
Abstract
Atherosclerosis is the leading cause of cardiovascular mortality and morbidity worldwide and is described as a complex disease involving several different cell types and their molecular products. Recent studies have revealed that atherosclerosis arises from a systemic inflammatory process, including the accumulation and activities of various immune cells. However, the immune system is a complicated network made up of many cell types, hundreds of bioactive cytokines, and millions of different antigens, making it challenging to readily define the associated mechanism of atherosclerosis. Nevertheless, we previously reported a potential persistent inflammatory process underlying atherosclerosis development, centered on a pathological humoral immune response between commensal microbes and activated subpopulations of substantial B cells in the vicinity of the arterial adventitia. Accumulating evidence has indicated the importance of gut microbiota in atherosclerosis development. Commensal microbiota are considered important regulators of immunity and metabolism and also to be possible antigenic sources for atherosclerosis development. However, the interplay between gut microbiota and metabolism with regard to the modulation of atherosclerosis-associated immune responses remains poorly understood. Here, we review the mechanisms by which the gut microbiota may influence atherogenesis, with particular focus on humoral immunity and B cells, especially the gut-immune-B2 cell axis.

Under high-fat and high-calorie conditions, signals driven by the intestinal microbiota via the TLR signaling pathway cause B2 cells in the spleen to become functionally active and activated B2 cells then modify responses such as antibody production (generation of active antibodies IgG and IgG3), thereby contributing to the development of atherosclerosis. On the other hand, intestinal microbiota also resulted in recruitment and ectopic activation of B2 cells via the TLR signaling pathway in perivascular adipose tissue (PVAT), and, subsequently, an increase in circulating IgG and IgG3 led to the enhanced disease development. This is a potential link between microbiota alterations and B cells in the context of atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerotic diseases comprise systemic disorders that represent a leading cause of mortality and morbidity worldwide. Although the molecular mechanisms responsible for the development of atherosclerosis are not completely understood, studies over the past decade have highlighted the critical role of the immune system in this process. In particular, cells, both the innate (macrophages) and adaptive (T cell and B lymphocytes) branches, of the immune system appear to play an important role in the development of this common condition [1,2,3]. In addition, recent studies have revealed that the gut microbiome exerts direct effects on the immune responses that regulate chronic inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease, and atherosclerosis [4, 5]. Moreover, abnormal cholesterol concentrations, an unhealthy diet, and alterations in the gut microbiota have been linked to atherosclerosis progression [4]. Accordingly, the intriguing relationship between commensal microbes and atherosclerosis has received increasing attention over the past few years. However, the specific mechanisms whereby commensal microbes regulate the development of atherosclerosis are just beginning to be elucidated [6, 7], with the role of the immune system in commensal microbe-derived atherosclerosis, i.e., metabolism-independent pathways, remaining largely unexplored. Therefore, the purpose of this review is to highlight current knowledge regarding the complex interplay between the microbiota and atherosclerosis via the immune system, with a particular focus on the associated roles played by humoral immunity, including both B1 and B2 cells.
Association of commensal microbiota with cardiovascular diseases
Atherosclerosis constitutes the main contributor to cardiovascular mortality, which is strongly associated with risk factors such as gender, age, genetic background, unhealthy diet, smoking, hypertension, diabetes mellitus, obesity, hyperlipidemia, and socioeconomic deprivation [8, 9]. However, minimizing such risk factors does not altogether protect against atherosclerosis. At least a 50% residual risk may remain, even in conjunction with high-potency statin therapy [10, 11]. This is because inflammation and hypercholesterolemia comprise the two key etiological factors for atherosclerosis [2, 12], whereas current therapeutic options for treating or preventing atherosclerosis mainly focus on lipid control alone, rather than resolving inflammation [13].
Bacterial infection has been proposed as a trigger of inflammation in atherosclerosis [13, 14]. To date, most epidemiological evidence has supported that a relationship between infection and atherosclerosis exists, based on associations between circulating antibacterial antibodies and atherosclerosis. For example, foreign antigenic stimuli, such as Porphyromonas gingivalis, Chlamydia pneumoniae, enterovirus, and cytomegalovirus, have been identified as potentially causative or bystander participants [15]. Furthermore, several studies have found that C. pneumoniae is present in the atherosclerotic lesions of patients with previous exposure and that infection with this bacterium may exacerbate atherosclerosis in animals [14, 16]. In addition to C. pneumoniae, almost 50 bacterial species have been detected in atherosclerotic plaques, whereas none was found in control tissues [5, 17].
Moreover, bacteria-host interactions have been associated with the initiation, perpetuation, and re-exacerbation of atherosclerotic lesions, eventually leading to thrombus formation and acute coronary syndromes or stroke [18,19,20,21,22]. Recent studies also showed that atheromas collect bacteria from the circulation and microbial molecular signatures have been detected at progressively higher frequencies in advanced lesions [7, 23]. In addition, some studies showed a correlation between aortic stiffness and blood level of soluble CD14, the main endotoxin receptor and defense against gram-negative bacteria, with high levels resulting in aortic stiffness [24, 25].
Although such evidence supports that bacterial infection may play a role in the atherosclerotic process from initial endothelial dysfunction to clinical manifestations, whether an infection initiates or augments atherosclerosis development remains uncertain. For example, only a few infectious agents such as Aggregatibacter actinomycetemcomitans, C. pneumoniae, Helicobacter pylori, and P. gingivalis have been shown to potentially contribute to atherosclerosis by increasing lesion areas in animal models [26]. Furthermore, several large, randomized clinical trials involving antibiotic therapy have shown no benefit, to date, regarding cardiovascular endpoints [27], with the recent suggestion that the organization of bacteria in antibiotic-resistant biofilms may have contributed to these negative results [28, 29]. This reflects the view that the total pathogenic infectious burden in any individual may be more important than any singular microbe as a risk factor for cardiovascular disease [30]. Therefore, an important endogenous bacterial source of infection, the ability of commensal microbes to potentially exert a substantial impact on atherosclerosis, has been recognized [7, 31].
