
Abstract
The Plasmodium falciparum P0 ribosomal phosphoprotein (PfP0) was identified for the first time by screening a cDNA expression library of P. falciparum parasites with sera from malaria-immune individuals. Due to its localization on the surface of different parasite life-cycle stages (merozoites and gametocytes) and its recognition by invasion-blocking antibodies, PfP0 has been considered a potential malaria-vaccine component. In this study, 16 20-mer-long synthetic peptides spanning the entire PfP0 sequence were evaluated by means of receptor–ligand assays with human red blood cells (RBCs) in order to determine the role played by these peptides in the invasion process. Four RBC high-activity binding peptides (HABPs), located mostly toward the N-terminal region, were identified: HABP 33898 (1MAKLSKQQKKQMYIEKLSSL20), HABP 33900 (41ASVRKSLRGKATILMGKNTRY60), HABP 33901 (61IRTALKKNLQAVPQIEKLLPY 80), and HABP 33906 (161LIKQGEKVTASSATLLRKFNY180). The binding pattern of HABPs 33898 and 33906 to enzyme-treated RBCs suggests receptors of protein nature for these two HABPs, one of which could correspond to a common 58-kDa RBC membrane protein, as indicated by results of cross-linking assays. Both HABPs exhibited high content of α-helical features and prevented P. falciparum merozoite invasion to RBCs in vitro by up to 91%. The invasion-blocking ability reported here for these PfP0 HABPs supports their inclusion in immunological studies with the aim of assessing their potential as candidates for a vaccine against P. falciparum malaria.
Similar content being viewed by others


Introduction
Malaria high morbidity and mortality rates impose an immense burden to public health systems worldwide. It has been estimated that around 300–500 million new malaria cases occur each year and that nearly three million people die as a result of this parasitic disease, being sub-Saharan African children under the age of five the most vulnerable population [1]. These alarming statistics highlight the urgent need to develop therapeutic and prophylactic methods capable of controlling the spread of this dreadful disease.
Studies by Cohen and colleagues [2], showing that nonimmune individuals could be protected against malaria by the passive transfer of antibodies from malaria-immune individuals, were crucial for establishing the importance of humoral mechanisms in the induction of an effector antiparasitic response. The next step consisted in determining the specificity of such antibodies by screening cDNA expression libraries of Plasmodium falciparum parasites with sera from acute malaria patients and malaria-immune individuals [3, 4]. Such immunoscreening studies found a λPf4 clone reacting exclusively and extensively with sera from immune individuals and whose sequence corresponded to the P. falciparum P0 ribosomal phosphoprotein (PfP0), a homolog of the mammalian P0 protein [5].
The human ribosomal phosphoprotein P0 associates with two other acidic ribosomal phosphoproteins, P1 and P2, to form the (P1)2–P0–(P2)2 pentameric protein complex that constitutes the stalk region at the GTPase center of the large ribosomal subunit [6]. In contrast to P1 and P2, P0 has been shown to be crucial for ribosomal activity and cell viability in Saccharomyces cerevisiae [7]. In P. falciparum, P0 is a 316-amino-acid-long protein (∼37 kDa) encoded by a single exon and whose sequence is conserved among different parasite strains. PfP0 is transcribed in all asexual intra-erythrocytic stages, showing a maximum transcription peak during trophozoite development [8].
Interestingly, even though PfP0 is located mainly in the cytoplasm, specific anti-PfP0 antibodies capable of inhibiting P. falciparum invasion in vitro have been found to react with merozoite and gametocyte surface epitopes [8, 9], thus raising questions about whether PfP0 is translocated or transported to merozoite surface during the invasion of red blood cells (RBCs). Evidence on behalf of this hypothesis has been gathered by studies showing P0 localization on the surface of other apicomplexan parasites, as well as on the surface of yeast cells and some mammalian cell lines [9–11]. However, the trafficking mechanism of P0 to cell surface has not been elucidated up to the moment due to the lack of hydrophobic sequences, transmembrane motifs, and Plasmodium exporting elements in its sequence [12]. A preliminary study suggests the existence of other membrane-trafficking mechanisms (not yet characterized), mediated via P0 interaction with hypothetical integral membrane proteins [12]. In fact, Sehgal et al. have recently reported the translocation of Toxoplasma gondii P0 (TgP0) to parasite surface despite the absence of important export sequences in this protein [11].
Antibodies directed against the C-terminal region of P0 have been detected in 10–15% of patients with autoimmune disorders such as systemic lupus erythematosus (SLE). The specificity of such antibodies has been narrowed to a 6-residue-motif (311 GFGLFD 316) common to all P ribosomal proteins [13, 14]. Immune responses against P0 have been also detected in patients with chronic Chagas’ disease and leishmaniasis [15, 16]. In all these cases, reactivity has been limited to the carboxy-terminal domain. Furthermore, several studies have revealed cross-reactivity between the C-terminal region of the human P0 protein (HuP0) and P0 proteins of Trypanosoma cruzi and P. falciparum parasites [17, 18]. Such cross-reactivity between PfP0 and HuP0 highlights the need to finely map PfP0 in order to identify nonautoimmune reactive short sequences suitable to be included as components of a synthetic antimalarial vaccine [9].
Studies on the immunogenicity and antigenicity of PfP0 have shown that antibodies targeting PfP0-N (residues 7–61) and PfP0-C (residues 61–316) terminal domains have low effect on the parasite’s intra-erythrocytic development, but specifically inhibit reinvasion of RBCs by P. falciparum merozoites, although the mechanism remains unclear [8, 19]. These antibodies have been also shown to protect mice against experimental challenge with the lethal Plasmodium yoelii 17XL strain [20]. In another study, a 16-amino acid-long region denoted as the P domain (301EEEEEEDGFMGFGMFD306) and having no cross-reactivity with HuP0 was found to induce protection against malaria in mice [21]. Additionally, antibodies against blood-stage antigens such as PfP0, merozoite surface protein 1 (MSP-1), and erythrocyte binding protein-175 (EBA-175) are frequently detected in newborn children from areas of high malaria endemicity. Three PfP0-derived peptides (33DNVGSNQMASVRKSLR48, 42SVRKSLRGKATILMGKNT59, and 285AKADEPKKEEAKKVE299) corresponding to the N- and C-termini of PfP0 have been shown to stimulate T cell-mediated responses in newborn infants [22].
Sharma and Pathak [23] have considered PfP0 an excellent target for an antimalarial vaccine due to its involvement as ligand in merozoite invasion to RBCs. Moreover, its surface localization and the protective efficacy shown by the results of passive and active immunization trials suggest that it is a potent immunogen.
In this study, we have identified four 20-amino-acid-long peptides in the PfP0 protein having high specific binding activity to RBCs, as determined by a sensitive and highly specific receptor–ligand methodology. These high activity binding peptides (HABPs) are located mostly toward the N-terminal and central regions and two of them strongly inhibit merozoite invasion to RBCs. No amino acid sequence variability was found in three out of the four HABPs being identified in this study, while HABP 33906 contained just one amino acid variation at its C-terminal portion in the P. falciparum 7G8 strain, compared to P. falciparum isolates from other geographical regions, as indicated by polymorphism studies. The results of this study support including PfP0 HABPs in additional studies aimed at designing a minimal subunit-based, multi-antigenic, multistage, chemically synthesized antimalarial vaccine.
