et al. 2016, 2020). Uga et al. (2003) reported that the QTL qSTL-4 on the chromosome 4 has the largest additive effect, and the allele from accession W1944 can increase the STL by 0.106 mm compared with Pei-kul. Thirty-nine QTLs have been identified for the SYL and they are located on chromosomes 1, 2, 3, 4, 6, 7, 9, 10, and 11 (Uga et al. 2003, 2010; Li et al. 2010; Marathi et al. 2015; Dang et al. 2016, 2020; Zhou et al. 2017). Uga et al. (2010) also reported that the QTL qSYL-3 on the chromosome 3 has the largest additive effect, and the allele from IR64 can increase the SYL by 0.102 mm compared with Kinandang Patong. Finally, 30 QTLs have been identified for the TSSL and they are distributed on chromosomes 1, 2, 3, 4, 6, 7, 9, and 12 (Li et al. 2010; Liu et al. 2015a; Marathi et al. 2015; Dang et al. 2016, 2020). The percentages of phenotypic variance explained by these QTLs for TSSL range from 2.9 to 20.0%. Li et al. (2010) reported the QTL qSSL-3 (SSL = TSSL here) on the chromosome 3 has the largest additive effect, and the allele from accession T821B can increase TSSL by 0.092 mm compared with G46B. Among all these QTLs, one candidate gene, LOC_Os03g14850 (originally named qSTL3), has been identified for controlling TSSL (Liu et al. 2015b). Two genes, OsSYL2 and OsSYL3, were identified for controlling the SYL (Dang et al. 2020). However, the underlying mechanism of action of these genes is still unclear.
In our previous study, a major QTL, qSTL3, was finely mapped to a 19.8-kb region in the short arm of the chromosome 3, and the LOC_Os03g14850 gene was validated as the gene of STL3 (Liu et al. 2015b). Because the STL reported by Liu et al. (2015b) actually included the total length of two parts (stigma and style) of the pistil, and no significant phenotypic difference in the STL was found between Nipponbare allele and Kasalath allele at this gene locus, we renamed the original QTL qSTL3 as qSYL3 in the present study to correctly reflect the real difference. In this study, we further verified that LOC_Os03g14850 is the SYL3 gene using transgenic complementation and overexpression experiments and determined the biological functions of the SYL3-k allele from the rice cultivar Kasalath. We also evaluated the potential importance of the SYL3-k allele in hybrid rice seed production utilising field experiments and elucidated the allelic polymorphism at the SYL3 locus in 136 accessions collected from different geographic zones.
Materials and methods
Rice materials and field planting
Rice cultivars Nipponbare, Kasalath, SSSL14, and 101 accessions in the core collection of rice were cultivated in a paddy field in Nanjing (118.6° E, 32.1° N). Thirty-five wild rice accessions were cultivated at the National Germplasm Guangzhou Wild Rice Nursery in Guangzhou (113.2° E, 23.1° N). The information of these accessions, including the name, latitude, and longitude, is listed in Supplementary Table S1. Rice cultivar SSSL14 is a single-segment substitution line with the genome background of Nipponbare and contains only one donor fragment from Kasalath in the middle of the short arm of chromosome 3. The transgenic plants were grown in the conditions prevalent at Nanjing and Hainan (110.0 °E, 18.5 °N). On the field, the rows were spaced 20 cm apart, and each plant was spaced at a distance of 17 cm according to standard agronomic management practices.
Trait measurement
At the heading stage, 10 target spikelets were collected from five plants of each accession prior to glume opening (at approximately 10:00 a.m.). For each spikelet, the STL, SYL, and TSSL of the fertile floret were measured using a stereo microscope (10 ×, MC50, Guangzhou, China), and the average values (in mm) of the 10 florets were taken for the accessions.
Sample preparation and observation under light microscopy
In order to investigate the cytologic reasons that affect the SYL, we conducted sheet pressing analysis using the styles of Nipponbare, SSSL14, Kasalath, transgenic complementation plants, and overexpression plants. At anthesis, the mature pistils of Nipponbare, SSSL14, Kasalath, and transgenic positive plants (including the complementation and overexpression plants) were observed under a light microscope (Olympus FV1000, Japan). The pistils were then sampled and fixed with 70% (v/v) alcohol, 38% (w/v) formaldehyde, and glacial acetic acid (90:5:5, by volume), washed with tap water three times, and placed in 5 μl of 6% NaOH between a cover glass and a microscope slide and a pressed plate. Longitudinal sections were then observed under a light microscope.
Application of exogenous phytohormones on the top second leaf blades of main stems
During the third or fourth stage of young panicle differentiation (according to the criteria reported by Itoh et al. (2005)), the second leaf blades from the top of the main stem of different Nipponbare and SSSL14 plants were smeared with 10 μM gibberellic acid (GA3; Shanghai Ryon Biotechnology Co., Ltd, Shanghai, China), 20 μM brassinosteroid (BR; Sigma-Aldrich Trading Company Ltd, Shanghai, China), and 1 μM indole-3-acetic acid (IAA; Beijing Solarbio Science & Technology Co., Ltd, Beijing, China) using a hygroscopic cotton ball. Control plants were treated with absorbent cottons containing the same volume of distilled water. The smear concentrations of GA3, BR, and IAA were determined according to a previous report (Zhao et al. 2010). Finally, we sampled the spikelets to measure the STL, SYL, and TSSL at the flowering stage.
Endogenous phytohormone content analysis using high performance liquid chromatography-tandem mass spectrometry (HPLC–MS/MS)
The content of endogenous plant hormones GA1, GA4, BR, and IAA in mature pistils at stage 8 of young panicle differentiation in the Nipponbare and SSSL14 plants was detected utilising the methods reported by Durgbanshi et al. (2005), Forcat et al. (2008) and Zhong et al. (2013).
The phytohormone content of GA1, GA4, and IAA was measured using HPLC (1260, Agilent technologies, USA) and MS (6420A, Agilent technologies, USA). The chromatographic conditions were: Poroshell 120-SB-C18 reversed phase column (150 × 2.1 mm, 2.7 μm); column temperature: 30℃; sample size: 2 μl; mobile phase: A:B = (methanol/0.1% formic acid): (water/0.1% formic acid); flow velocity: 0.3 ml/min; and gradient elution mode: 0–1 min, 20% A; 1–3 min, 20–50% A; 3–9 min, 50–80% A; 9–10.5 min, 80% A; 10.5–10.6 min, 80–20% A; 10.6–13.6 min, 20% A. The mass spectrometry conditions were: ESI− negative ion mode; scan type: multiple-reaction monitoring; air curtain: 15 psi; spray voltage: − 4000 v; atomising pressure: 65 psi; auxiliary pressure: 70 psi; and atomisation temperature: 400 °C.