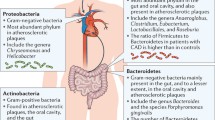
Consistent with this observation, numerous studies have reported the detection of bacterial DNA in atherosclerotic lesions, as well as in human atherosclerotic plaques [7, 17, 32]. In particular, pyrosequencing results revealed that the bacteria in lesions are derived from the gut and oral cavity [7], suggesting the possible involvement of oral and gut microbiota in the development of the disease.
Furthermore, fecal samples from healthy individuals and patients with symptomatic atherosclerosis were found to differ by several species. For example, the genus Collinsella was enriched in patients with symptomatic atherosclerosis, whereas Eubacterium and Roseburia were enriched in healthy controls [33]. Moreover, in patients with coronary artery disease, the number of Lactobacillales and the ratio of Firmicutes to Bacteroidetes increased, along with the levels of Escherichia coli, Klebsiella spp., Enterobacter aerogenes, Ruminococcus gnavus, Eggerthella lenta, Streptococcus spp., Lactobacillus salivarius, Solobacterium moorei, and Atopobium parvulum. In comparison, the number of Bacteroidetes, Roseburia intestinalis, Faecalibacterium prausnitzii, Bacteroides spp., Prevotella copri, and Alistipes shahii decreased [34, 35].
Accordingly, treatment with systemic antibiotics in humans showed no reduction in cardiovascular event rates, possibly owing to the specific eradication of gram-positive strains by azithromycin, whereas gram-negative (LPS-containing) intestinal bacteria remained unaffected [28]. Moreover, the approach of using broad-spectrum antibiotics to deplete the gut microbial population also causes a reduction in numerous beneficial products derived from commensal microbes [6, 36].
Gut dysbiosis and inflammation in atherosclerosis
The specific mechanisms whereby commensal microbes may regulate the development of atherosclerosis are just beginning to be elucidated. However, numerous studies have identified the ability of commensal microbe-derived metabolites to act as hormones or bioactive metabolites modulating cardiovascular disease risk. These have focused on metabolism-dependent mechanisms, including the gut microbe-derived trimethylamine N-oxide (TMAO) pathway [37], the short-chain fatty acids (SCFA) pathway, and the primary and secondary bile acids pathways. In contrast, metabolism-independent pathways, particularly the role of the immune system in commensal microbe-derived atherosclerosis, remain largely unexplored.
It is considered possible that the intestinal microbiota can regulate atherosclerosis development via bacterial wall compounds, such as endotoxin and lipopolysaccharide (LPS), or indirectly through the regulation of innate immunity and chronic inflammatory tone by bacterial products [38]. For example, in a 5-year epidemiological study of 516 middle-aged men and women, those with plasma LPS levels over 50 pg/mL exhibited a threefold increased risk of developing atherosclerosis, whereas the subpopulation of smokers or ex-smokers with the same LPS level evinced a 13-fold increase [39]. Nevertheless, to our knowledge, intervention studies to lower LPS plasma levels and thereby subsequently decrease cardiovascular disease risk have not been conducted, even though such results would verify the importance of LPS in the etiology of cardiovascular disease [39]. Furthermore, microRNA components of intestinal microbiota [40] can affect macrophage function and subsequent inflammatory tone. In addition, lack of microbiota reduced plasma LPS levels, along with pro-inflammatory cytokine gene expression in macrophages and the aorta, during atherosclerosis development [41].
The interaction of microbes, as well as the components of the bacterial cell, with the immune system was previously considered to be most active in the distal gut [42, 43]. However, recent studies have found that low levels of microbiota can also enter the bloodstream to cause chronic low-grade inflammation systemically [6, 44]. The phenomenon whereby low level of gut-derived bacteria can appear in the circulation is commonly referred to as “metabolic endotoxaemia” and it has been found to be prevalent in atherosclerosis [44].
Generally, the gut barrier plays a critical role in preventing the translocation of bacterial components. This barrier is efficient when the microbiota is complex and stable, whereas under some conditions, such as diets high in fat and cholesterol or certain diseases, major changes could be induced in the host microbiota composition, which in turn has been associated with increased intestinal permeability [45,46,47]. For example, we and others have found that mice fed a high-fat diet exhibited increased intestinal permeability and decreased expression of genes encoding tight junction proteins, including zonula occludens-1 (ZO-1), claudin-1, and occludin, whereas the administration of antibiotics in conjunction with the high-fat diet effectively ameliorated these negative effects [48, 49]. In follow-up experiments, it was confirmed that the obese mice exhibited the highest levels of intestinal permeability; moreover, obesity-prone rats were also found to exhibit increased gut permeability, plasma LPS, and inflammation, albeit with reduced epithelial barrier function as compared with obesity-resistant rats [50,51,52]. In addition, our group also found that lubiprostone attenuates the development of atherosclerotic lesions by ameliorating leaky gut syndrome-induced inflammation through the restoration of the intestinal barrier [53]. Consistent with these observations, individuals with inflammatory bowel disease were at higher risk of developing coronary artery disease, despite having lower rates of traditional risk factors than their age-matched controls, in a longitudinal cohort study [39]. Targeted sampling studies have shown that LPS levels are higher in blood samples recovered from the hepatic vein as opposed to the systemic circulation (direct sampling from the ventricles), providing direct evidence that LPS can be translocated from the gut [54]. Therefore, owing to the compromise of the intestinal barrier, commensal microbes or commensal microbe-derived molecules, such as LPS or peptidoglycan, can readily enter the bloodstream and exert systemic effects, including the induction of infection or chronic low-grade inflammation and immunoreaction, affecting multiple immune populations.