Materials and methods
Peptide synthesis and radiolabeling
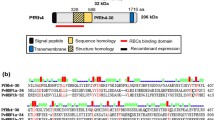
Sixteen nonoverlapping 20-amino-acid-long peptides covering the entire sequence of PfP0 reported in the 7G8 strain (accession no. 4191737) [5] were synthesized using t-Boc amino acids (Bachem) and MBHA resin (0.5 meq/g), following the solid-phase multiple peptide synthesis methodology [24]. The sequences of these chemically synthesized PfP0 peptides are shown in Fig. 1, indicating the name assigned to each peptide according to our institute’s serial numbering system. Peptides were cleaved by the low–high hydrogen fluoride technique [25] and analyzed by reversed-phase high-performance liquid chromatography and matrix assisted laser desorption/ionization time-of-flight mass spectrometry. A tyrosine residue was added to the carboxyl-terminus of those peptides not containing this residue in their sequence to enable radiolabeling with Na125I [26]. Briefly, peptides were individually radiolabeled using 5 μL Na125I (100 mCi/mL; MP Biomedicals) and 15 μL chloramine-T (2.8 mg/mL) as oxidizing agent. The reaction was stopped after 15 min by adding 15 μL sodium metabisulfite (2.3 mg/mL), and radiolabeled peptides were separated by eluting the reaction mixture through a Sephadex G-10 column (Pharmacia, Uppsala, Sweden). Each eluted fraction was then analyzed in a gamma counter (Auto Gamma Cobra II Packard).

a Binding profile of PfP0 synthetic peptides to RBCs. Black bars represent percentage of binding activity defined as the amount of peptide (picomoles) binding specifically per added peptide (picomoles). Peptides having a binding percentage ≥2 were considered HABPs according to previously described criteria [26]. Each assay was carried out in triplicate. The last amino acids in peptide 33913 highlighted in bold types correspond to the human P0 protein motif reported to be recognized by antibodies in the sera of autoimmune patients. A tyrosine residue was added at the C terminus of those peptides not containing such residue in their sequence to enable radiolabeling. b Binding profile of each PfP0-HABP to HeLa cells and WBCs. Data were analyzed under the same conditions as in a. c Saturation curves of HABPs 30900, 30901, and 30906. Increasing amounts of radiolabeled peptide were added in the presence or absence of unlabeled peptide. The curves represent specific binding. In the Hill plot (inset), the x-axis corresponds to log F = free peptide and the y-axis to log (B/Bmax − B), where B is the amount of bound peptide and Bmax the maximum amount of bound peptide
Binding assays to RBCs
A sensitive and specific receptor–ligand interaction assay thoroughly described elsewhere [26] was used to finely map the RBC binding sequences of PfP0. In brief, 2 × 107 RBCs obtained from healthy donors were incubated with increasing quantities of radiolabeled peptide (0, 200, 400, 600, and 800 nM) in the absence (total binding) or presence (nonspecific binding) of an excess of unlabeled peptide (20 μM). Peptide samples were incubated at room temperature for 90 min and washed twice with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffered saline (HBS), before measuring cell-associated radioactivity in a gamma counter. Assays were carried out in triplicate.
Each PfP0-HABP was also assessed in binding assays with 1 × 106 white blood cells (WBCs) isolated from peripheral blood samples of healthy donors by Ficoll–Hypaque density gradient centrifugation (Pharmacia Biotech Inc), or with 1.2 × 106 HeLa cells. For both types of assays, 96-well cell culture plates were loaded with cells in a final volume of 120 μL and incubated for 90 min at 4°C with increasing quantities of radiolabeled peptide (0, 200, 400, 600, and 800 nM) in the presence or absence of nonradiolabeled peptide. A 100-μL aliquot of this solution was passed through a 60:40 dioctyl phthalate–dibutyl phthalate cushion (density 1.015 g/mL) and centrifuged at 8,000×g for 5 min. Cell-associated radioactivity was quantified as described above.
Saturation assays
Modified binding assays were carried out to determine the binding constants describing binding of PfP0 HABPs to RBCs. Briefly, 1.5 × 107 RBC suspensions were incubated with increasing concentrations of radiolabeled peptide starting at 0 and increasing up to 1,900 nM in the absence or presence of unlabeled peptide (20 μM) in a final volume of 255 μL. Samples were incubated for 90 min at room temperature and then washed twice with HBS before measuring cell-associated radioactivity in a gamma counter [26].
Enzymatic treatment
PfP0 HABPs were assessed with enzyme-treated RBCs in order to determine the effect that each enzymatic treatment had on their ability to bind to RBCs. Human RBC suspensions (60% hematocrit) were treated with one of the following enzyme solutions: 150 μU/mL neuraminidase (ICN 9001-67-6), 1 mg/mL trypsin (Sigma T-1005), or 1 mg/mL chymotrypsin (Sigma C-4129) in HBS buffer (pH 7.4). Cell suspensions were incubated with enzyme solutions for 60 min at 37°C, washed twice with HBS, and assessed same as in binding assays to RBCs. Binding to untreated RBCs was used as control of total RBC binding.
Peptide cross-linking
PfP0 HABPs were tested in cross-linking assays with human RBCs in order to identify their possible RBC receptors. Briefly, each radiolabeled peptide was individually incubated with a 7% RBC suspension for 90 min at room temperature in the absence or presence of unlabeled peptide. Excess of unbound peptide was removed by washing cells twice with HBS before cross-linking bound peptides by incubation with bis-sulfosuccinimidyl suberate (BS3; 1 mg/mL, Sigma-Aldrich) at 4°C. The reaction was stopped by adding 15 μL Tris-HCl buffer (pH 7.4) after 60 min had elapsed. Cells were washed with HBS by centrifuging samples at 2,000×g for 5 min and discarding the supernatant. Each sample was then added a lysis buffer containing 5 mM Tris-HCl, 7 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.1 mM phenylmethylsulfonyl fluoride, and Laemmli buffer and centrifuged at 15,000×g for 15 min. Proteins in the cell lysate were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 12% gel. Cross-linked proteins were detected by exposing gels on a BioRad Imaging Screen K (BioRad Molecular Imager FX; BioRad Quantity One, Quantitation Software) for 5–8 days. The apparent molecular weight of each protein cross-linked to radiolabeled peptides was estimated by comparing migration distances with molecular weight markers (Fermentas Life Sciences). An additional cross-linking assay was carried out with PfP0 HABPs and enzyme-treated RBCs, following the same experimental conditions described above.
Immunization of rabbits with PfP0 peptides
Three pairs of New Zealand rabbits were immunized with PfP0 HABPs. Rabbits were selected based on their Western blot reactivity to P. falciparum trophozoite and merozoite lysate proteins. Each rabbit group was subcutaneously inoculated on day 0 with a mixture containing 250 µg of each monomeric PfP0 HABP, as follows: Group 1 was inoculated with HABPs 33898 and 33900 (rabbits 63 and 72), group 2 with HABPs 33901 and 33906 (rabbits 59 and 60), and group 3 (rabbits 76 and 78) with peptides 33912 (281PASAAKADEPKKEEAKKVEEY 300) and 33913 (297KVEEEEEEEEDGFMGFGMFDY 316), which were identified as B cell epitopes of PfP0 by an analysis carried out with the ANTHEPROT software available at http://antheprot-pbil.ibcp.fr/ [27]. Each peptide was diluted to a 250 μg/mL and emulsified in Freund’s complete adjuvant containing 0.5 mg of the FIS T-helper cell epitope, a peptide from sperm-whale myoglobin encompassing residues 106–118 (FISEAIIHVLHSR) [28]. Rabbits received booster doses emulsified in incomplete Freund’s adjuvant on days 20 and 40. One rabbit was immunized with phosphate buffered saline (PBS) as negative control. All animals were bled on day 60 and serum samples were collected for further analysis. Immunizations and bleedings were carried out according to the guidelines established by the Colombian Ministry of Health for handling live animals with research or experimentation purposes.
