
Abstract
Diabetes mellitus (DM) has become a growing concern to global public health, being at the forefront of acute disorders and causes of mortality across the globe. Clinically approved drugs that are currently being used are faced with severe side effects, consequently necessitating the development of new drugs with no/fewer side effects and improved pharmacological potency. Herein, we report a rapid and efficient synthesis of thiazolidinone Schiff bases (2a-2t) from benzylidenehydrazines and thioglycolic acid under neat conditions through ultra-sonication. All the synthesized compounds were obtained in exceptional yields (89–95%) and confirmed by 1D and 2D nuclear magnetic resonance (NMR) spectroscopy, as well as High-resolution mass spectrometry (HRMS). The synthesized compounds were then evaluated for their antidiabetic activity through α-glucosidase and α-amylase inhibitory potentials and their antioxidant activity through Nitric Oxide (NO), 2,2′-diphenyl-1-picrylhydrazyl (DPPH), and Ferric reducing antioxidant power (FRAP) assays. Among them, 2q (IC50 = 96.63 μM) and 2h (IC50 = 125.27 μM) emerged as the most potent derivatives against α-amylase relative to reference drug acarbose (IC50 = 131.63 µM), respectively. Antioxidant evaluation further revealed that the synthesized derivatives were excellent NO scavengers disclosing 2n (IC50 = 44.95 µM) as the most potent derivative. Moreover, in silico ADME calculations predicted these compounds to have excellent drug-like properties. Kinetic studies disclosed the mode of α-amylase inhibition as competitive while molecular docking studies of the most active derivatives performed into the binding active site of human pancreatic α-amylase enzyme deciphered their ligand-protein interactions that explicated their observed experimental potencies.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) is a chronic metabolic disease characterized by high blood glucose levels, known as hyperglycemia. The elevated blood glucose level is a consequence of either insufficient insulin secretion by the pancreas (Type 1 diabetes mellitus-T1DM) or resistance to produced insulin by cells (Type 2 diabetes mellitus-T2DM) [1, 2]. It is currently estimated that in 2021, 537 million people globally were affected by diabetes with this number expected to increase to 700 million by the year 2045 [3]. Approximately 90% of all cases of diabetes that have been documented are T2DM [4]. Detrimental complications associated with DM include nephropathy, neuropathy, retinopathy, eyesight, and cardiovascular complications. Another alternative for controlling DM is consuming healthy food and frequent workout activities [5, 6].
Over and above, targeting key enzymes (α-amylase and α-glucosidase) in the gastrointestinal tract that are responsible for carbohydrate hydrolysis is one of the focal approaches for T2DM management as inhibition of these enzymes will decrease postprandial hyperglycemia. In order to fulfill its role, α-amylase hydrolyzes carbohydrates’ α-1,4-glycosidic linkages, resulting in the production of dextrin and maltose, which is then converted to glucose by α-glucosidase and raises blood glucose levels [7, 8]. Miglitol, voglibose, and acarbose (Fig. 1) are clinically authorized inhibitors of glucosidase and amylase that help moderate postprandial hyperglycemia. Even though these inhibitors have been successful in lowering postprandial hyperglycemia, their use is associated with unpleasant side effects like gastrointestinal disturbances, diarrhea, and abdominal pain. This suggests that there is an urgent need for the development of new α-amylase and α-glucosidase inhibitors with minimal/no side effects for long-term use [9, 10].

Antidiabetic drugs and reported α-amylase and α-glucosidase inhibitors containing thiazolidinone
DM is also noted to participate in the overproduction of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), hydroxyl radical (•OH), and superoxide (O2•‒) resulting in oxidative stress which is one of the main processes in which DM progresses [11, 12]. Moreover, oxidative stress has been associated with the pathogenesis of several diseases such as angiocardiopathy, stroke, arteriosclerosis, and cirrhosis [13, 14]. Consequently, research into the development of active compounds having dual antidiabetic and antioxidant potential is a promising approach for DM treatment.