The BR content was measured using HPLC (1290, Agilent technologies, USA) and tandem MS (SCIEX-6500 Qtrap, Allen-Bradley, USA). The chromatographic conditions were: Poroshell 120-SB-C18 reversed phase column (150 × 2.1 mm, 2.7 μm); column temperature: 35 °C; sample size: 2 μl; mobile phase: A:B = (methanol): (water/0.1% ammonia); flow velocity: 0.35 ml/min; and gradient elution mode: 0–2 min, 80% A; 2–3.5 min, 80–95% A; 3.5–6 min, 95% A; 6–6.1 min, 95–80% A; 6.1–10 min, 80% A. The mass spectrometry conditions were: ESI− positive ion mode; scan type: multiple-reaction monitoring; air curtain: 15 psi; spray voltage: + 4500 v; atomising pressure: 65 psi; auxiliary pressure: 70 psi; and atomisation temperature: 350 ℃.
For the calculation of hormone content in the samples, we used the following formula: hormone content in the sample (ng/g fresh weight) = detection concentration (ng/ml) × volume coefficient (ml)/mass coefficient (g), where the volume coefficient is the volume of the solution used in the final dissolution of the sample and the mass coefficient is the mass of the sample.
Plasmid construction and plant transformation
To produce the complementation construct pCAMIA1300-SYL3, a 4844-kb SYL3 genomic DNA fragment including the 2221-kb upstream sequence, full-length 2223-kb SYL3 sequence, and 400-kb downstream sequence was amplified from Kasalath and cloned into the plant binary vector pCAMBIA1300 using In-Fusion™ Advantage PCR Cloning Kits (Takara, Japan). To generate the overexpression constructs, the full-length coding sequence (CDS) of SYL3 was amplified from Kasalath and cloned into the plant binary vectors pBWA(V)HS with a 35S promoter. A 2321-kb DNA fragment upstream of the SYL3 start codon was amplified from Kasalath and cloned into the pBWD(LB)1C–GUS plus vector to generate the plasmid PROSYL3:GUS.
We used the site-directed mutation method to construct two single nucleotide polymorphism (SNP) constructs (H2 and H3, the allele-specific transformation vectors). Firstly, the SYL3-n CDS was used as the template to be modified by site-directed mutagenesis based on the primers containing the mutated nucleotides. For the H2 vector, the base G in position S1 was replaced by the base A. For the H3 vector, the base G in position S2 was replaced by the base A. The PCR products were cloned into the plasmid pBWA(V)BS using the homologous recombination method. The genotype of SSSL14 is H1, and the genotype of Nipponbare is H4. All the site-directed mutagenesis transgenes were driven by the SYL3-n promoter. The primer sequences used for the vector construct are listed in Supplementary Table S2.
We verified all the resultant constructs by sequencing, and then, the binary vectors were electroporated into the Agrobacterium tumefaciens strain EHA105 and transformed into Nipponbare (e.g. empty vector, complementation vector, pSYL3-k::GUS, H2, and H3), SSSL14 (e.g. empty vector and overexpression vector), and 7001S (e.g. complementation vector) using the method described by Hiei et al. (1994). Positive pSYL3-k::GUS transgenic plants were selected based on the antibiotic resistance, and GUS histochemical staining analysis was conducted using the method described by Jefferson et al. (1987). Images were captured using a stereo light microscope (Leica DFC 420, Leica Microsystems, Germany).
Subcellular localisation
The SYL3-k-RFP in-frame fusion protein construct and the nuclear marker NLS-mCherry construct were co-expressed transiently in rice leaf protoplasts using the polyethylene glycol method (Chiu et al. 1996; Chen et al. 2006). The corresponding amino acid sequence of the NLS protein was MDPKKKRKV. We observed the fluorescence of the samples above at an excitation wavelength of 588 nm and an emitting wavelength of 635 nm under a confocal laser-scanning microscope (Olympus FV1000). The primer sequences are listed in Supplementary Table S2.
Quantitative RT-PCR analysis
Using the RNApure Plant Kit (Beijing CoWin Biotech Co. Ltd, Beijing, China), total RNA was extracted from roots, culms, leaf blades, leaf sheaths, young panicle from stage 3–8, and young pistils at stage 8 of inflorescence developmental course (Ikeda et al. 2004; Itoh et al 2005) sampled from Nipponbare and SSSL14. RNase-free DNase I was used to remove the genomic DNA in the sample. cDNA at a volume of 20 μl was synthesised using 1 μg RNA using the HiScript II 1st Strand cDNA synthesis Kit (Vazyme biotech co., Ltd, Nanjing, China). Quantitative RT-PCR (20 μl reaction volume) was carried out using 0.4 μl of cDNA, 0.4 μM of each gene-specific primer, and the AceQTM qPCR Kit (Vazyme) in the Roche Applied Science LightCycler™ 480 (Roche diagnostics Ltd., Germany). The ubiquitin gene was used as the internal controls. The primers used for qRT-PCR are listed in Supplementary Table S2. The corresponding PCR programme was pre-denaturation at 95 °C for 5 min, denaturation at 94 °C for 10 s, annealing at 60 °C for 30 s, and a total of 40 cycles. The relative expression of the target gene was calculated using the following formula: Exp = 2−ΔCt, where ΔCt = Cttarget gene − Ctinternal control (Livak and Schmittgen 2001).
Pollen fertility observation
We stained the mature pollen grains of variety 9311, 7001SSYL3−n, and 7001SSYL3−k using a 1% I2-KI water solution and examined them under a light microscope (Olympus BH-2) under 100× magnification.
Phylogenetic tree construction
The amino acid sequences of the conserved parts of the MADS-box proteins in Arabidopsis and rice were obtained from the studies conducted by Alvarez-Buylla et al. (2000) and Parenicová et al. (2003) and the Plant Transcription Factor Database (http://plntfdb.bio.uni-potsdam.de/v3.0/). The amino acid sequences of the conserved parts of the MADS-box proteins in wheat and barley were obtained from http://www.ncbi.nlm.nih.gov/. Then, sequence alignment and construction of the neighbour-joining tree were performed using the MEGA5 software (Tamura et al. 2011) based on the maximum likelihood method.