Enzyme-linked immunosorbent assay
Ninety-six-well plates coated with 10 μg/mL of each peptide were incubated at 37°C for 1 h, left overnight at 4°C, and incubated at 37°C for an additional hour. Plates were washed five times with PBS-Tween 0.05% and incubated with 0.5% skimmed milk diluted in PBS-Tween 0.05% for 1 h at room temperature. An initial 1:200 sera dilution collected from immunized rabbits was added and serially diluted by twofold along the entire row of wells. Plates were incubated for 1 h at 37°C and washed five times with PBS-Tween 0.05% to remove excess of unbound antibody. Wells were then loaded with 100 μL of peroxidase-coupled goat antirabbit IgG antibody diluted 1:5,000 and incubated for 1 h at 37°C. Excess of unbound antibody was removed by washing wells thrice with PBS-Tween 0.05% and immunoreactivity was revealed by using the TMB Microwell Peroxidase Substrate System kit (KPL Laboratories, WA, USA), according to the manufacturer’s instructions. Rabbit antibody titers were determined by successive twofold primary antibody dilutions until reaching an A 620 value equal to the control value ± 2 SD.
IgG isolation from anti-PfP0 rabbit sera
IgG proteins were isolated from serum samples of each immunized rabbit group by using the Protein A IgG purification kit (PIERCE). Briefly, the column was equilibrated with 3–5 mL of binding buffer, loaded with 5 mL of antibody sample, and then washed by passing 5–15 mL volumes of binding buffer through the column. Rabbit IgG was eluted by passing 5–10 mL volumes of elution buffer (pH 2.8) through the column. Fractions of 1 mL were collected and neutralized with 100 μL of binding buffer. Each fraction was analyzed by measuring absorbance at 280 nm in order to determine which fractions contained antibodies. Protein content was quantified by the bicinchoninic acid method.
SDS-PAGE and Western blot
Proteins in a lysate of mature P. falciparum trophozoites and schizonts (FCB-2) were separated by SDS-PAGE in a discontinuous gel using a 7.5–15% (w/v) acrylamide gradient. Briefly, 500 μg/mL of lysate were loaded per gel, separated by SDS-PAGE, and transferred to nitrocellulose membrane (Hybond 203c, Pharmacia) using the semidry blotting technique. The nitrocellulose membrane was blocked using 5% skimmed milk diluted in Tris-buffered saline with 0.05% Tween (TBS-T) and washed thrice with TBS-T. Pre-immune and final bleeding purified serum samples were diluted 1:100 in blocking solution and incubated individually with membrane strips. After five washes with TBS-T, strips were incubated for 1 h with 1:5,000 alkaline phosphatase-conjugated antigoat IgG antibody (ICN Biomedicals). The immunoreaction was detected using NBT/BCIP (KPL, Gaithersburg, MA, USA).
Indirect immunofluorescence assays
Schizonts obtained from a sorbitol-synchronized culture of the P. falciparum FCB-2 strain (5–7% parasitemia) were used for PfP0-localization assays. Parasites were washed thrice with PBS (pH 7.2–7.4), then centrifuged at 2,500×g for 4 min, and suspended in a 1:1 PBS–fetal bovine serum solution. This solution was seeded on eight-well multitest slides and let to dry at room temperature for 24 h. Slides were blocked by adding 30 μL PBS-1% skimmed milk per well for 10 min, washed twice with PBS-T, then incubated with 10 μL of rabbit serum dilution 1:100 for 30 min, washed again thrice, and finally incubated with a 1:20 dilution of fluorescein-conjugated goat antirabbit IgG antibody (Vector Laboratories CA 94010). Immunofluorescence was read on an Olympus BX51 fluorescence microscope.
Invasion inhibition assays with PfP0 HABPs and anti-PfP0 HABPs
Samples of late schizonts obtained from P. falciparum (FCB-2 strain) synchronized cultures (5% parasitemia and 5% hematocrit) [29] were incubated with either 50, 100, and 200 µM concentrations of each HABP or with different concentrations of IgG purified from rabbit sera raised against PfP0 HABPs and B cell epitopes (rabbits 60, 63, and 78), as described above. After incubating samples for 18 h at 37°C in a 5% O2, 5% CO2, and 90% N2 atmosphere, culture supernatants were harvested and cells were labeled by incubation with 15 µg/mL hydroethidine for 30 min at 37°C. Cells were washed thrice with PBS and analyzed in a FacsCalibur flow cytometer (FACsort, FL2 channel) equipped with CellQuest software [30]. Infected erythrocytes and ethylene glycol tetraacetic acid (EGTA)/chloroquine-treated uninfected erythrocytes were used as controls. A low activity binding peptide (LABP) and purified rabbit pre-immune sera were used as negative binding controls. All assays were carried out in triplicate.
Circular dichroism
Secondary structure elements in PfP0 HABPs were examined by circular dichroism (CD) [31]. HABPs (5 μM) were analyzed a 1-cm light pass length quartz cell thermostated at 20°C using 30% (v/v) 2,2,2-trifluroetanol (TFE) as cosolvent, which has been shown to stabilize nascent structures [32]. Spectra were obtained in a nitrogen-flushed Jasco J-810 spectrometer at room temperature by averaging three sweeps taken from 260 to 190 nm at a scan rate of 20 nm/min and a 1-nm bandwidth. Data were collected by the Spectra Manager Software and analyzed using the SELCON3, CONTINLL, and CDSSTR software [31, 33].
Extraction and purification of P. falciparum genomic DNA
Human RBCs parasitized with the P. falciparum FCB-2, FVO, and PAS-2 strains were obtained from an asynchronous culture and maintained as described elsewhere [29]. Genomic DNA (gDNA) was extracted from 200 μL aliquots of each parasite culture (30% parasitemia) by using 0.2% saponin and then purified using the UltraClean DNA Blood Isolation kit (MO BIO, Carlsbad, CA, USA).
PCR amplification and sequencing
The regions encoding PfP0 HABPs 33898, 33900, 33901, and 33906 in the P. falciparum FCB-2, FVO, and PAS-2 strains were amplified by PCR using genomic DNA of each parasite strain (2 µL) as template. The PfP0-f (5′-ATTTTTTCGCGCATTGTTCC-3′) and PfP0-r (5′-GCATCATAAATTACACCATCA-3′) primers were designed using Gene Runner v3.05 software based on the genome of the P. falciparum 3D7 reference strain (GenBank access number PF11_0313) [34]. The PfP0-f was designed 62 bp upstream the initiation codon. The region encoding HABP 33577 from the P. falciparum integral membrane protein Pf25-IMP was amplified using DIR1 and REV1 primers to be used as a positive PCR control [35].
DNA regions were amplified in 50 μL amplification reaction mixtures containing 1 U Taq polymerase (Bioline, Taunton, MA, USA), 1× Taq polymerase reaction buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, and 0.4 µM of each primer. The thermocycling conditions were as follows: an initial denaturing step at 95°C for 5 min followed by 35 cycles consisting of 1 min annealing at 56°C for PfP0-f/PfP0-r or 58°C for DIR1/REV1, 1 min extension step at 72°C, and 1 min denaturing at 95°C, followed by a final extension cycle at 72°C for 5 min. A reaction containing water instead of DNA was used as negative control. Amplification products were visualized in 1% agarose gels stained with SYBR® safe (Invitrogen, Eugene, OR, USA). Amplification products were purified using the Wizard PCR preps kit (Promega, Madison, WI, USA) and sequenced using PfP0-f and PfP0-r.