Heterocyclic compounds are well established in the literature and play a pivotal role in medicinal chemistry as they are conferred with several pharmacological potentials [15, 16]. As per available statistics, more than 85% of all biologically active agents are heterocyclic [17]. Among many heterocyclic scaffolds, thiazolidinone is a privileged pharmacophore with diverse biological activities including antidiabetic and antioxidant activities [18, 19]). Epalrestat and pioglitazone (Fig. 1) are some of the drugs containing thiazolidinone core which are clinically approved as antidiabetic agents [20, 21]. These drugs have been implicated in severe side effects such as diarrhea, vomiting, nausea, increased levels of liver enzymes, heart failure, pedal edema, weight gain, and bone loss [22, 23]. Other studies have also shown the potential of thiazolidinone core as having antidiabetic properties. For instance, Ullah et al. [24] reported that compound A (IC50 = 0.40 µM, Fig. 1) showed more potency against α-amylase compared with acarbose (IC50 = 0.90 µM). Ramandeep et al. [25] also reported compound B to be a more potent inhibitor of α-glucosidase (IC50 = 42.80) than acarbose (IC50 = 478.07 µM). Finally, Singh et al. [26] reported compound C as a potent α-amylase inhibitor (IC50 = 19.67 µM) when compared with acarbose (IC50 = 22.94 µM).
The Schiff base, also known as imine (-C = N-) is another prominent pharmacophore that has drawn the attention of medicinal chemists due to its enormous biological potential [27]. The pharmacophore is recognized to establish binding affinity towards electrophilic and nucleophilic groups in the active pocket of the enzyme hence leading to enzyme inhibition [28]. Compounds containing Schiff bases in their structural unit have also been explored for their antidiabetic potentials; for example, Shamim et al. [28] reported that derivative D (Fig. 2) showed excellent α-glucosidase inhibition (IC50 = 9.43 µM) in comparison to standard drug acarbose (IC50 = 750 µM). Taha et al. [29] also reported Schiff bases E and F to possess excellent potency against α-glucosidase (IC50 = 1.10 µM) and α-amylase (IC50 = 0.90 µM) compared to acarbose with IC50 of 100.30 µM and 9.8 µM, respectively. Furthermore, derivative G [30] also exhibited potent α-glucosidase (IC50 = 1.40 µM) and α-amylase (IC50 = 1.10 µM) inhibitions compared to acarbose (IC50 = 12.20 µM and 11.50 µM, respectively).

Reported compounds as α-amylase and α-glucosidase inhibitors containing Schiff base
Green and eco-friendly methods for the synthesis of organic compounds for drug development have over the years been receiving significant attention from the scientific community [31]. The ultrasound technique is one of these methods that is described to significantly reduce reaction times, and afford high yields, and pure products that require easy workup over conventional methods [32, 33]. In continuation of our pursuit to discover potent antidiabetic and antioxidant agents [34, 35], we have prepared twenty thiazolidinone Schiff bases (2a-t) under solvent and catalyst-free conditions using ultrasound irradiation at 70 °C in excellent yields. Out of these twenty scaffolds, ten compounds viz. 2c, 2f-2k, 2m, 2s, and 2t have not been synthesized before using any conventional approach. The antidiabetic potential of 2a-t was investigated by evaluating their inhibitory activity toward α-amylase and α-glucosidase, while antioxidant potentials were evaluated against DPPH, FRAP, and NO. In silico molecular docking simulations were subsequently performed to comprehend the binding mode of selected compounds toward the active pocket of human pancreatic α-amylase enzyme (PDB ID: 2QV4).
Results and discussion
Chemistry
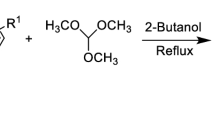
The synthetic approach employed for the synthesis of thiazolidinone Schiff bases 2a-t is shown in Scheme 1. Initially, different substituted benzaldehydes were treated with hydrazine hydrate to afford dibenzylidenehydrazine derivatives (1a-t) which underwent Knoevenagel condensation in the presence of thioglycolic acid under ultrasound conditions to afford target compounds (2a-t).

Synthetic pathway for thiazolidinone Schiff bases (2a-2t)
The preparation of thiazolidinone derivatives using imines and thioglycolic acid is documented in the literature. Conventional methods exploit harsh solvents such as toluene, dioxane, pyridine, and DMF at high temperatures and longer reaction times [36,37,38,39]. Herein we have successfully synthesized thiazolidinones bearing iminic unit in significantly reduced time (30 min to 1 hour) and high yields (89–95%) using no solvent (neat conditions).