Furthermore, it has become clear that microbiota-derived bioactive compounds can signal to distant organs, contributing to the development of cardiovascular disease states [55]. For example, outer membrane components of gut microbiota such as LPS, other virulence factors, and pathogen-associated molecular patterns (PAMPs) can be detected in human tissues and trigger local and systemic inflammatory responses [56,57,58]. In particular, increased intestinal microbiota-derived LPS load from the colon lumen was shown to be associated with various metabolic abnormalities, including the induction of adipose inflammation. In addition, LPS-induced inflammatory cytokines in perivascular adipose tissue (PVAT), which surrounds nearly all blood vessels, can act in a paracrine manner to exacerbate vascular inflammation and atherosclerosis. Similarly, through the circulation, bacteria can reach visceral fat or atheromas, directly promoting local inflammatory cascades or eliciting a specific immune response [59, 60], thereby indirectly influencing host metabolism and systemic inflammation [15, 61].
Commensal microbe–induced atherosclerosis via immune response
It has long been understood that our immune system can sense various types of bacterial components, such as LPS and peptidoglycan, via cognate pattern receptors located on immune cells [62], and then activate several inflammatory pathways. In general, these pathways involve Toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)–like receptors (NLRs) [63]. In particular, circulating LPS, derived from different gut microbial species, are believed to confer their deleterious effects on developing atherosclerosis primarily through the TLRs and their receptors, for example, cluster of differentiation 14 (CD14), with TLR receptor activation downstream signaling cascades including nuclear factor kappa B (NF-κB) and c-Jun N-terminal kinase pathways. Activation of the NF-κB pathway promotes gene expression that recruits and activates inflammatory cells and downstream molecules such as cytokines, including the pro-inflammatory factors interleukin-6 (IL-6), IL-1, IL-27, tumor necrosis factor alpha (TNF-α), inducible nitric oxide synthase, and leukocyte adhesion molecules [64]. Similarly, activation of the c-Jun N-terminal kinase pathway leads to the upregulation of stress response genes and is implicated in pathological cardiac events [54].
For example, through the use of TLR and low-density lipoprotein (LDL) receptor double knockout mice, several studies have demonstrated that TLRs may be contributors to atherosclerosis development [65]. Indeed, Ding et al. [66] found that a TLR deficiency reduced atherosclerosis without any effect on inflammation. Moreover, inactivation of the TLR pathway by deletion of TLRs or the downstream cytosolic adaptor, myeloid differentiation factor-88 (Myd88), reduces aortic lesions in apolipoprotein E–deficient (ApoE−/−) and LDL receptor–deficient (Ldlr−/−) mice [67]. Notably, all these models showed a reduction of lesion area and regional lipid content without any significant alteration of plasma cholesterol levels. Consistent with these findings, clinical investigations have revealed that the upregulation of TLRs was associated with inflammatory activation in human atherosclerosis and promoted the development of atherosclerosis [68,69,70].
Additionally, another gut microbial PAMP, peptidoglycan, was also found to be associated with atherosclerosis via NLRs. Through peptidoglycan recognition, NLRs promote intracellular bacteria clearance through a program involving NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways [71]. Recently, knockout of NOD1 in mice was shown to significantly reduce the development of atherosclerotic lesions [63]. Moreover, some NOD2-knockout mouse studies revealed that NOD2 represents a critical regulator of intestinal bacterial immunity and helps to maintain the integrity of the gut barrier [72]. In addition, other PAMPs have been identified that may promote atherosclerosis development through NLR protein 3 (NLRP3)-inflammasome-caspase-dependent signaling pathways, causing the conversion of pro-IL-1 beta and pro-IL-18 into active cytokines and subsequent induction of inflammation [73,74,75,76].
Through these pathways, the microbiota activate the innate and adaptive immunity via receptors on endothelial cells, innate lymphoid cells (ILCs), dendritic cells (DCs), other myeloid cells, and lymphocytes [77,78,79]. In turn, this provides stimuli for the activation of leukocytes and arterial cells within atheromas [80].
Moreover, microbial antigens are also associated with the molecular mechanism termed “molecular mimicry” [81], as self-peptides such as heat shock proteins (e.g., mycobacteria, Chlamydia) have also been found to be associated with atherosclerosis [82]. For example, Binder et al. showed that pneumococcal vaccination decreases atherosclerotic lesion formation through a molecular mimicry mechanism between Streptococcus pneumoniae and oxidized LDL [83]. In addition, a recent study has reported that auto-antibodies produced by B lymphocytes are present in plaques, which may cross-react with outer membrane proteins of bacteria, as well as with a cytoskeleton protein involved in atherogenesis [60]. These findings demonstrated that, in human atherosclerotic plaques, a local cross-reactive immune response may occur, wherein antibodies cross-react with a bacterial antigen and a self-protein. These results also illustrated that antibodies and B lymphocytes could play an important role in the disease process [60, 84].
Crosstalk between microbiota and B cells
As an important component of the immune system, B cells play a critical role in inflammation through their ability to detect and process antigens, terminally differentiate into plasma cells, and produce antibodies or cytokines [85]. B cells can also affect atherosclerosis development via production of atherogenic antibodies [86] and secretion of pro-inflammatory cytokines, including TNFα, which represent T cell–independent pathways [87]. However, in contrast to the ability of macrophages and specific T cell subsets to promote inflammation in the vessel wall during atherosclerosis [88,89,90], B cells may have a more complex role in atherosclerosis development through antibody production, which is not yet fully elucidated [91]. For instance, B1 cell–derived natural IgM antibodies have consistently been shown to be athero-protective [92, 93], while B2 cell responses may promote atherogenesis by supporting pro-atherogenic T cells [94].