The amino acid sequences of the region encoding HABPs 33898, 33900, 33901, and 33906 in the FCB2, FVO, and PAS-2 strains were aligned to the reported PlasmoDB sequence of the 3D7 (available at http://www.plasmodb.org/plasmo/home.jsp), HB3 (available at the Broad Institute website: http://www.broad.mit.edu/), and 7G8 strains [5] using Clustal W software [36].
Results
N-terminal PfP0 peptides bind specifically to RBCs
Sixteen 20-mer-long nonoverlapping peptides spanning the complete PfP0 protein amino acid sequence were synthesized to map its RBC binding regions. The specific binding curve of each peptide was obtained by determining the difference between total binding (binding in the absence of nonradiolabeled peptide) and unspecific binding (binding in the presence of nonradiolabeled peptide) curves. Peptides showed three distinct binding behaviors: nonbinding to RBCs (low total binding and unspecific binding), unspecific binding to RBCs (high and comparable total binding and unspecific binding), and specific binding (high specific binding and low unspecific binding). Peptides binding specifically to RBCs had specific binding curve with ≥0.02 slopes and thereby were considered as HABPs [26, 37]. Only the following four PfP0 peptides satisfied such criteria: 33898 (1MAKLSKQQKKQMYIEKLSSL20), 33900 (41ASVRKSLRGKATILMGKNTRY 60), and 33901 (61IRTALKKNLQAVPQIEKLLPY 80), all of which are located in the N-terminal region, whereas HABP 33906 (161LIKQGEKVTASSATLLRKFNY180) is located in the protein’s central region. No HABPs were found in the C-terminal domain (Fig. 1a).
Binding of PfP0-HABPs to other human cell types was also assessed in order to determine whether binding to RBCs was specific (Fig. 1b). The results showed that binding of PfP0-HABPs to WBCs and HeLa cells is low and unspecific, finding binding activities close to 0.1% and 0.5%, respectively.
Saturation assays
Dissociation constants (K d), Hill coefficients (n H), and the approximate number of binding sites per cell were calculated for each HABP based on saturation curves and Hill analysis [38]. K d for HABPs 33900, 33901, and 33906 were 360, 530, and 310 nM, respectively. All HABPS showed n H larger than one (1.5, 1.3, and 1.1, respectively), indicating positive cooperativity. Binding sites per cell for each of these HABPs were 281,000, 48,000, and 34,500, respectively (Fig. 1b). Binding of HABP 33898 was not saturable at the experimental conditions followed with all the other PfP0 HABPs.
Enzymatic treatment
Binding assays were carried out using RBCs treated with either neuraminidase, chymotrypsin, or trypsin and compared with binding to untreated RBCs. Table 1 shows changes in the specific binding activity of each HABP. Specific binding of HABP 33900 diminished by more than 86% when RBCs were treated with trypsin, but binding of the other HABPs was not affected by this treatment. Specific binding of HABPs 33900 and 33906 diminished by 42% and 55%, respectively, when RBCs were treated with chymotrypsin while bindings of HABPs 33901 and 33898 were not affected by such treatment. Treatment with neuraminidase showed no considerable effect on the specific binding activity of any PfP0 HABP (>50%).
Cross-linking assays
As it can be seen in Fig. 2a, all HABPs bound to a RBC membrane protein with an apparent molecular weight of 58 kDa, which suggests a common receptor for the four PfP0 HABPs. The molecular weight of this protein points toward glycophorin A as the probable receptor since this is one of the most abundant proteins on the surface of RBCs [39]. Binding to this receptor was inhibited by the presence of unlabeled peptide, thus demonstrating the specificity of this receptor–ligand interaction. HABPs 33901 and 33898 bound also to a ∼25-kDa RBC receptor as well as to a ∼17-kDa RBC membrane protein, which could be suggesting degradation of RBC membrane proteins or binding of these HABPs to low molecular weight RBC membrane proteins as well as to some isoforms of glycophorins.

a Autoradiogram of cross-linking assays between RBCs and PfP0 HABPs. Lanes 1, 3, 5, and 7 correspond to total binding while lanes 2, 4, 6, and 8 show binding inhibited by the presence of unlabeled peptide. All HABPs detected a common band, while HABPs 33898 and 33901 recognized two bands of about 25 and 17 kDa. b Cross-linking of PfP0-HABPs with enzyme-treated RBCs. Lane 1 HABP 33898 was incubated with neuraminidase-treated RBCs (N). Lane 2 HABP 33900 cross-linked to trypsin-treated RBCs (T). Lane 3 HABP 33900 cross-linked to chymotrypsin-treated RBCs (C). Lane 4 HABP 33906 cross-linked to chymotrypsin-treated RBCs. HABP 33901 was not included in the assay since its binding was not susceptible to any enzymatic treatment
An additional assay was performed in order to corroborate the results of cross-linking assays and the sensitivity profile found for the RBC receptor of PfP0-HABPs (Fig. 2b). Each HABP was incubated with enzyme-treated RBCs depending on its enzymatic profile, i.e., HABP 33900 was incubated with RBCs pretreated with trypsin or chymotrypsin, HABP 33906 was incubated with chymotrypsin-treated RBCs, and HABP 33898 was incubated with neuraminidase-treated RBCs. No cross-linking bands were detected for HABPs 33901 and 33906, while HABP 33898 strongly cross-linked to a band of ∼17 kDa, which is similar to the weight of the band detected in cross-linking assays with nonenzyme-treated RBCs.
Determination of antipeptide antibody titers by ELISA
Anti-HABP IgG titers raised in each immunized rabbit were determined by enzyme-linked immunosorbent assay. As shown in Fig. 3a, all four PfP0 HABPs induced very high antibody titers. Antibody titers induced by HABP 33900 (in rabbit 63), HABP 33906 (in rabbit 60), and peptide 33913 (in rabbit 78) were 102,400. HABP 33901 (in rabbit 60) and peptide 33912 (in rabbit 78) induced antibody titers of 204,800 while HABP 33898 induced an antibody titer >409,600 (in rabbit 63).

a Antibody production induced in rabbits by immunization with PfP0 HABPs. The recognition of each individual peptide by the sera of immunized rabbits was assessed in enzyme-linked immunosorbent assays. The figure shows antibody titers induced by each peptide after administering the pre-immune (Pre) and postthird doses (Post). Rabbit antibody titers were determined by successive twofold primary antibody dilutions until reaching an A620 value equal to the control value ±2 SD. b Localization of PfP0 in late schizonts as determined by indirect immunofluorescence assays. Purified final-bleeding sera were used as primary antibody. PfP0 showed a grape-like fluorescence pattern typical of merozoite surface proteins. c Recognition of PfP0 in a late P. falciparum schizont lysate by Western blot using anti-PfP0 antibodies. Pre-immune sera (lanes 1, 2, and 3), hyperimmune sera (lanes 4, 5, and 6). Lanes 1 and 4 correspond to sera obtained from group 1 immunized with peptides 33898 and 33900. Lane 2 and 5 correspond to sera obtained from group 2 immunized with peptides 33901 and 33906. Lanes 3 and 6 to sera from group 3 immunized with peptides 33912 and 33913. The molecular weight of the band being recognized at ∼37 kDa agrees with the molecular weight predicted for PfP0
Immunofluorescence assays and Western blot analysis
The IgG antibodies purified from sera of rabbits immunized with PfP0 HABPs, as well as from rabbits being immunized with PfP0 peptides 33912 and 33913 (B cell predicted epitopes), were assessed by Western blot in order to determine whether they recognized PfP0 in a P. falciparum schizont lysate (FCB-2 strain) and in intact schizonts. These antibodies recognized specifically a 37-kDa protein (Fig. 3c), agreeing with the molecular weight predicted for PfP0. No bands on the parasite lysate were recognized by pre-immune sera.