F1 hybrid seed production potential evaluation for the SYL3 alleles in the paddy field
In order to evaluate the potential of the SYL3 gene in the production of F1 hybrid rice seeds, we conducted an actual F1 seed production experiment using isogenic lines 7001SSYL3−n (short SYL) and 7001SSYL3−k (long SYL) as the female parents and variety 9311 as the common male parent in the paddy fields of Jiangpu Experimental station, Nanjing. 7001SSYL3−n is a long day-sensitive male sterile line used in commercial F1 seed production in Eastern China, and 7001SSYL3−k is an isogenic line obtained in this study. The male and female parents were grown in a ratio of 2:6:2, i.e. 4 lines of 9311 plants were planted around 6 lines of female plants. For each combination, 30 m2 of land was utilised for planting. Before artificial pollination at the flowering time, we observed the fertility of the pollen of 7001SSYL3−n and 7001SSYL3−k under a light microscope to ensure that the pollen of the female parents was sterile. We performed artificial supplementary pollination twice per day during pollen dispersal. Thirty days after artificial supplementary pollination, we harvested the seeds from the female plants individually. The potential of the SYL3 allele for hybrid rice seed production was evaluated according to the weight of rice grains harvested from the male sterile plants per 1.5 m2 of area.
Geographical distribution and genetic diversity analyses of the qSYL3 alleles
In order to analyse the molecular variation in the SYL3 alleles, we collected 136 rice accession resources and designed primers to amplify the SYL3 coding regions of these varieties (Supplementary Table S1). The sequencing was completed at Nanjing TsingKe biological technology Co., Ltd (Nanjing, China). The genetic pedigree diagram of the SYL3 alleles in cultivated and wild rice was developed using the median-joining model of NETWORK version 5.0 (Bandelt et al. 1999). After sequencing, the nucleotide diversity of the cultivated rice groups (indica, japonica, and javanica) and wild rice group in 20 genes around SYL3 (Supplementary Table S3) was analysed using the DnaSP 5.0 software (Librado and Rozas 2009).
Coalescent simulations
Coalescent simulation was used to model the bottleneck impact on sequence diversity using Hudson’s ms programme (Hudson 2002). We modelled the divergence of the cultivated rice (O. sativa) population and wild rice (O. rufipogon and O. nivara), with a population bottleneck in the cultivated rice. We used the bottleneck model reported by Zhu et al. (2007) in this study (Supplementary Figure S1). We combined O. rufipogon and O. nivara as a single population as the progenitor of O. sativa following the report by Londo et al. (2006). Based on the bottleneck model, we assumed that a single ancestral population of size Na experienced an instantaneous size shift to a bottlenecked population of size Nb at time t2 generation ago and the bottleneck population expanded instantaneously to the present population of size Np at time t1 generation ago. The parameter d, the duration of the bottleneck, and the parameter Nb were used for the bottleneck. The parameter K, which is the ratio of Nb and d, was used to describe the severity of the bottleneck in domestication (Wright and Gaut 2005).
The parameter d (200, 500, 1000, 1500, 2000, and 3000) was set as per the reports by Zhu et al. (2007) and Asano et al. (2011). We first set Na to be 120,000 to perform the simulation. A grid of 20 K values is listed in Supplementary Table S4, and the corresponding Nb, which is K × d, is also shown. We simulated the average number of SNP patterns in a 100-kb region corresponding with the region from genes 6–17 listed in Supplementary Table S3.
Results
Verification of LOC_Os03g14850 as the gene SYL3 using transgenic complementation and overexpression experiments
Within the 19.8-kb chromosome region harbouring the qSYL3 locus, three annotated genes, namely LOC_Os03g14850, LOC_Os03g14860, and LOC_Os03g14880, were found according to the rice genome annotation database. Among these annotated genes, LOC_Os03g14850 was determined as the candidate gene of SYL3 according to the results of the qRT-PCR and T-DNA insertion (Liu et al. 2015b). LOC_Os03g14850 is predicted to encode a MADS-domain family (MADS is named for yeast MCM1, plant AGAMOUS and DEFICIENS, and mammal Serum Response Factor) SRF-like protein. According to the results of the neighbour-joining tree, all the MADS-box proteins are divided into two groups (Type I and Type II) with more than 95% supports, and Type I is further divided into three subgroups (Mα, Mβ, and Mγ). LOC_Os03g14850 is in the subgroup Mα in the Type I group and shares 78% amino acid identity with gene LOC_Os06g22760 (Supplementary Figure S2A). As shown in Figure S2B, LOC_Os03g14850 contains one M domain (8–57 aa) at its N-terminal region. We also found that the structure of LOC_Os03g14850 is different from that of the other members in the MADS-box family because it contained the least number of exons (Supplementary Fig. S2C).
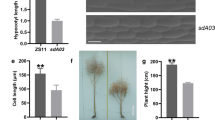
Three SNPs were identified in the CDS of LOC_Os03g14850 between the Nipponbare allele (designated SYL3-n) and Kasalath allele (designated SYL3-k) (Fig. 1A). A 4223-bp genomic fragment from Kasalath was able to fully increase TSSL and SYL and had no effect on the STL in the transgenic complementation lines comparing with the Nipponbare plants with the empty vector (EV-N) (Fig. 1B–E). Furthermore, the transgenic overexpression plants with increased expression of SYL3-k showed longer TSSL and SYL and no change in the STL compared with the SSSL14 plants with the empty vector (EV-S) (Fig. 1F–I). Thus, we concluded that LOC_Os03g14850 was the SYL3 gene.

Gene structure of SYL3 and the phenotypes identification of SYL3 complementary and overexpression strains. A Gene structure of SYL3 and natural variation between the Nipponbare (SYL3-n) and Kasalath (SYL3-k) alleles. The black boxes denote exons, and the blue line denotes introns. B Pistil morphology of four independent transgenic complementary lines and Nipponbare with empty vector (EV-N) as a control. Scale bar, 1 mm. C–E Comparison of the phenotype between transgenic complementary lines and EV-N. Data represent means ± SD (n = 43 independent plants). F Pistil morphology of four independent transgenic overexpression lines and SSSL14 with empty vector (EV-S) as a control. Scale bar, 1 mm. G–I Comparison of the phenotype between transgenic overexpression lines and EV-S. Data represent means ± SD (n = 52 independent plants). *P < 0.05, **P < 0.01, Student’s t-test. TSSL, the sum of stigma and style length; STL, stigma length; SYL, style length
Figure 2A shows the pistil morphology of Nipponbare, SSSL14, and Kasalath. As reported by Liu et al. (2015a, b), there were no significant differences in the STL among Nipponbare, SSSL14, and Kasalath. The SYL of Nipponbare was significantly shorter than that of SSSL14 and Kasalath (Table 1). The cell length of the style in Nipponbare (3.53 ± 0.61 μm) was significantly shorter than that of SSSL14 (9.67 ± 1.56 μm) and Kasalath (12.63 ± 1.95 μm) (P < 0.01) (Table 1; Fig. 2B, C). We also found that the cell length in the style of the complementation and overexpression lines was significantly longer than that of their respective controls (Fig. 2D–G). These results indicated that the SYL3-k allele increased SYL via the elongation of cell length in style.