In general, commensal microbes or commensal microbe-derived LPS or peptidoglycan can be selectively recognized by the hosts’ innate immune TLRs in B cells [95,96,97]. Similar to other immune cells, B cells also exhibit variations in TLR expression patterns; specifically, the signaling via MyD88 is able to modify B cell responses, such as antibody production, antigen presentation, and cytokine secretion [98,99,100]. Notably, recent studies have demonstrated that activation of TLRs, via MyD88 signaling in B cells, is necessary for antibody responses to T cell–dependent (TD) antigens and to influence B cell tolerance, which leads to pathogenic autoantibody production [96, 97, 100]. However, the role of the TLR signaling pathway in B2 cells during atherosclerosis development is not fully elucidated. Nevertheless, in our previous study, we found that under hyperlipidemic conditions, signals driven by the microbiota via the TLR signaling pathway may cause B2 cells to become functionally active, potentially leading to the generation of active antibodies, cytokines, and chemokines, thereby providing a mechanism in which they may be contributing to atherosclerosis development [1, 8, 53].
Extensive studies of the peripheral blood, peritoneal cavity, and other lymphoid tissues have identified B1 and B2 cells as the two main B cell subsets in mice, based on their developmental origin [101]. In addition, the B2 cells that represent the vast majority of B cells, including follicular (FO) as well as marginal zone (MZ) B cells [102] respond to antigen presentation in a TD manner undergoing proliferation, affinity maturation, and isotype class switching to produce a large amount of highly specific antibodies against foreign pathogens [102].
B2 cells reside mainly in the spleen, accounting for 60% of the total number of splenic lymphocytes. Splenic B2 cells comprise approximately 80% FO B2 cells and 10% MZ B2 cells [102]. In general, FO B2 cells predominantly participate in TD antibody responses to highly specific determinants that are usually associated with microbial proteins [103]. In contrast, as MZ B2 cells are peripheral to the FO B2 cells and reside in the marginal sinuses of the spleen [104], the location of the interface between the spleen and the circulation, they are thus located at the first line of defense against blood-borne antigens [103, 105]. MZ B2 cells predominantly give rise to rapid T cell–independent (TI) antibody responses to highly conserved carbohydrate and glycolipid determinants associated with microbes, producing TI antibodies such as the IgM response that bridges the gap between infection and the production of TD antibodies [106,107,108,109,110,111,112,113,114]. Moreover, the high expression of MZ B2 cells of antigen-presenting CD1d molecules, which bond lipids and glycolipids, allows them to act as antigen processing cells for the activation of natural killer T cells (NKT cells) [115].
In parallel, commensal microbes stimulate the homing of DCs, along with neutrophils, to the MZ of the spleen, which has an important role in the activation of MZ B2 cells [107]. In addition, recent findings show that neutrophils occupy peri-MZ areas of the spleen in the absence of infection, being recruited via a non-inflammatory pathway that originates during fetal life and accelerates after birth, a time that coincides with the colonization of mucosal surfaces by bacteria [114]. Moreover, serum natural IgG levels are severely reduced, whereas serum natural IgM levels are normal in germ-free animals [116]. These results indicate the essential role of commensal microbes in the activation of MZ B2 cells and B2 cell–mediated IgG antibody production. Additionally, unique roles were reported for MZ B2 cells in atherosclerosis development, involving enhanced pro-atherogenic T cell responses in mice [117].
Intestinal commensal microbes have come to be accepted as an important antigen source for the activation of specific splenic B2 cells in association with arteriosclerosis. The main pathways by which host and commensal microbiota interact are when commensal microbiota or their metabolites enter the hosts’ circulation. As with other infection antigens, the innate immune system is capable of sensing various types of commensal microbiota components via TLRs, which then signal via MyD88-dependent pathways to activate NF-κB-driven pro-inflammatory signaling, subsequently leading to an adaptive immune response [100, 118]. For example, the results of our recent study collectively demonstrated that under hyperlipidemic conditions, signals driven by the intestinal microbiota, via the TLR signaling pathway, cause B2 cells in the spleen to become functionally active. Subsequently, the activated B2 cells then modify responses such as antigen presentation and antibody production, thereby potentially contributing to the development of atherosclerosis [1].
In addition, Hamze et al. [119] used laser capture microdissection to analyze individual lymphocytes in dissected coronary arteries, finding that the majority of B cells were present in the adventitia of these arteries and that they primarily expressed markers associated with the activated plasmoblast phenotype, suggesting the cells were active at the sites of disease. Moreover, the presence of B cells in the aortic adventitia has been supported by considerable evidence in human and murine models of atherosclerosis, configuring artery tertiary lymphoid organs (ATLO) in blood vessels [120, 121]. Recent studies further support that B cell activation in the adventitia is important for regulating atherosclerosis [119, 122]. In comparison, the PVAT, which is intimately associated with the adventitial layer of the vessel wall, has been implicated, through paracrine effects on the vasculature, to play a pivotal role in the pathogenesis of atherosclerosis [123]. PVAT constitutes a complex mixture of various cell types including immune cells, such as macrophages, T cells, and B cells [124, 125] that histologically form fat-associated lymphoid clusters (FALCs) [126]. Due to the close interaction between PVAT and adventitia, immune cells in the PVAT likely contribute to the development of atherosclerosis. Consistent with this conjecture, we have demonstrated that, under hyperlipidemic conditions, intestinal microbiota may enter the blood owing to the reduced intestinal mucosal barrier capacity. This may then result in the recruitment and ectopic activation of B2 cells, especially FO B2 cells, via the TLR signaling pathway in PVAT, and, subsequently, in an increase in circulating IgG and IgG3, ultimately leading to enhanced disease development [1].