Immunofluorescence assays with anti-PfP0 rabbit sera revealed a grape-like fluorescence pattern similar to the one shown by merozoite surface proteins in schizont stages (Fig. 3b). Such immunofluorescence pattern agrees with the localization of PfP0 reported in other studies [8, 9].
Invasion inhibition assays
The functional role of PfP0 HABPs during the parasite’s erythrocytic cycle was evaluated by assessing their ability to inhibit RBC invasion in vitro. Late schizonts obtained from synchronized cultures were exposed to the different HABPs and to a LABP (negative binding control). The results of these assays (see Table 2) showed that the invasion inhibition ability of HABPs 33898 and 33906 depended on the peptide concentration, since a higher inhibition percentage was obtained at 200 μM (67% and 91%, respectively). On the contrary, low inhibition percentages were obtained with HABPs 33900 and 33901, which were similar to the percentages obtained with the LABP.
In an independent inhibition assay, different concentrations of purified IgG were used to assess the ability of anti-PfP0 HABP antibodies to inhibit merozoite invasion of RBCs. All anti-PfP0 HABP purified antibodies (rabbits 60 and 63) inhibited invasion by up to 90% in a concentration-dependent manner, with concentrations ranging between 110 and 88 µg/mL (Table 2), while pre-immune serum samples showed no inhibitory ability. Antibodies induced by immunization with PfP0 B cell epitopes (33912 and 33913) inhibited invasion by 92%.
Circular dichroism studies
Data regarding the secondary structure of each PfP0 HABP were obtained by analyzing their corresponding CD spectra. Spectra of HABPs 33898 and 33906 showed the presence of α-helical features, as shown by a molar ellipticity maximum at 190 nm and two minima at 209 and 220 nm. On the contrary, spectra obtained for HABPs 33900 and 33901 indicated less ordered structures (Fig. 3). Data deconvolution of each CD spectrum revealed an 80% α-helicity for HABPs 38898 and 33906, whereas HABPs 33900 and 33901 presented 50% α-helicity (Fig. 4).

CD spectra of PfP0 HABPs. Spectra were obtained by averaging three scans taken in aqueous TFE (30% v/v) solution. The results were expressed as mean residue ellipticity [Θ] in degrees square centimeter per decimole according to the \( \left[ \Theta \right] = \Theta_{\lambda } /\left( {100 \times l \times c \times n} \right) \) function where \( \Theta_{\lambda } \) represents measured ellipticity, l is the optical path length, c peptide concentration, and n the number of amino acid residues in the sequence
DNA amplification and polymorphism analysis
The amplification product of the region encoding HABPs 33898, 33900, 33901, and 33906 was visualized on agarose gels, detecting a single band of ∼664 bp. Amino acid sequences of the three analyzed strains (FCB2, FVO, and PAS-2) were aligned to the reported 3D7 sequence using Clustal W software [36]. The region encoding PfP0 HABPs shared 100% amino acid identity in the three studied strains. No synonymous substitutions were identified when the corresponding nucleotide sequences were aligned, since they shared 100% identity (data not shown).
Additionally, when the amino acid sequences of the PAS-2, FVO, and FCB-2 strains were aligned to the reported HB3 and 7G8 strains, five substitutions and one insertion were found in the 7G8 strain compared to the other strains. Substitutions occurred in the following positions, according to the amino acid numbering of the 7G8 strain: 108 (P → S), 110 (A → H), 139 (F → C), 140 (E → Q), and 177 (Q → R), being the later substitution located in the C terminus of HABP 33906. An S insertion was detected in position 109 in the 7G8 strain.
Discussion
In the search for developing a fully effective antimalarial vaccine, we have focused mainly on the identification of the amino acid sequences establishing intimate molecular interactions with host cells during parasite invasion. The merozoite stage has been an important target for vaccine development studies since it is responsible for the invasion and subsequent rupture of RBCs, two phenomena that are directly associated with the clinical manifestations of malaria. Due to the large number of proteins involved in the invasion process to RBCs (∼58 to 90 according to transcriptome studies) [40], we have long been devoted to the identification of HABPs in P. falciparum proteins crucial for the completion of the parasite’s life cycle, seeking to include such minimal protein subunits in the design of a multi-epitope, multistage, chemically synthesized antimalarial vaccine, an endeavor thoroughly described previously elsewhere [26]. Polymorphism analysis of the HABPs found in these invasion-relevant proteins have shown that a large number of these sequences are conserved among the different parasite strains. Moreover, when these HABPs have been tested in the experimental Aotus monkey model, they have been found to be neither immunogenic nor protective. Nevertheless, when the amino acid sequences of these HABPs are modified according to previously reported rules [41], their immunological properties improved drastically, as indicated by the induction of high long-lasting antibody titers associated with protection against experimental challenge with a 100% infective dose of P. falciparum parasites [42–44]. Currently, our search for specific sequences in other parasite proteins involved in invasion to target cells (RBCs and hepatocytes) continues, seeking to gather a pool of HABPs from the different parasite-stage proteins to design a fully effective antimalarial vaccine.
PfP0 is expressed in asexual erythrocytic stages of the parasite, mainly trophozoites, in which it has a predominant localization in the cytoplasmic where it fulfills an important role in the assembly of the ribosomal stalk [8]. However, various studies have reported PfP0 expression on the surface of merozoite forms [9], as corroborated by the immunofluorescence data presented in this study, suggesting an additional role of PfP0 in parasite’s invasion to RBCs. Additionally, PfP0 has been implicated in other cellular functions besides ribosomal assembly. In Drosophila melanogaster, P0 has been shown to possess DNA-binding activity, specifically apurinic/apyrimidinic endonuclease activity [45], and has been also implicated as a regulatory protein in apoptosis and carcinogenesis of mammalian cells [46, 47].
In this study, we identified the RBC binding regions of PfP0 by dividing its complete sequence into 20-mer-long nonoverlapping peptides and assessing each peptide’s specific binding to RBCs. Four PfP0 HABPs were identified: HABP 33898 (1MAKLSKQQKKQMYIEKLSSL20), HABP 33900 (41ASVRKSLRGKATILMGKNTRY 60), HABP 33901 (61IRTALKKNLQAVPQIEKLLPY 80), and HABP 33906 (161LIKQGEKVTASSATLLRKFNY180). Binding of these HABPs to RBCs was highly specific and selective since PfP0-HABPs displayed low and unspecific binding to WBCs and HeLa cells when they were assessed under the same experimental conditions as those used with RBCs.