Morphology display of pistil and cell in style part for Nipponbare, SSSL14 and Kasalath. A Pistil morphology viewed by stereomicroscopy. Scale bar, 1 mm. B Cell morphology of style. Scale bar, 10 μm. C Comparison of cell length among Nipponbare, SSSL14 and Kasalath. Data represent means ± SD (n = 40 independent plants). D Cell morphology for EV-N and transgenic complementary line #3-11-17, #12-15-2 and #15-17-8. Scale bar, 10 μm. E Comparison of cell length between EV-N and #3-11-17, #12-15-2 and #15-17-8. Data represent means ± SD (n = 24 independent plants). F Cell morphology for EV-S and #4-2-8, #10-3-7 and #14-5-11. Scale bar, 10 μm. G Comparison of cell length between EV-S and #4-2-8. Data represent means ± SD (n = 30 independent plants). **P < 0.01, Student’s t-test
In addition, significant differences were observed between the EV-N plants and complementation lines in the plant height (P < 0.01) (Supplementary Fig. S3A), panicle exertion length (P < 0.05) (Supplementary Fig. S3B), and panicle length (P < 0.01) (Supplementary Fig. S3C). We also observed significant differences between the EV-S plants and overexpression lines in terms of plant height (P < 0.01) (Supplementary Fig. S3D), panicle exertion length (P < 0.05) (Supplementary Fig. S3E), and panicle length (P < 0.05) (Supplementary Fig. S3F). From these results, we speculated that LOC_Os03g14850 had a pleiotropic effect to control plant height, panicle length, and panicle exertion length.
qSYL3 is constitutively expressed gene and expresses in nuclear
To examine the spatiotemporal expression patterns of SYL3-k, we detected GUS signals in the root, culm, leaf blade, leaf sheath, and pistil of the pSYL3-k::GUS transgenic plants of Nipponbare and found that all the examined tissues showed a significant GUS signal (Fig. 3A–F). Furthermore, we performed qRT-PCR analysis of the endogenous SYL3 transcripts using the total RNA isolated from different tissues and same tissues at different development stages of plants Nippponbare and SSSL14, respectively. As shown in Fig. 3G, SYL3 transcripts were detected in all the tissues examined, but its expression was most abundant in the young panicle and pistil at stage 8 of inflorescence differentiation in Nippponbare and SSSL14. SSSL14 (SYL3-k) is more highly expressed in pistil tissues compared to Nippponbare (SYL3-n). These results indicated that SYL3 is a constitutively expressed gene.

Expression pattern of SYL3. A–F The GUS expression pattern in the SYL3-k:GUS transgenic plant. A Root, Scale bar, 2 cm; B stem, Scale bar, 1 cm; C leaf blade, scale bar, 1 cm; D leaf sheath, scale bar, 1 cm; E spikelet, Scale bar, 1 mm; F pistil, Scale bar, 1 mm. The experiment was repeated 3 times independently with similar results. G Tissue-specific expression pattern of SYL3-k revealed by qRT-PCR. The ubiquitin gene was used as an internal control. Data represent means ± SD. (n = 3 biologically independent samples). *P < 0.05, **P < 0.01, Student’s t-test
To determine the subcellular localisation of SYL3, we fused the full-length CDS of SYL3-k with that of RFP. The nuclear localisation signal and mCherry fusion protein were used as the nuclear marker. The transient expression experiments in rice leaf protoplasts showed that RFP-SYL3 was specifically localised in the nucleus (Supplementary Fig. S4), providing evidence that SYL3 expresses in nuclear.
The effect of SYL3-k on style elongation is associated with higher GA4 levels in pistil, which might be caused by higher expression level of OsGA3ox2
We examined GA1, GA4, BR, and IAA levels in the pistil tissues of Nipponbare and SSSL14 to explore the cause of cell length elongation in the style. The results showed that the GA4 content in the pistils of the Nipponbare plants was 7.01 ng/g, which was significantly lower than that in SSSL14 plants (9.19 ng/g) (Table 2). There were no significant differences in the GA1 content in the pistils between the Nipponbare and SSSL14 plants (Table 2). There was no BR detected in the pistils of both the Nipponbare and SSSL14 plants. The IAA content in the pistils of Nipponbare was not significantly different from that of SSSL14 (Table 2). Thus, we concluded that the cell length elongation in the style tissues resulted from the increase in the GA4 content in the pistil.
To understand the mechanism of action of SYL3 in the regulation of GA4 content, the expression patterns of 20 key genes in the gibberellin (GA) biosynthesis pathway in rice pistils at stage 8 of young panicle differentiation were investigated using qRT-PCR. When the ubiquitin gene was used as the internal control, the qRT-PCR results showed that the expression level of OsCPS1, OsKS3, and OsGA3ox2 in SSSL14 and transgenic 35S::SYL3-k overexpression line was significantly higher than those in Nipponbare (Fig. 4), but the expression level of the other 17 genes showed no significant differences. Considering the study that demonstrated that the expression level of OsCPS1 and OsKS3 did not affect the content of bioactive GAs (Sakamoto et al. 2004), we speculated that the higher expression level of OsGA3ox2 in the plants with SYL3-k allele is likely to be related to the increasing content of GA4 in the SSSL14 plants.

qRT-PCR analysis of genes associated with GA synthesis pathway in Nipponbare, SSSL14 and transgenic 35S::SYL3-k overexpression line. The relative expression level of each gene in pistil at stage 8 of young panicle differentiation of Nipponbare, SSSL14 and transgenic 35S::SYL3-k overexpression line were analysed by qRT-PCR and normalised using the ubiquitin gene as an internal control. Data are means ± SD (n = 3). Asterisks indicate statistically significant differences compared with the wild type at P < 0.01 by Student’s t test
To determine whether SYL3-k affects GA biosynthesis or signal transduction, we applied exogenous hormone GA3 solution to smear the flag leaf blade surface at stage 5 of inflorescence differentiation in Nipponbare and SSSL14 plants. The results showed that there were significant differences in the SYL, but not in the STL between the plants treated with water and those treated with GA3 in Nipponbare (Fig. 5). Additionally, no significant differences were found in the SYL and STL between the plants treated with water, and those treated with BR or IAA in Nipponbare (Fig. 5). Similar results were obtained in the SSSL14 plants (Fig. 5). These results suggested that SYL3 might be involved in GA biosynthesis, but not in signal transduction.