Conclusions and outstanding questions
New insights regarding how atherosclerosis can be affected by commensal microbiota have been provided over the past decade; however, the underlying molecular and cellular mechanisms remain largely unexplored. In this review, we have provided an overview of the metabolism-independent pathways in atherosclerosis development and discussed the possible mechanisms involving splenic B2-B cell activation following commensal microbe dysbiosis and translocation into the bloodstream. As inflammation constitutes a key etiological factor for atherosclerosis, future research must seek to pinpoint the specific immune response mediated by microbiota. In particular, the impact of the microbiota on immune cells and its consequences for atherogenesis await further elucidation.
Finally, various studies have revealed the roles of athero-protective B1 cells and athero-promoting B2 cells, and the disruption of the balance between B1 cells and B2 cells may lead to the progression of atherosclerosis. Therefore, targeting activated B2 cells or induced athero-protective B1 cells might be one of the therapeutic procedures for the subjects with atherosclerosis. Furthermore, a potential translational extension of the current research would be to better characterize the specific humoral immunity in individuals with atherosclerosis. We sought to confirm the existence of antibodies specific for antigens derived against commensal microbes and to develop in vitro diagnostic procedures for assessing the current immunological status of atherosclerosis patients. For instance, we found an IgG3 class of antibodies specific for bacterial antigens in these patients that may prove useful as a translation tool in clinical settings (unpublished observation). Also of clinical relevance, probiotics, and not antibiotics, may effectively alter the state of dysbiosis in subjects with atherosclerosis, while also reducing the specific effect of commensal microbes in the development of atherosclerosis. Additionally, interventional approaches can also be applied to enhance the intestinal function of subjects with atherosclerosis. For instance, patients with coronary heart disease and constipation might present interventional opportunities involving the use of laxative agents for improving intestinal commensal microbiota [53, 127, 128].
References
Chen L, Ishigami T, Nakashima-Sasaki R, Kino T, Doi H, Minegishi S, Umemura S (2016) Commensal microbe-specific activation of B2 cell subsets contributes to atherosclerosis development independently of lipid metabolism. EBioMedicine 13:237–247
Libby P (2002) Inflammation in atherosclerosis. Nature 420:868–874
Wick G, Knoflach M, Xu Q (2004) Autoimmune and inflammatory mechanisms in atherosclerosis. Annu Rev Immunol 22:361–403
Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, Thovarai V, Badger JH, Vats R, Sundd P, Tang HY et al (2018) An interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity 49(943–957) e949:943–957.e9
Jonsson AL, Backhed F (2017) Role of gut microbiota in atherosclerosis. Nat Rev Cardiol 14:79–87
Brown JM, Hazen SL (2015) The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu Rev Med 66:343–359
Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Backhed F (2011) Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A 108(Suppl 1):4592–4598
Chen L, Ishigami T (2016) Intestinal microbiome and atherosclerosis - authors’ reply. EBioMedicine 13:19–20
Matziouridou C, Marungruang N, Nguyen TD, Nyman M, Fak F (2016) Lingonberries reduce atherosclerosis in Apoe(-/-) mice in association with altered gut microbiota composition and improved lipid profile. Mol Nutr Food Res 60:1150–1160
Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG et al (2008) Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 359:2195–2207
Tang WH, Hazen SL (2014) The contributory role of gut microbiota in cardiovascular disease. J Clin Invest 124:4204–4211
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17:1410–1422
Li J, Lin S, Vanhoutte PM, Woo CW, Xu A (2016) Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in apoe-/- mice. Circulation 133:2434–2446
Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobe EP, Carlquist JF (1998) Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azithromycin prevents it in a rabbit model. Circulation 97:633–636
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM et al (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63
Thomas M, Wong Y, Thomas D, Ajaz M, Tsang V, Gallagher PJ, Ward ME (1999) Relation between direct detection of Chlamydia pneumoniae DNA in human coronary arteries at postmortem examination and histological severity (Stary grading) of associated atherosclerotic plaque. Circulation 99:2733–2736
Ott SJ, El Mokhtari NE, Musfeldt M, Hellmig S, Freitag S, Rehman A, Kuhbacher T, Nikolaus S, Namsolleck P, Blaut M et al (2006) Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation 113:929–937
Angiolillo DJ, Liuzzo G, Pelliccioni S, De Candia E, Landolfi R, Crea F, Maseri A, Biasucci LM (2003) Combined role of the Lewis antigenic system, Chlamydia pneumoniae, and C-reactive protein in unstable angina. J Am Coll Cardiol 41:546–550
Clayton TC, Thompson M, Meade TW (2008) Recent respiratory infection and risk of cardiovascular disease: case-control study through a general practice database. Eur Heart J 29:96–103
Grau AJ, Urbanek C, Palm F (2010) Common infections and the risk of stroke. Nat Rev Neurol 6:681–694
Ruggeri ZM (2002) Platelets in atherothrombosis. Nat Med 8:1227–1234
Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P (2004) Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med 351:2611–2618
Ford PJ, Gemmell E, Chan A, Carter CL, Walker PJ, Bird PS, West MJ, Cullinan MP, Seymour GJ (2006) Inflammation, heat shock proteins and periodontal pathogens in atherosclerosis: an immunohistologic study. Oral Microbiol Immunol 21:206–211