All HABPs were located at the N terminus and the central region of PfP0 (Fig. 1a), which have been implicated in cellular immunity to P. falciparum merozoites by studies finding cytokine production against PfP0 peptides in umbilical cord-blood samples from newborn children of malaria-infected mothers, being such cellular responses in average higher than the ones elicited by major P. falciparum antigens such as MSP-1 and EBA-175 [22]. Additionally, antibodies raised against the N-terminal domain of PfP0 have been shown to inhibit in vitro growth of parasite asexual blood stages [8], which agrees with our results for HABPs 33898 and 33906. On the contrary, our results showed that the PfP0 P peptide (301EEEEEEDGFMGFGMFD306) reported to induce protection against experimental challenge with P. yoelii in mice [21] has no specific binding activity to RBCs (Fig. 1).
Interestingly, no HABPs were found in the C-terminal portion of PfP0. Cross-reactivity studies with sera from SLE patients have reported a region shared between PfP0 and the human P0 protein and antibodies in such cross-reacting sera have been shown to inhibit P. falciparum growth in vitro. In this study, peptide 33913 297KVEEEEEEEEDGFMGFGMFD 316 containing this shared sequence was found to have no binding activity to RBCs. Therefore, based on the cross-reactivity between PfP0 and HuP0 and the nonbinding capacity of peptide 33913, we conclude that the long C-terminal domain of PfP0 is not a suitable target for an antimalarial vaccine.
HABPs 33900, 33901, and 33906 bound strongly and with high affinity to RBC membrane receptors, as indicated by their K d values within the submicromolar range (310 to 530 nM; Fig. 1b). The number of binding sites per cell for PfP0 HABPs ranged within 34,500 to 281,000, suggesting a variable concentration of these binding sites per cell on erythrocyte membrane and, in some cases, intermediate K d values. These can be explained by the relation existing between the variables in the saturation curve equation: B = [B max] [F]/K d + [F]. Binding of HABP 33898 was unsaturable under the experimental conditions followed in the assay, suggesting a larger number of copies of RBC membrane receptors or interaction with different binding sites on one or several receptors.
The possible nature of RBC surface receptors was analyzed by testing the ability of all HABPs to bind to enzyme-treated RBCs in modified binding assays. The diminished capacity of HABP 33900 to bind trypsin-treated RBCs points toward proteins exposed on RBC surface or even to the not yet identified X receptor as the possible receptors for this HABP [48, 49]. On the other hand, the reduction in the binding of HABP 33906 to chymotrypsin-treated RBCs suggests a possible sialic acid-independent interaction, possibly with RBC surface proteins sensitive to chymotrypsin treatment such as, for example, band 3 protein [50]. However, additional studies are needed in order to precise the identity of the RCB receptors for PfP0 HABPs.
The identity of the possible RBC receptors for these HABPs was further studied by determining the molecular weight of the RBC receptor of each HABP in cross-linking assays (Fig. 2). All HABPs showed a strong recognition of a ∼58-kDa protein, which agrees with the molecular weight of glycophorin A (∼60 kDa) [39, 51, 52]. Even though different binding profiles were found in binding assays with enzyme-treated RBCs, it is feasible that PfP0 HABPs are recognizing different portions of the same protein and that the receptor-binding sites recognized by each HABP are removed by the enzymatic treatment. For instance, trypsin removes glycophorins A and C, which are the most abundant glycophorins on RBC surface [39, 48], while neuraminidase removes sialic acid groups on sialoglycoproteins. On the other hand, HABP 33901, whose binding was not affected by any of the treatments being applied, and HABP 33898, whose binding was slightly sensitive to treatment with neuraminidase, recognized two additional receptors of ∼25 and ∼17 kDa with high affinity.
This suggests that treatment of RBC with these different enzymes exposes cryptic residues on membrane proteins, but can also be indicating that the receptor for both HABPs is being degraded since it has been determined that some RBC proteins are quite vulnerable to proteolytic degradation [53]. However, several studies suggest the existence of other not yet characterized receptors such as type X, Y, Z, and E receptors [54–56]. Additionally, the different binding behaviors shown by each PfP0 HABP to enzyme-treated RBCs correlate with the alternative invasion pathways that P. falciparum merozoites can employ to invade RBCs. This has been observed in some proteins, including members of the erythrocyte-binding antigen and P. falciparum reticulocyte-binding homolog families, where changes in the expression and/or use of these proteins is associated with the use of other RBC surface molecules as binding receptors for P. falciparum invasion to RBCs [55, 57–60].
The results of cross-linking assays were corroborated with the sensitivity profile found for the RBC receptor of PfP0-HABPs, by performing an additional cross-linking assay with enzyme-treated RBCs (Table 1; Fig. 2). As shown in Fig. 2b, no bands were detected by HABPs 33900 or 33906 when RBCs were treated with chymotrypsin or trypsin. These data suggest that the receptor for these PfP0 HABPs is sensitive to the enzymatic cleavage by these two proteases and that both HABPs bind specifically to an RBC surface receptor of ∼58 kDa.
Interestingly, the ∼58- and 25-kDa bands cross-linked by HABP 33898 in nonenzyme-treated RBCs were no longer detected when the assay was carried out with neuraminidase-treated RBCs. On the contrary, the ∼17-kDa band detected in nonenzyme-treated RBCs was also recognized in neuraminidase-treated RBCs (Fig. 2a). These data suggest that binding of HABP 33898 is not completely sialic acid dependent, which is in agreement with the enzymatic profile found for this HABP showing that binding was only diminished to 63%.
The specificity of the HABP–RBC interaction was far more evident in the invasion inhibition in vitro assays carried out with each PfP0 HABP. HABPs 33898 and 33906 inhibited merozoite invasion to RBCs by a higher percentage compared to the inhibition percentage shown by LABP 33911. A similar inhibitory effect of around 90% was observed when using anti-PfP0 HABP antibodies, strongly suggesting the involvement of these HABPs and hence of PfP0 in P. falciparum invasion to RBCs. PfP0 anti-B cell epitope antibodies showed a 92% inhibitory activity, suggesting that B cell epitopes may lie contiguous to HABPs in the folded PfP0 and thereby that antibodies targeting B cell epitopes may inhibit RBC invasion due to a steric blockage effect. Additionally, structures of these HABPs were found to contain a large percentage of α-helical structure stabilized in 30% (v/v) TFE aqueous solution, which is relevant since previous studies have shown that the structural modification of some α-helical HABPs induces protective immune responses in Aotus monkeys [42, 61, 62].
Another important aspect to be consider when searching for suitable antimalarial vaccine candidates is their degree of polymorphism among parasite strains, since this has been one of the major constraints for inducing a long-lasting strain-transcending protective immune response and sequence variations have been associated with distraction of the host’s immune response toward strains-specific epitopes. For this reason, it was of key importance for us to assess the degree of polymorphism in the region containing HABPs 33898, 33900, 33901, and 33906 in three P. falciparum strains: FCB-2 (the same strain used in our inhibition assays), FVO, and PAS-2, as well as in the HB3 and 3D7 reference strains whose genome sequences are available at the Broad Institute and the PlasmoDB databases, respectively. All HABP sequences were highly conserved among the different strains compared to the 3D7 strain. However, HABP 33906 presented an amino acid variation in the 7G8 strain that consisted in the substitution of an uncharged polar amino acid (glutamine Q) for a positively charged polar residue (arginine R) in the C-terminal region. These physicochemical changes are important aspects to take into consideration when designing an antimalarial vaccine, as has been reported previously [41].