Pistil morphological display and length comparison between control and treatment of GA3, BR and IAA in Nipponbare and SSSL14
To further explore whether SYL3 can be involved in GA signal transduction, four key genes for GA signal transduction were selected for qRT-PCR analysis. Previous studies revealed that genes GID1, GID2, OsSPY, and SLR1 up-regulated GA signal transduction, but there were no significant expression level differences among Nipponbare, SSSL14 and transgenic 35S::SYL3-k overexpression line (Supplementary Figure S5). These results indicated that SYL3-k was not involved in GA signal transduction mediated by genes GID1, GID2, OsSPY, and SLR1.
Yield of the F1 hybrid seeds harvested from the male sterile line with the SYL3-k allele was significantly higher than that with the SYL3-n allele
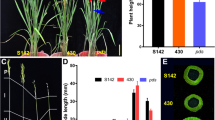
The SYL of 7001SSYL3−k was significantly longer than that of 7001SSYL3−n (Fig. 6A–D). To further evaluate the potential of the SYL3-k allele in hybrid rice seed production, we performed a field experiment using the two combinations, 7001SSYL3−n × 9311 and 7001SSYL3−k × 9311. The pollen of plants in 7001SSYL3−n and 7001SSYL3−k could not be stained by a 1% solution of I2-KI and was completely sterile (Fig. 6E). Therefore, the F1 seeds were obtained by artificial supplementary pollination at the flowering stage (Fig. 6F). The stigma exertion rate of 7001SSYL3−k was 25.8%, which is significantly higher than that of 7001SSYL3−n (16.3%). The outcrossing seed setting rate of the 7001SSYL3−k × 9311 combination was 57.4%, which is significantly (P < 0.05) higher than that of the 7001SSYL3−n × 9311 combination (37.2%) (Fig. 6G–I). The weight of the F1 seeds harvested from the female parents in the 1.5 m2 area for the combination of 7001SSYL3−k × 9311 was 581.07 g, which is significantly (P < 0.05) higher than that of the combination of 7001SSYL3−n × 9311 (501.98 g) in the same area of land. These results indicate that the SYL3-k allele could significantly (P < 0.05) increase the yield of the F1 hybrid seeds via enhancing the outcrossing rate of the male sterile lines.

Hybrid rice seed production evaluation of SYL3 gene. A Pistil morphology of 7001SSYL3−n and 7001SSYL3−k. Scale bar, 1 mm. B Comparison of the TSSL of 7001SSYL3−n and 7001SSYL3−k. C Comparison of the STL of 7001SSYL3−n and 7001SSYL3−k. D Comparison of the SYL of 7001SSYL3−n and 7001SSYL3−k. Data represent means ± SD (n = 40 independent plants), **P < 0.01, Student’s t-test. E Pollen viability test. Fertile pollen grains of 9311 could be stained by 1% I2-KI, while abortive pollen grains of 7001SSYL3−n and 7001SSYL3−k could not be stained. Scale bar, 100 μm. F Operation of artificial supplementary pollination with a bamboo pole. G Field performance of 7001SSYL3−n and 7001SSYL3−k. H Panicle morphology of 7001SSYL3−n and 7001SSYL3−k. Scale bar, 5 cm. I Comparison of the seed setting percentage of 7001SSYL3−n and 7001SSYL3−k. Data represent means ± SD (n = 30 independent plants), **P < 0.01, Student’s t-test
Identification of functional nucleotide polymorphisms (FNPs) in SYL3
To explore the molecular basis of the functional differences between the SYL3-n and SYL3-k alleles in regulating SYL, the CDS of SYL3-n and SYL3-k were compared. Following the comparisons, three SNP sites at positions 1082 (S1: G/A), 1154 (S2: G/A), and 2142 (S3: G/A) were found (Fig. 1A). For S1, the base G in the SYL3-n allele was changed to base A in the SYL3-k allele, leading to a change of the amino acid from Arg to His (R100H). For S2, the base G in the SYL3-n allele was changed to base A in the SYL3-k allele, leading to a change of the amino acid from Arg to Gln (R124Q). For S3, the base G in the SYL3-n allele was changed to base A in SYL3-k allele, leading to a change of the amino acid from Trp to the stop codon (W158 Stop codon) (Fig. 1A). We then compared the SYL3 coding region sequences of 101 cultivated rice accessions and did not find additional polymorphisms (Supplementary Table S1). The simple association between the three SNPs and the pistil traits of the 101 cultivated rice accession resources was analysed. Our results indicate that SNP positions S1 and S2 are both significantly associated with the SYL (Supplementary Table S5).
To verify the effects of S1 and S2, we identified the pistil phenotypes of the transgenic plants of four haplotypes, namely H1, H2, H3, and H4 (Supplementary Fig. S6A). The H1 (S1A/S2A) and H4 (S1G/S2G) constructs are identical to SSSL14 and Nipponbare, respectively. H2 is the S1G/S2A recombinant CDS, whereas H3 is the S1A/S2G recombinant CDS (Supplementary Figure S6A). We found that the transgenic positive plants and the transgenic negative plants followed the segregation ratio of 3:1 by detecting the T2 generation transgenic plant population. Except for the transgenic plants of H4, the transgenic positive plants of H1, H2, and H3 had an increase in SYL, among which the transgenic positive plants of H1 demonstrated the strongest effect (Supplementary Figure S6B–F). In conclusion, S1 and S2 are the FNPs that regulate the SYL.
Geographic distribution and genetic diversity analyses of the SYL3 alleles
To examine the evolutionary origin of the SYL3 alleles, we performed a network analysis based on the sequences of SYL3 coding regions in 35 wild rice accessions and 101 cultivated rice accessions (Supplementary Fig. S6G). The wild rice accessions, which were distributed at latitudes S16°–N28°, included the H1, H2, and H3 haplotypes (Supplementary Table S1). The indica rice accessions, which were distributed at N5°–N38°, included the H1, H2, H3, and H4 haplotypes (Supplementary Table S1). The javanica rice accessions, which were distributed at S12°–N21°, included the H3 and H4 haplotypes (Supplementary Table S1). The japonica rice accessions, which were distributed at N22°–N45°, included the H4 haplotype (Supplementary Table S1). During the domestication of rice from the south to north, haplotypes H1, H2, and H3 gradually disappeared after the artificial selections and the haplotype H4 remained. The H4 haplotype plays an important role for the northward expansion of japonica to regions with naturally long days in Asia.