Amar J, Ruidavets JB, Bal Dit Sollier C, Bongard V, Boccalon H, Chamontin B, Drouet L, Ferrieres J (2003) Soluble CD14 and aortic stiffness in a population-based study. J Hypertens 21:1869–1877
Vlachopoulos C, Dima I, Aznaouridis K, Vasiliadou C, Ioakeimidis N, Aggeli C, Toutouza M, Stefanadis C (2005) Acute systemic inflammation increases arterial stiffness and decreases wave reflections in healthy individuals. Circulation 112:2193–2200
Rosenfeld ME, Campbell LA (2011) Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost 106:858–867
Cleland JG, Huan Loh P, Freemantle N, Clark AL, Coletta AP (2004) Clinical trials update from the European Society of Cardiology: SENIORS, ACES, PROVE-IT, ACTION, and the HF-ACTION trial. Eur J Heart Fail 6:787–791
Grayston JT, Kronmal RA, Jackson LA, Parisi AF, Muhlestein JB, Cohen JD, Rogers WJ, Crouse JR, Borrowdale SL, Schron E, Knirsch C (2005) Azithromycin for the secondary prevention of coronary events. N Engl J Med 352:1637–1645
Muhlestein JB, Anderson JL, Carlquist JF, Salunkhe K, Horne BD, Pearson RR, Bunch TJ, Allen A, Trehan S, Nielson C (2000) Randomized secondary prevention trial of azithromycin in patients with coronary artery disease: primary clinical results of the ACADEMIC study. Circulation 102:1755–1760
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352:1685–1695
Singh V, Yeoh BS, Vijay-Kumar M (2016) Gut microbiome as a novel cardiovascular therapeutic target. Curr Opin Pharmacol 27:8–12
Pessi T, Karhunen V, Karjalainen PP, Ylitalo A, Airaksinen JK, Niemi M, Pietila M, Lounatmaa K, Haapaniemi T, Lehtimaki T et al (2013) Bacterial signatures in thrombus aspirates of patients with myocardial infarction. Circulation 127(1219–1228):e1211–e1216
Karlsson FH, Fak F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Backhed F, Nielsen J (2012) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 3:1245
Emoto T, Yamashita T, Sasaki N, Hirota Y, Hayashi T, So A, Kasahara K, Yodoi K, Matsumoto T, Mizoguchi T, Ogawa W, Hirata KI (2016) Analysis of gut microbiota in coronary artery disease patients: a possible link between gut microbiota and coronary artery disease. J Atheroscler Thromb 23:908–921
Jie Z, Xia H, Zhong SL, Feng Q, Li S, Liang S, Zhong H, Liu Z, Gao Y, Zhao H, Zhang D, Su Z, Fang Z, Lan Z, Li J, Xiao L, Li J, Li R, Li X, Li F, Ren H, Huang Y, Peng Y, Li G, Wen B, Dong B, Chen JY, Geng QS, Zhang ZW, Yang H, Wang J, Wang J, Zhang X, Madsen L, Brix S, Ning G, Xu X, Liu X, Hou Y, Jia H, He K, Kristiansen K (2017) The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun 8:845
Jernberg C, Lofmark S, Edlund C, Jansson JK (2010) Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 156:3216–3223
Matsuzawa Y, Nakahashi H, Konishi M, Sato R, Kawashima C, Kikuchi S, Akiyama E, Iwahashi N, Maejima N, Okada K, Ebina T, Hibi K, Kosuge M, Ishigami T, Tamura K, Kimura K (2019) Microbiota-derived trimethylamine N-oxide predicts cardiovascular risk after STEMI. Sci Rep 9:11647
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
Harris K, Kassis A, Major G, Chou CJ (2012) Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J Obes 2012:879151–879114
Swaminathan G, Rossi F, Sierra LJ, Gupta A, Navas-Martin S, Martin-Garcia J (2012) A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS Pathog 8:e1002937. https://doi.org/10.1371/journal.ppat.1002937
Kasahara K, Tanoue T, Yamashita T, Yodoi K, Matsumoto T, Emoto T, Mizoguchi T, Hayashi T, Kitano N, Sasaki N, Atarashi K, Honda K, Hirata KI (2017) Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res 58:519–528
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229–241
Wells JM, Rossi O, Meijerink M, van Baarlen P (2011) Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A 108(Suppl 1):4607–4614
Neves AL, Coelho J, Couto L, Leite-Moreira A, Roncon-Albuquerque R Jr (2013) Metabolic endotoxemia: a molecular link between obesity and cardiovascular risk. J Mol Endocrinol 51:R51–R64
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563
Ghosh SS, Bie J, Wang J, Ghosh S (2014) Oral supplementation with non-absorbable antibiotics or curcumin attenuates western diet-induced atherosclerosis and glucose intolerance in LDLR-/- mice--role of intestinal permeability and macrophage activation. PLoS One 9:e108577. https://doi.org/10.1371/journal.pone.0108577
Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M, Frenette PS (2015) Neutrophil ageing is regulated by the microbiome. Nature 525:528–532
Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM (2007) Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50:2374–2383
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57:1470–1481
Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM et al (2009) Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58:1091–1103
de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE (2010) Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299:G440–G448
Muccioli GG, Naslain D, Backhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:392
Arakawa K, Ishigami T, Nakai-Sugiyama M, Chen L, Doi H, Kino T, Minegishi S, Saigoh-Teranaka S, Sasaki-Nakashima R, Hibi K, Kimura K, Tamura K (2019) Lubiprostone as a potential therapeutic agent to improve intestinal permeability and prevent the development of atherosclerosis in apolipoprotein E-deficient mice. PLoS One 14:e0218096. https://doi.org/10.1371/journal.pone.0218096
Tang WHW, Li DY, Hazen SL (2018) Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol 16:137–154
Caesar R, Reigstad CS, Backhed HK, Reinhardt C, Ketonen M, Lunden GO, Cani PD, Backhed F (2012) Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 61:1701–1707
Ellis TN, Kuehn MJ (2010) Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev 74:81–94
Jager J, Marwitz S, Tiefenau J, Rasch J, Shevchuk O, Kugler C, Goldmann T, Steinert M (2014) Human lung tissue explants reveal novel interactions during Legionella pneumophila infections. Infect Immun 82:275–285
Nakao R, Hasegawa H, Ochiai K, Takashiba S, Ainai A, Ohnishi M, Watanabe H, Senpuku H (2011) Outer membrane vesicles of Porphyromonas gingivalis elicit a mucosal immune response. PLoS One 6:e26163. https://doi.org/10.1371/journal.pone.0026163