The multiple sequence alignment between P. falciparum isolates from different geographical regions (FCB2 from Colombia, FVO from Vietnam, 3D7 isolated in The Netherlands, HB3 isolated in Honduras, PAS-2 of unknown origin, and 7G8 isolated in Brazil) showed high amino acid sequence identity and similarity among the evaluated HABPs, highlighting the importance of these HABPs as immune targets capable of controlling the parasite’s enormous variability and therefore as promising candidates for an effective nonstrain-specific antimalarial vaccine.
Immunogenicity studies determined that PfP0 is highly immunogenic, as shown by the high antibody titers induced in rabbits immunized with mixtures of PfP0 HABPs (even >409,600 in some rabbits). Interestingly, studies carried out by Chatterjee and colleagues [19, 20] found that several boosting doses are needed in order to induce >10,000 anti-N and C-terminal PfP0 antibody titers and that at least 12 weeks were necessary in order to trigger a specific antibody response. Such findings demonstrate that immunizations with regions as large as the two PfP0 domains (the N terminus spans from amino acid 17 to 61 and the C terminus from amino acid 61 to 316) are not appropriate targets for inducing an effective immune response, in contrast to the immune responses elicited by 20-mer long peptides, given the high identity shared with HuP0 and the potential cross-reactivity that this would imply.
Even though previous studies have found that PfP0 has a predominant intracellular localization due to its active role in ribosome assembly [8], the results of our immunological assays (Western blot and immunofluorescence assays) indicate that PfP0 is expressed specifically during the schizont stage in the FCB-2 strain as conclusively shown by the recognition of a 37-kDa band in schizont lysate (Fig. 3c), the fluorescence pattern typically found in merozoite surface-exposed proteins (Figs. 3b and 4) and the recognition of the native PfP0 expressed by the parasite by anti-PfP0 HABP antibodies induced in rabbits. These data strongly suggest PfP0 involvement in RBC–merozoite interactions during the parasite’s invasion process. Besides being detected on parasite surface, PfP0 has been also identified as a protective surface protein due to its exclusive recognition by sera from malaria-immune individuals [3]. These data, together with the results of this study showing that PfP0 HABPs located at the N-terminal domain inhibit merozoite invasion in vitro, support the inclusion of these PfP0 HABPs in further studies aimed at designing a minimal subunit-based, multicomponent, multistage, chemically synthesized antimalarial vaccine.
References
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI (2005) The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434:214–217
Cohen S, Mc GI, Carrington S (1961) Gamma-globulin and acquired immunity to human malaria. Nature 192:733–737
Lobo CA, Kar SK, Ravindran B, Kabilan L, Sharma S (1994) Novel proteins of Plasmodium falciparum identified by differential immunoscreening using immune and patient sera. Infect Immun 62:651–656
Kemp DJ, Coppel RL, Cowman AF, Saint RB, Brown GV, Anders RF (1983) Expression of Plasmodium falciparum blood-stage antigens in Escherichia coli: detection with antibodies from immune humans. Proc Natl Acad Sci U S A 80:3787–3791
Goswami A, Chatterjee S, Sharma S (1996) Cloning of a ribosomal phosphoprotein P0 gene homologue from Plasmodium falciparum. Mol Biochem Parasitol 82:117–120
Rich BE, Steitz JA (1987) Human acidic ribosomal phosphoproteins P0, P1, and P2: analysis of cDNA clones, in vitro synthesis, and assembly. Mol Cell Biol 7:4065–4074
Santos C, Ballesta JP (1994) Ribosomal protein P0, contrary to phosphoproteins P1 and P2, is required for ribosome activity and Saccharomyces cerevisiae viability. J Biol Chem 269:15689–15696
Goswami A, Singh S, Redkar VD, Sharma S (1997) Characterization of P0, a ribosomal phosphoprotein of Plasmodium falciparum antibody against amino-terminal domain inhibits parasite growth. J Biol Chem 272:12138–12143
Singh S, Sehgal A, Waghmare S, Chakraborty T, Goswami A, Sharma S (2002) Surface expression of the conserved ribosomal protein P0 on parasite and other cells. Mol Biochem Parasitol 119:121–124
Koren E, Reichlin MW, Koscec M, Fugate RD, Reichlin M (1992) Autoantibodies to the ribosomal P proteins react with a plasma membrane-related target on human cells. J Clin Invest 89:1236–1241
Sehgal A, Kumar N, Carruthers VB, Sharma S (2003) Translocation of ribosomal protein P0 onto the Toxoplasma gondii tachyzoite surface. Int J Parasitol 33:1589–1594
Aruna K, Chakraborty T, Nambeesan S, Mannan AB, Sehgal A, Balachandara SR, Sharma S (2004) Identification of a hypothetical membrane protein interactor of ribosomal phosphoprotein P0. J Biosci 29:33–43
Elkon K, Skelly S, Parnassa A, Moller W, Danho W, Weissbach H, Brot N (1986) Identification and chemical synthesis of a ribosomal protein antigenic determinant in systemic lupus erythematosus. Proc Natl Acad Sci U S A 83:7419–7423
Mahler M, Kessenbrock K, Raats J, Williams R, Fritzler MJ, Bluthner M (2003) Characterization of the human autoimmune response to the major C-terminal epitope of the ribosomal P proteins. J Mol Med (Berlin, Germany) 81:194–204
Skeiky YA, Benson DR, Elwasila M, Badaro R, Burns JM Jr, Reed SG (1994) Antigens shared by Leishmania species and Trypanosoma cruzi: immunological comparison of the acidic ribosomal P0 proteins. Infect Immun 62:1643–1651
Levin MJ, Vazquez M, Kaplan D, Schijman AG (1993) The Trypanosoma cruzi ribosomal P protein family: classification and antigenicity. Parasitol Today (Personal ed) 9:381–384
Kaplan D, Ferrari I, Bergami PL, Mahler E, Levitus G, Chiale P, Hoebeke J, Van Regenmortel MH, Levin MJ (1997) Antibodies to ribosomal P proteins of Trypanosoma cruzi in Chagas disease possess functional autoreactivity with heart tissue and differ from anti-P autoantibodies in lupus. Proc Natl Acad Sci U S A 94:10301–10306
Singh S, Chatterjee S, Sohoni R, Badakere S, Sharma S (2001) Sera from lupus patients inhibit growth of P. falciparum in culture. Autoimmunity 33:253–263
Chatterjee S, Singh S, Sohoni R, Kattige V, Deshpande C, Chiplunkar S, Kumar N, Sharma S (2000) Characterization of domains of the phosphoriboprotein P0 of Plasmodium falciparum. Mol Biochem Parasitol 107:143–154
Chatterjee S, Singh S, Sohoni R, Singh NJ, Vaidya A, Long C, Sharma S (2000) Antibodies against ribosomal phosphoprotein P0 of Plasmodium falciparum protect mice against challenge with Plasmodium yoelii. Infect Immun 68:4312–4318
Rajeshwari K, Patel K, Nambeesan S, Mehta M, Sehgal A, Chakraborty T, Sharma S (2004) The P domain of the P0 protein of Plasmodium falciparum protects against challenge with malaria parasites. Infect Immun 72:5515–5521
Malhotra I, Mungai P, Muchiri E, Ouma J, Sharma S, Kazura JW, King CL (2005) Distinct Th1- and Th2-Type prenatal cytokine responses to Plasmodium falciparum erythrocyte invasion ligands. Infect Immun 73:3462–3470