We examined the four haplotypes composed of S1 and S2 sites of functional SNPs in the CDS sequence of SYL3 gene locus of 108 male sterile lines. It was found that 21 japonica male sterile lines (16 of which were cytoplasmic nuclear interaction male sterile lines and 5 were photosensitive male sterile lines) were all H4 haplotypes, while among 87 indica male sterile lines (45 of which were cytoplasmic nuclear interaction male sterile lines and 42 were photo-thermo-sensitive male sterile lines), three haplotypes, i.e. H2, H3, and H4, were detected (Supplementary Table S6). This result is similar to the identification result of the natural population composed of 101 cultivated varieties. In fact, the cytoplasmic nuclear interaction male sterile line is produced by replacing the normal fertile cytoplasm in the maintainer line with the sterile cytoplasm; in contrast, photo-thermo-sensitive genic male sterile line is produced by introducing photo-thermo-sensitive genic male sterile gene into the nuclear genome of normal fertile varieties. Therefore, the nuclear genome of maintainer lines (or excellent varieties to be transformed into photo-thermo-sensitive male sterile lines) basically determines the nuclear genome of male sterile lines. Because maintainer lines (or excellent varieties) were selected from the population of bred varieties, the corresponding alleles at SYL3 locus are inevitably artificially selected.
To test the genetic differences between indica and japonica subpopulations, we sequenced and calculated the nucleotide diversity for 20 genes around the SYL3 locus. The results showed that the average nucleotide diversity of the 20 genes for japonica (πjaponica = 0.017) was lower than that for indica rice (πindica = 0.069), javanica rice (πjavanica = 0.036), and wild rice (πwild rice = 0.141). We also investigated the nucleotide diversity values (π) and FST ratio (japonica rice accessions/indica rice accessions) values in the SYL3 coding sequence, flanking region (surrounding 40-kb, referring to Gao et al. 2019; Yang et al. 2019; Gao et al. 2020), and the whole genome using a diversity panel of the 3 K rice genome project. The results show that the π values of the SYL3 locus CDS in japonica rice accessions were lower than those in indica rice accessions and wild rice rufipogon accessions and were also lower than those in its flanking regions (Supplementary Fig. S7A). Meanwhile, the FST value in SYL3 was obviously above the genome-wide threshold 0.087 (Supplementary Fig. S7B), suggesting the genetic difference in SYL3 locus and its flanking regions between indica and japonica subpopulations.
In order to further elucidate that the low genetic diversity of japonica rice is caused by artificial selection rather than a domestication bottleneck effect, a coalescent simulation was performed with two derived populations (wild rice and cultivated rice). After the simulation, the K value for the 20 genes around SYL3 was 0.01, which is much lower than the K value (0.2) reported by Zhu et al. (2007). These results suggested that the low gene diversity surrounding SYL3 could not be simply explained by the domestication bottleneck effect. The selective sweep around the gene SYL3 provides additional crucial evidence that SYL3 may experience artificial selection during the domestication or improvement in japonica rice.
Discussion
In this study, we found that there were no significant differences in the STL among Nipponbare, SSSL14, and Kasalath. The SYL of SSSL14 and Kasalath was significantly longer than that of Nipponbare (Table 1). The finding that the SYL, rather than the STL, varies with the latitude has an important significance in biology and agriculture. The main reasons for this are as follows: First, the stigma is an organ receiving/capturing pollen and is critical for seed setting and reproduction. The STL does not change easily. The longer the STL, the better it is at capturing pollen. Second, the style is merely a passage that allows the pollen tube to reach the ovary. The SYL can be long or short. However, a short SYL can save the energy of the plant, which is beneficial. The shorter the SYL, the better chances of the pollen tube reaching the ovary. Therefore, the change in TSSL is mainly caused by the change of the SYL, which is kind of an environmental adaptation of rice cultivation towards the Northern Hemisphere, and is a new discovery brought to light by this study.
Previous studies indicated that endogenous phytohormones, such as GAs, BR, and auxin IAA, play many important roles throughout plant development like stem and cell elongation and pollen tube growth (Kobayashi et al. 1988; Clouse and Sasse 1998; Steber and McCourt 2001; Ozga and Reinecke 2003; Martinelli et al. 2009; Pomares-Viciana et al. 2017). According to the reports by Kobayashi et al. (1994) and Yamaguchi (2008), we know that GA1 and GA4 are the major bioactive GAs in regulating the growth of rice, there is no endogenous GA3 in rice plants (Kobayashi et al. 1994), and GA3 and GA7 mainly exist in the fungus Gibberella fujikuroi (Tudzynski et al. 2003; Yamaguchi 2008). Therefore, we examined GA1, GA4, BR, and IAA levels in the pistil tissues of Nipponbare and SSSL14 to explore the cause of cell length elongation in the style. We found that the change in the SYL is caused by a change in the GA4 content in pistils, which is another novel discovery made in this study. We also found that the increase in GA4 content could elongate the cell length in the style to increase the SYL, and thus TSSL, which was beneficial in increasing the exertion rate of the stigma and increasing the yield of hybrid rice seed production.
Based on the endogenous phytohormone detection, we found that the content of GA4 in SSSL14 was significantly higher than that in Nipponbare (Table 2). Through the exogenous GA3 smears, it was found that SYL of Nipponbare and SSSL14 could be significantly elongated (Fig. 5). These results suggested that SYL3-k was related to GA biosynthesis, but not related to the GA signal transduction. Furthermore, we investigated the expression patterns of 20 key genes in the GA biosynthesis pathway in rice pistils and found that the expression levels of OsCPS1, OsKS3, and OsGA3ox2 in SSSL14 and 35S::SYL3-k were significantly higher than those in Nipponbare. Sakamoto et al. (2004) reported that the expression level of OsCPS1 and OsKS3 did not affect the content of bioactive GAs. It is reported that BR greatly induces the expression of OsGA3ox2, one of the GA biosynthesis genes, leading to increased GA1 levels in rice seedlings (Tong et al. 2014). The growth of reproductive organs is regulated mainly by GA4, and the vegetative growth is regulated by GA1 (Kobayashi et al. 1988, 1989). However, in this study, we did not detect BR in rice pistils. Therefore, we speculated that the higher expression of OsGA3ox2 in the SYL3-k isogenic line was more likely to be related to increasing the GA4 content.
In this study, we also found a significant difference in SYL3 locus and its flanking regions between indica and japonica subpopulation. First, based on the correlation analysis between the alleles and traits and site-directed mutation tests, we demonstrated that the two SNPs, S1 (1082:G/A) and S2 (1154:G/A), were the FNPs, which have four haplotypes (H1, H2, H3, and H4). Out of the four haplotypes, three haplotypes (H1, H2, and H3) had strong effects on the SYL and mainly existed in wild rice, and indica and javanica rice varieties, which grow in low latitude areas (Supplementary Fig. S6G). However, haplotype H4 had no effects on the SYL and mainly existed in japonica, which is planted in high latitude areas of the Northern Hemisphere (Supplementary Fig. S6G). Secondly, the nucleotide diversity (π) values of the SYL3 locus coding sequence in japonica rice accessions are lower than those in indica rice accessions and wild rice rufipogon accessions and also lower than those in its the flanking regions (Supplementary Fig. S7A). Meanwhile, the FST value in SYL3 was obviously above the genome-wide threshold 0.087 (Supplementary Fig. S7B).