Burioni R, Canducci F, Saita D, Perotti M, Mancini N, De Marco D, Clementi N, Chieffo A, Denaro M, Cianflone D et al (2009) Antigen-driven evolution of B lymphocytes in coronary atherosclerotic plaques. J Immunol 183:2537–2544
Canducci F, Saita D, Foglieni C, Piscopiello MR, Chiesa R, Colombo A, Cianflone D, Maseri A, Clementi M, Burioni R (2012) Cross-reacting antibacterial auto-antibodies are produced within coronary atherosclerotic plaques of acute coronary syndrome patients. PLoS One 7:e42283. https://doi.org/10.1371/journal.pone.0042283
Caesar R, Fak F, Backhed F (2010) Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med 268:320–328
Mukherjee S, Karmakar S, Babu SP (2016) TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis 20:193–204
Ma J, Li H (2018) The role of gut microbiota in atherosclerosis and hypertension. Front Pharmacol 9:1082
Hennessy EJ, Parker AE, O’Neill LA (2010) Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 9:293–307
Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ (2002) Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 105:1158–1161
Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK (2010) High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5:e12191
Sallam T, Ito A, Rong X, Kim J, van Stijn C, Chamberlain BT, Jung ME, Chao LC, Jones M, Gilliland T, Wu XH, Su GL, Tangirala RK, Tontonoz P, Hong C (2014) The macrophage LBP gene is an LXR target that promotes macrophage survival and atherosclerosis. J Lipid Res 55:1120–1130
Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT et al (2004) Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med 10:416–421
Bjorkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, Ordija CM, Dowley NE, Golenbock DT, Freeman MW (2004) The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics 19:319–330
Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A 101:10679–10684
Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE (2014) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14:9–23
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307:731–734
Mukherjee S, Joardar N, Sengupta S, Sinha Babu SP (2018) Gut microbes as future therapeutics in treating inflammatory and infectious diseases: lessons from recent findings. J Nutr Biochem 61:111–128
Prochnicki T, Latz E (2017) Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab 26:71–93
Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481:278–286
van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA (2011) Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol 32:110–116
Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D (2012) Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37:158–170
Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, Umesaki Y, Mathis D, Benoist C, Relman DA, Kasper DL (2012) Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149:1578–1593
Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D (2012) Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336:1321–1325
Libby P, Loscalzo J, Ridker PM, Farkouh ME, Hsue PY, Fuster V, Hasan AA, Amar S (2018) Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. J Am Coll Cardiol 72:2071–2081
Bachmaier K, Neu N, de la Maza LM, Pal S, Hessel A, Penninger JM (1999) Chlamydia infections and heart disease linked through antigenic mimicry. Science 283:1335–1339
Riley E, Dasari V, Frishman WH, Sperber K (2008) Vaccines in development to prevent and treat atherosclerotic disease. Cardiol Rev 16:288–300
Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ (2003) Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9:736–743
Ishigami T, Abe K, Aoki I, Minegishi S, Ryo A, Matsunaga S, Matsuoka K, Takeda H, Sawasaki T, Umemura S, Endo Y (2013) Anti-interleukin-5 and multiple autoantibodies are associated with human atherosclerotic diseases and serum interleukin-5 levels. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27:3437–3445
LeBien TW, Tedder TF (2008) B lymphocytes: how they develop and function. Blood 112:1570–1580
Karper JC, de Jager SC, Ewing MM, de Vries MR, Bot I, van Santbrink PJ, Redeker A, Mallat Z, Binder CJ, Arens R et al (2013) An unexpected intriguing effect of Toll-like receptor regulator RP105 (CD180) on atherosclerosis formation with alterations on B-cell activation. Arterioscler Thromb Vasc Biol 33:2810–2817
Kyaw T, Tipping P, Bobik A, Toh BH (2017) Opposing roles of B lymphocyte subsets in atherosclerosis. Autoimmunity 50:52–56
Hansson GK, Hermansson A (2011) The immune system in atherosclerosis. Nat Immunol 12:204–212
Lahoute C, Herbin O, Mallat Z, Tedgui A (2011) Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol 8:348–358
Tabas I (2010) Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10:36–46
Kyaw T, Tipping P, Toh BH, Bobik A (2011) Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis. Curr Opin Lipidol 22:373–379
Sage AP, Mallat Z (2014) Multiple potential roles for B cells in atherosclerosis. Ann Med 46:297–303
Tsiantoulas D, Sage AP, Mallat Z, Binder CJ (2015) Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb Vasc Biol 35:296–302
Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vre E, Esposito B, Vilar J et al (2010) B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 207:1579–1587
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ (2010) TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 184:1840–1848
Pasare C, Medzhitov R (2005) Control of B-cell responses by Toll-like receptors. Nature 438:364–368
Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderon Gomez E, Sweenie CH, Hao Y, Freitas AA, Steinhoff U, Anderton SM et al (2008) TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol 180:4763–4773
Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, Kuhl AA, Loddenkemper C, Haury M, Nedospasov SA et al (2010) Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity 33:777–790
Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A (2012) Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol 12:282–294