Sharma S, Pathak S (2008) Malaria vaccine: a current perspective. J Vector Borne Dis 45:1–20
Merrifield RB (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc 85:2149–2154
Tam JP, Heath WF, Merrifield RB (1983) SN 1 and SN 2 mechanisms for the deprotection of synthetic peptides by hydrogen fluoride. Studies to minimize the tyrosine alkylation side reaction. Int J Pept Protein Res 21:57–65
Rodriguez LE, Curtidor H, Urquiza M, Cifuentes G, Reyes C, Patarroyo ME (2008) Intimate molecular interactions of P. falciparum merozoite proteins involved in invasion of red blood cells and their implications for vaccine design. Chem Rev 108:3656–3705
Deleage G, Combet C, Blanchet C, Geourjon C (2001) ANTHEPROT: an integrated protein sequence analysis software with client/server capabilities. Comput Biol Med 31:259–267
Prieto I, Hervas-Stubbs S, Garcia-Granero M, Berasain C, Riezu-Boj JI, Lasarte JJ, Sarobe P, Prieto J, Borras-Cuesta F (1995) Simple strategy to induce antibodies of distinct specificity: application to the mapping of gp120 and inhibition of HIV-1 infectivity. Eur J Immunol 25:877–883
Trager W, Jenson JB (1978) Cultivation of malarial parasites. Nature 273:621–622
Wyatt CR, Goff W, Davis WC (1991) A flow cytometric method for assessing viability of intraerythrocytic hemoparasites. J Immunol Methods 140:23–30
Sreerama N, Venyaminov SY, Woody RW (1999) Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci 8:370–380
Roccatano D, Colombo G, Fioroni M, Mark AE (2002) Mechanism by which 2, 2, 2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc Natl Acad Sci U S A 99:12179–12184
Compton LA, Johnson WC Jr (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal Biochem 155:155–167
PlasmoDB. The Plasmodium Genome Resource. http://www.plasmodb.org/plasmo/home.jsp
Curtidor H, Arevalo G, Vanegas M, Vizcaino C, Patarroyo MA, Forero M, Patarroyo ME (2008) Characterization of Plasmodium falciparum integral membrane protein Pf25-IMP and identification of its red blood cell binding sequences inhibiting merozoite invasion in vitro. Protein Sci 17:1494–1504
Combet C, Blanchet C, Geourjon C, Deleage G (2000) NPS@: network protein sequence analysis. Trends Biochem Sci 25:147–150
Urquiza M, Rodriguez LE, Suarez JE, Guzman F, Ocampo M, Curtidor H, Segura C, Trujillo E, Patarroyo ME (1996) Identification of Plasmodium falciparum MSP-1 peptides able to bind to human red blood cells. Parasite Immunol 18:515–526
Yamamura HI, Enna SJ, Kuhar MJ (1978) Neurotransmitter receptor binding. Raven, New York
Perkins M (1981) Inhibitory effects of erythrocyte membrane proteins on the in vitro invasion of the human malarial parasite (Plasmodium falciparum) into its host cell. J Cell Biol 90:563–567
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol 1:E5
Patarroyo ME, Patarroyo MA (2008) Emerging rules for subunit-based, multiantigenic, multistage chemically synthesized vaccines. Acc Chem Res 41:377–386
Salazar LM, Alba MP, Curtidor H, Bermudez A, Luis EV, Rivera ZJ, Patarroyo ME (2004) Changing ABRA protein peptide to fit into the HLA-DRbeta1*0301 molecule renders it protection-inducing. Biochem Biophys Res Commun 322:119–125
Salazar LM, Alba MP, Torres MH, Pinto M, Cortes X, Torres L, Patarroyo ME (2002) Protection against experimental malaria associated with AMA-1 peptide analogue structures. FEBS Lett 527:95–100
Torres MH, Salazar LM, Vanegas M, Guzman F, Rodriguez R, Silva Y, Rosas J, Patarroyo ME (2003) Modified merozoite surface protein-1 peptides with short alpha helical regions are associated with inducing protection against malaria. Eur J Biochem / FEBS 270:3946–3952
Yacoub A, Kelley MR, Deutsch WA (1996) Drosophila ribosomal protein PO contains apurinic/apyrimidinic endonuclease activity. Nucleic Acids Res 24:4298–4303
Brockstedt E, Rickers A, Kostka S, Laubersheimer A, Dorken B, Wittmann-Liebold B, Bommert K, Otto A (1998) Identification of apoptosis-associated proteins in a human Burkitt lymphoma cell line. Cleavage of heterogeneous nuclear ribonucleoprotein A1 by caspase 3. J Biol Chem 273:28057–28064
Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita M, Tanaka K, Shuda M, Yamamoto M (1999) Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res 59:4990–4996
Pasvol G (1984) Receptors on red cells for Plasmodium falciparum and their interaction with merozoites. Philos Trans R Soc Lond 307:189–200
Baum J, Maier AG, Good RT, Simpson KM, Cowman AF (2005) Invasion by P. falciparum merozoites suggests a hierarchy of molecular interactions. PLoS Pathog 1:e37
Goel VK, Li X, Chen H, Liu SC, Chishti AH, Oh SS (2003) Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc Natl Acad Sci U S A 100:5164–5169
Perkins ME (1984) Surface proteins of Plasmodium falciparum merozoites binding to the erythrocyte receptor, glycophorin. J Exp Med 160:788–798
Perkins ME (1984) Binding of glycophorins to Plasmodium falciparum merozoites. Mol Biochem Parasitol 10:67–78
Steck TL (1974) The organization of proteins in the human red blood cell membrane. A review. J Cell Biol 62:1–19
Mayer DC, Kaneko O, Hudson-Taylor DE, Reid ME, Miller LH (2001) Characterization of a Plasmodium falciparum erythrocyte-binding protein paralogous to EBA-175. Proc Natl Acad Sci U S A 98:5222–5227
Rayner JC, Vargas-Serrato E, Huber CS, Galinski MR, Barnwell JW (2001) A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J Exp Med 194:1571–1581
Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH (1994) Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science (New York, NY) 264:1941–1944
Triglia T, Thompson JK, Cowman AF (2001) An EBA175 homologue which is transcribed but not translated in erythrocytic stages of Plasmodium falciparum. Mol Biochem Parasitol 116:55–63
Triglia T, Duraisingh MT, Good RT, Cowman AF (2005) Reticulocyte-binding protein homologue 1 is required for sialic acid-dependent invasion into human erythrocytes by Plasmodium falciparum. Mol Microbiol 55:162–174
Perkins ME, Holt EH (1988) Erythrocyte receptor recognition varies in Plasmodium falciparum isolates. Mol Biochem Parasitol 27:23–34
Mitchell GH, Hadley TJ, McGinniss MH, Klotz FW, Miller LH (1986) Invasion of erythrocytes by Plasmodium falciparum malaria parasites: evidence for receptor heterogeneity and two receptors. Blood 67:1519–1521
Espejo F, Bermudez A, Torres E, Urquiza M, Rodriguez R, Lopez Y, Patarroyo ME (2004) Shortening and modifying the 1513 MSP-1 peptide's alpha-helical region induces protection against malaria. Biochem Biophys Res Commun 315:418–427
Lozano JM, Patarroyo ME (2007) A rational strategy for a malarial vaccine development. Microbes Infect 9:751–760
Acknowledgments
Special thanks go to Nora Martinez for translating this manuscript.
Conflict of interest statement
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arevalo-Pinzon, G., Curtidor, H., Reyes, C. et al. Fine mapping of Plasmodium falciparum ribosomal phosphoprotein PfP0 revealed sequences with highly specific binding activity to human red blood cells. J Mol Med 88, 61–74 (2010). https://doi.org/10.1007/s00109-009-0533-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0533-5