In conclusion, our work revealed that the variation in SYL was caused by the variation in GA4 level in pistil, which is closely related to the variation in the expression level of OsGA3ox2. These results greatly improve our understanding of the causes of pistil length variation and provide the basis for the breeding of male sterile lines with high stigma exertion rate to increase the yield of F1 seed production.
Data availability
Data related to this manuscript are available within this paper and its Supplementary data.
References
Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, de Pouplana LR, Martinez-Castilla L, Yanofsky MF (2000) An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci 97:5328–5333. https://doi.org/10.1073/pnas.97.10.5328
Asano K, Yamasaki M, Takuno S et al (2011) Artificial selection for a green revolution gene during japonica rice domestication. Proc Natl Acad Sci 108:11034–11039. https://doi.org/10.1073/pnas.1019490108
Bandelt HJ, Forster P, Röhl A (1999) Median-Joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Chen S, Tao LZ, Zeng LR, Vega-Sanchez ME, Umemura K, Wang GL (2006) A highly efficient transient protoplast system for analyzing defence gene expression and protein-protein interactions in rice. Mol Plant Pathol 7:417–427. https://doi.org/10.1111/j.1364-3703.2006.00346.x
Cheng SH, Zhuang JY, Cao LY, Chen SG, Peng YC, Fan YY, Zhan XD, Zheng KL (2004) Molecular breeding for super rice hybrids. Chin J Rice Sci 5:377–383. https://doi.org/10.16819/j.1001-7216.2004.05.001
Chiu WL, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6:325–330. https://doi.org/10.1016/s0906-9822(02)00483-9
Clouse SD, Sasse JM (1998) Brassinosteroids, essential regulator of plant growth and development. Annu Rev Plant Physiol Plant Mol Biol 49:427–451. https://doi.org/10.1146/annurev.arplant.49.1.427
Dang XJ, Liu EB, Liang YF, Liu QM, Breria CM, Hong DL (2016) QTL detection and elite alleles mining for stigma traits in Oryza sativa by association mapping. Front Plant Sci 7:1188. https://doi.org/10.3389/fpls.2016.01188
Dang XJ, Yang Y, Zhang YQ et al (2020) OsSYL2AA, an allele identified by gene-based association, increases style length in rice (Oryza sativa L.). Plant J 104:1491–1503. https://doi.org/10.1111/TPJ.15013
Durgbanshi A, Arbona V, Pozo O, Miersch O, Sancho JV, Gómez-Cadenas A (2005) Simultaneous determination of multiple phytohormones in plant extracts by liquid chromatography-electrospary tandem mass spectrometry. J Agric Food Chem 53:8437–8442. https://doi.org/10.1021/jf050884b
Forcat S, Bennett MH, Mansfield JW, Grant MR (2008) A rapid and robust method for simultaneously measuring changes in the phytohormones ABA, JA and SA in plants following biotic and abiotic stress. Plant Methods 4:16. https://doi.org/10.1186/1746-4811-4-16
Gao ZY, Wang YF, Chen G et al (2019) The indica nitrate reductase gene OsNR2 allele enhances rice yield potential and nitrogen use efficiency. Nat Commun 10:5207. https://doi.org/10.1038/s41467-019-13110-8
Gao YH, Xu ZP, Zhang LJ, Li SC, Wang SG, Yang HL, Liu XL, Zeng DL, Liu QQ, Qian Q, Zhang BC, Zhou YH (2020) MYB61 is regulated by GRF4 and promotes nitrogen utilization and biomass production in rice. Nat Commun 11:5219. https://doi.org/10.1038/s41467-020-19019-x
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282. https://doi.org/10.1046/j.1365-313x.1994.6020271.x
Hudson RR (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338. https://doi.org/10.1093/bioinformatics/18.2.337
Ikeda K, Sunohara H, Nagato Y (2004) Developmental course of inflorescence and spikelet in rice. Breeding Sci 54:147–156. https://doi.org/10.1270/jsbbs.54.147
Itoh J, Nonomura K, Ikeda K, Yamaki S, Inukai Y, Yamagishi H et al (2005) Rice plant development: from zygote to spikelet. Plant Cell Physiology 46:23–47. https://doi.org/10.1093/pcp/pci501
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907. https://doi.org/10.1002/j.1460-2075.1987.tb02730.x
Kobayashi M, Yamaguchi I, Murofushi N, Ota Y, Takahashi N (1988) Fluctuation and localization of endogenous gibberellins in rice. Agric Biol Chem 52:1189–1194. https://doi.org/10.1080/00021369.1988.10868799
Kobayashi M, Sakurai A, Saka H, Takahashi N (1989) Quantitative analysis of endogenous gibberellins in normal and dwarf cultivars of rice. Plant Cell Physiol 30:963–969. https://doi.org/10.1093/oxfordjournals.pcp.a077841
Kobayashi M, Gaskin P, Spray CR, Phinney BO, MacMillan J (1994) The metabolism of gibberellin A20 to gibberellin A1 by tall and dearf mutants of Oryza sativa and Arabidopsis thaliana. Plant Physiol 106:1367–1372. https://doi.org/10.1104/pp.106.4.1367
Li HB, Gao FY, Zeng LH, Li QX, Lu XJ, Li ZH, Ren JS, Su XW, Ren GJ (2010) QTL analysis of rice stigma morphology using an introgression line from Oryza longistaminata. Mol Plant Breed 8:1082–1089
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Liu QM (2015a) QTL dissection of out-crossing related traits and fine mapping of a stigma length QTL qSTL3.1 in rice (Oryza sativa L.). Dissertation, Nanjing Agricultural University. https://kns-cnki-net-s.vpn2.njau.edu.cn:8118/kcms/detail/detail.aspx?dbcode=CDFD&dbname=CDFDLAST2017&filename=1017042034.nh&v=wSSgb99IlPl3KzkcmbCUtkJ0TyvUufAJd61%25mmd2BlPjaB4aqEjSBDA0Lo3%25mmd2FPpflXxkgy
Liu QM, Qin JC, Li TW et al (2015b) Fine mapping and candidate gene analysis of qSTL3, a stigma length-conditioning locus in rice (Oryza sativa L.). PLoS ONE 10:e0127938. https://doi.org/10.1371/journal.pone.0127938
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Londo JP, Chiang YC, Hung K, Chiang TY, Schaal BA (2006) Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci 103:9578–9583. https://doi.org/10.1073/pnas.0603152103