Perry HM, Bender TP, McNamara CA (2012) B cell subsets in atherosclerosis. Front Immunol 3:373
Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ (2014) B cells and humoral immunity in atherosclerosis. Circ Res 114:1743–1756
Martin F, Kearney JF (2002) Marginal-zone B cells. Nat Rev Immunol 2:323–335
Victora GD (2014) ILCs in the zone. Nat Immunol 15:313–314
Liu YJ, Oldfield S, MacLennan IC (1988) Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur J Immunol 18:355–362
Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM (2004) B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity 21:379–390
Balazs M, Martin F, Zhou T, Kearney J (2002) Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity 17:341–352
Cariappa A, Mazo IB, Chase C, Shi HN, Liu H, Li Q, Rose H, Leung H, Cherayil BJ, Russell P, von Andrian U, Pillai S (2005) Perisinusoidal B cells in the bone marrow participate in T-independent responses to blood-borne microbes. Immunity 23:397–407
Choi YS, Baumgarth N (2008) Dual role for B-1a cells in immunity to influenza virus infection. J Exp Med 205:3053–3064
Guinamard R, Okigaki M, Schlessinger J, Ravetch JV (2000) Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat Immunol 1:31–36
Kruetzmann S, Rosado MM, Weber H, Germing U, Tournilhac O, Peter HH, Berner R, Peters A, Boehm T, Plebani A, Quinti I, Carsetti R (2003) Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med 197:939–945
Martin F, Oliver AM, Kearney JF (2001) Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 14:617–629
Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, Plebani A, Kumararatne DS, Bonnet D, Tournilhac O, Tchernia G, Steiniger B, Staudt LM, Casanova JL, Reynaud CA, Weill JC (2004) Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 104:3647–3654
Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G, Knowles DM, Tam W, Chiu A, Bussel JB, Serrano S, Lorente JA, Bellosillo B, Lloreta J, Juanpere N, Alameda F, Baró T, de Heredia CD, Torán N, Català A, Torrebadell M, Fortuny C, Cusí V, Carreras C, Diaz GA, Blander JM, Farber CM, Silvestri G, Cunningham-Rundles C, Calvillo M, Dufour C, Notarangelo LD, Lougaris V, Plebani A, Casanova JL, Ganal SC, Diefenbach A, Aróstegui JI, Juan M, Yagüe J, Mahlaoui N, Donadieu J, Chen K, Cerutti A (2011) B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol 13:170–180
Zietara N, Lyszkiewicz M, Krueger A, Weiss S (2011) ICOS-dependent stimulation of NKT cells by marginal zone B cells. Eur J Immunol 41:3125–3134
Pereira P, Forni L, Larsson EL, Cooper M, Heusser C, Coutinho A (1986) Autonomous activation of B and T cells in antigen-free mice. Eur J Immunol 16:685–688
Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, Weller S, Tsiantoulas D, Raffort J, Marcus D, Finigan A, Kitt L, Figg N, Schirmbeck R, Kneilling M, Yeo GSH, Binder CJ, de la Pompa JL, Mallat Z (2017) Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med 23:601–610
Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM (2006) Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation 114:2482–2489
Hamze M, Desmetz C, Berthe ML, Roger P, Boulle N, Brancherau P, Picard E, Guzman C, Tolza C, Guglielmi P (2013) Characterization of resident B cells of vascular walls in human atherosclerotic patients. J Immunol 191:3006–3016
Srikakulapu P, Hu D, Yin C, Mohanta SK, Bontha SV, Peng L, Beer M, Weber C, McNamara CA, Grassia G, Maffia P, Manz RA, Habenicht AJR (2016) Artery tertiary lymphoid organs control multilayered territorialized atherosclerosis B-cell responses in aged mice. Arterioscler Thromb Vasc Biol 36(6):1174–1185
Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, Spanbroek R, Lippert B, Reardon CA, Getz GS, Fu Y-X, Hehlgans T, Mebius RE, van der Wall M, Kruspe D, Englert C, Lovas A, Hu D, Randolph GJ, Weih F, Habenicht AJR (2009) Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J Exp Med 206(1):233–248
Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, Weih F, Weber C, Gerdes N, Habenicht AJ (2014) Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res 114:1772–1787
Rajsheker S, Manka D, Blomkalns AL, Chatterjee TK, Stoll LL, Weintraub NL (2010) Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol 10:191–196
Kawahito H, Yamada H, Irie D, Kato T, Akakabe Y, Kishida S, Takata H, Wakana N, Ogata T, Ikeda K, Ueyama T, Matoba S, Mori Y, Matsubara H (2013) Periaortic adipose tissue-specific activation of the renin-angiotensin system contributes to atherosclerosis development in uninephrectomized apoE-/- mice. Am J Physiol Heart Circ Physiol 305:H667–H675
Srikakulapu P, Upadhye A, Rosenfeld SM, Marshall MA, McSkimming C, Hickman AW, Mauldin IS, Ailawadi G, Lopes MBS, Taylor AM, McNamara CA (2017) Perivascular adipose tissue harbors atheroprotective IgM-producing B cells. Front Physiol 8:719
Newland SA, Mohanta S, Clément M, Taleb S, Walker JA, Nus M, Sage AP, Yin C, Hu D, Kitt LL, Finigan AJ, Rodewald H-R, Binder CJ, McKenzie ANJ, Habenicht AJ, Mallat Z (2017) Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat Commun 8(1):15781
Kato T, Honda Y, Kurita Y, Iwasaki A, Sato T, Kessoku T, Uchiyama S, Ogawa Y, Ohkubo H, Higurashi T, Yamanaka T, Usuda H, Wada K, Nakajima A (2017) Lubiprostone improves intestinal permeability in humans, a novel therapy for the leaky gut: a prospective randomized pilot study in healthy volunteers. PLoS One 12:e0175626. https://doi.org/10.1371/journal.pone.0175626
Mishima E, Fukuda S, Mukawa C, Yuri A, Kanemitsu Y, Matsumoto Y, Akiyama Y, Fukuda NN, Tsukamoto H, Asaji K, Shima H, Kikuchi K, Suzuki C, Suzuki T, Tomioka Y, Soga T, Ito S, Abe T (2017) Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int 92:634–645
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Ishigami, T., Doi, H. et al. Gut microbiota and atherosclerosis: role of B cell for atherosclerosis focusing on the gut-immune-B2 cell axis. J Mol Med 98, 1235–1244 (2020). https://doi.org/10.1007/s00109-020-01936-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-020-01936-5