Lu ZM, Hong DL (1999) Advances in hybrid rice seed production techniques. In: AS Basra (ed.), Heterosis and hybrid seed production in agronomic crops. New York: Food Products Press, an imprint of the Haworth Press, 65–79
Marathi B, Ramos J, Hechanova SL, Oane RH, Jena KK (2015) SNP genotyping and characterization of pistil traits revealing a distinct phylogenetic relationship among the species of Oryza. Euphytica 201:131–148. https://doi.org/10.1007/s10681-014-1213-2
Martinelli F, Uratsu SL, Reagan RL, Chen Y, Tricoli D, Fiehn O, Rocke DM, Gasser CS, Dandekar AM (2009) Gene regulation in parthenocarpic tomato fruit. J Exp Bot 60:3873–3890. https://doi.org/10.1093/jxb/erp227
Ozga JA, Reinecke DM (2003) Hormonal interactions in fruit development. J Plant Growth Regul 22:73–81. https://doi.org/10.1007/s00344-003-0024-9
Parenicová L, De Folter S, Kieffer M et al (2003) Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15:1538–1551. https://doi.org/10.1105/tpc.011544
Pomares-Viciana T, Die J, Del Rio-Celestino M, Roman B, Gomez P (2017) Auxin signalling regulation during induced and parthenocarp fruit set in zucchini. Mol Breed 37:56. https://doi.org/10.1007/s11032-017-0661-5
Sakamoto T, Miura K, Itoh H et al (2004) An overview of gibberellin metabolism enzyme genes and their related mutants in rice. Plant Physiol 134:1642–1653. https://doi.org/10.1104/pp.103.033696
Steber CM, McCourt P (2001) A role for brassinosteroids in germination in Arabidopsis. Plant Physiol 125:763–769. https://doi.org/10.2307/4279701
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tong HN, Xiao YH, Liu DP, Gao SP, Liu LC, Yin YH, Jin Y, Qian Q, Chu CC (2014) Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 26:4376–4393. https://doi.org/10.1105/tpc.114.132092
Tudzynski B, Mihlan M, Rojas MC, Linnemannstons P, Gaskin P, Hedden P (2003) Characterization of the final two genes of the gibberellin biosynthesis gene cluster of Gibberella fujikuroi: des and P450–3 encode GA4 desaturase and the 13-hydroxylase, respectively. J Biol Chem 278:28635–28643. https://doi.org/10.1074/jbc.M301927200
Uga Y, Fukuta Y, Cai HW, Iwata H, Ohsawa R, Morishima H, Fujimura T (2003) Mapping QTLs influencing rice floral morphology using recombinant inbred lines derived from a cross between Oryza sativa L. and Oryza rufipogon Griff. Theor Appl Genet 107:218–226. https://doi.org/10.1007/s00122-003-1227-y
Uga Y, Siangliw M, Nagamine T, Ohsawa R, Fujimura T, Fukuta Y (2010) Comparative mapping of QTLs determining glume, pistil and stamen sizes in cultivated rice (Oryza sativa L.). Plant Breed 129:657–669. https://doi.org/10.1111/j.1439-0523.2009.01765.x
Wang Y, Hua Z, Zhang Z, Li Q, Li R, Su Y, Yao J, Wang Z (2008) Breeding and application of japonica male sterile lines with high stigma exsertion rate in rice. Hybrid Rice 23:4–8. https://doi.org/10.3724/SP.J.1005.2008.01083
Wright SI, Gaut BS (2005) The effects of artificial selection on the maize genome. Science 308:1310–1314. https://doi.org/10.1126/science.1107891
Yamaguchi S (2008) Gibberellin metabolism and its regulation. Ann Rev Plant Biol 59:225–251. https://doi.org/10.1146/annurev.arplant.59.032607.092804
Yan WG, Li Y, Agrama HA, Luo DG, Gao FY, Lu XJ, Ren GJ (2009) Association mapping of stigma and spikelet characteristics in rice (Oryza sativa L.). Mol Breed 24:277–292. https://doi.org/10.1007/s11032-009-9290-y
Yang BH (1997) Studies on stigma exsertion rate and outcrossing rate of CMS lines in rice. Hybrid Rice 12:13–15. https://doi.org/10.16267/j.cnki.1005-3956.1997.01.005
Yang YH, Guo M, Sun SY, Zou YL, Yin SY, Liu YN, Tang SZ, Gu MH, Yang ZF, Yan CJ (2019) Natural variation of OsGluA2 is involved in grain protein content regulation in rice. Nat Commun 10:1949. https://doi.org/10.1038/s41467-019-09919-y
Yu T, Zhang L, Hu ZL, Song WZ, Liu SJ, Zhang ZH, Zhu YG (2003) Genetic analysis of floral characters in a DH population derived from an indica /japonica cross of rice. J Wuhan Botany Res 21:459–463
Zhao SQ, Hu J, Guo LB, Qian Q, Xue HW (2010) Rice leaf inclination 2, a VIN3-like protein, regulates leaf angle through modulating cell division of the collar. Cell Res 20:935–947. https://doi.org/10.1038/cr.2010.109
Zhong DL, Ding M, Tang FB, Mo RH, Teng Y (2013) Determination of four endogenous phytohormones in bamboo shoots by liquid chromatography-tandem mass spectrometry. Chin J Anal Chem 41:1739–1743. https://doi.org/10.3724/SP.J.1096.2013.30610
Zhou H, Li PB, Xie WB et al (2017) Genome-wide association analyses reveal the genetic basis of stigma exsertion in rice. Mol Plant 10:634–644. https://doi.org/10.1016/j.molp.2017.01.001
Zhu QH, Zheng XM, Luo JC, Gaut BS, Ge S (2007) Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24:875–888. https://doi.org/10.1093/molbev/msm005
Acknowledgements
We thank Dr. Jianbo He for software assistance. We thank Prof. Shirong Zhou and Donglei Yang for technical assistance. This work was supported by a grant from the National Natural Science Foundation of China (31571743, 31601374 and 31671658) and Cyrus Tang Crop Seed Innovation Center, Nanjing Agricultural University.
Author information
Authors and Affiliations
Contributions
DH and JJ designed the research; XD, YZ, YL, SC, and EL performed most of experiments and analysed the data. XD, YZ, YL, EL, SC, BF, QL, DS, ZD, ZF, DL, HW, SZ, XH, and YL conducted the collection of rice germplasm and planted them to investigate phenotypic data; XD wrote the manuscript; DH and JJ revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Communicated by Hai-Chun Jing.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dang, X., Zhang, Y., Li, Y. et al. SYL3-k increases style length and yield of F1 seeds via enhancement of endogenous GA4 content in Oryza sativa L. pistils. Theor Appl Genet 135, 321–336 (2022). https://doi.org/10.1007/s00122-021-03968-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03968-y