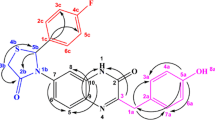
The synthesized compounds were characterized using NMR and MS spectroscopy. For example, in representative derivative 2a, thiazolidinone moiety displayed three proton resonances viz. a singlet peak at δH 6.56 which corresponds to H-5, and two doublets which correspond to diastereoisomeric methylene protons H-3 appeared at δH 4.07 (J = 16.08 Hz) and δH 4.03 (J = 16.06 Hz). The iminic proton H-2” appeared downfield at δH 8.26. The resonances at δH 7.63 to 7.36 are ascribed to aromatic protons. Furthermore, the successful formation of the target products was confirmed by heteronuclear multiple bond coherence (HMBC). For example, in derivative 2a, methine proton H-5 (δH 6.56) showed correlations with C-2 (δC 168.08), C-3 (δC 30.52), C-1’ (δC 151.11) and C-6’/C-1’ (δC 126.47) (Fig. 2) Diastereoisomeric methylene protons H-3 showed correlations with C-2 (δC 168.08) and C-5 (δC 60.90) while the imine proton 2” showed correlations with C-3” (δC 134.10) and C-5”/C-9” (Fig. 3).

HMBC (1H → 13C) correlation of compound 2a
Antidiabetic activities
Digestive enzymes viz. α-glucosidase and α-amylase are involved in polysaccharides hydrolysis and therefore play a crucial part in glucose regulation. Studies show that inhibition of these enzymes is significant for the management of T2DM [40, 41]. Accordingly, the synthesized compounds 2a-2t were evaluated for their inhibitory activities against α-glucosidase and α-amylase enzymes using acarbose as a standard inhibitor. The inhibitory potencies represented as IC50 values are shown in Table 1. The obtained results indicated that the synthesized compounds were poor inhibitors of α-glucosidase enzyme with compound 2o bearing para nitro group showing IC50 value of 494.67 µM in comparison to standard acarbose which showed IC50 value of 108.10 µM.
More significantly, evaluation against α-amylase presented good to moderate varied inhibitory potencies with IC50 values ranging from 96.63 to 559.25 µM in comparison to standard acarbose (IC50 = 131.63 µM). The structure-activity relationship (SAR) analysis for α-amylase inhibition was subsequently explored by firstly comparing different substituted derivatives at the same position of the phenyl ring and secondly comparing derivatives containing the same substituents at different positions.
It is significant to note that derivative 2q containing nitro group at ortho-position exhibited the highest potency against α-amylase in the series (IC50 = 91.63 µM) which is 1.4 fold more potent than acarbose (IC50 = 131.63 µM). Also 2q showed greater inhibitory activity when compared to its ortho-substituted counterparts, ortho-fluoro derivative 2h (IC50 = 125.527 µM) the second potent derivative, ortho-trifluoromethyl 2k (IC50 = 181.31 µM), ortho-chloro derivative 2d (IC50 > 600 µM) and ortho-bromo 2n (IC50 > 600 µM). The difference in the inhibitory potencies may be caused by the strength of the electron-withdrawing power of the substituents with the most electronegative substituted derivatives displaying the strongest α-amylase inhibition. This is probably attributed to the increased polarization in the molecule that leads to stronger electrostatic interactions with the target protein.
The positioning of different para substituents also affected the α-amylase inhibitory potencies. For instance, the replacement of trifluoromethyl in 2j (IC50 = 181.31 µM) with different halogens decreases its potency as observed in 2f (IC50 = 467.10 µM), 2b (IC50 = 555.90 µM), and 2l (IC50 > 600 µM). However, the introduction of the NO2 group at the para position improved the activity as seen in derivative 2o (IC50 = 230.52 µM). It is then deducible that the inhibitory potency depends on the strength of the electron-donating groups as well as the bulkiness of the halogens. The higher potency of derivative 2k could be due to the presence of three more electronegative fluorine atoms that can lower the electron density of the aromatic ring thereby enhancing its acidity and influencing the electrostatic interactions with the substrate [42]. The order of potency in the case of meta-substituted compounds was F > Cl > NO2 > Br with IC50 values of 167.10 µM for 2g, 234.68 µM for 2c, 272.81 µM for 2p and 559.25 µM for 2m. Noticeably, the compounds containing electron-donating substituents exhibited moderate potency as shown by 2r (IC50 = 318.77 µM) and 2s (IC50 = 338.40 µM) which was more than bromo substituted derivative 2m (IC50 = 559.25 µM).
In terms of comparing derivatives containing the same substituents, amongst the nitro-containing derivatives, ortho-substituted derivative 2q (IC50 = 96.63 µM) emerged as the most active, followed by para derivative 2o IC50 = 230.52 µM) and meta derivative 2p (IC50 = 272.81 µM). A comparison of the fluoro-substituted derivatives revealed that ortho-substituted derivative 2h (IC50 = 125.27 µM) displayed the highest potency which was slightly superior to the standard drug, acarbose (IC50 = 131.63 µM). This was followed by meta substituted derivative (IC50 = 167.10 µM), ortho-para derivative 2i (IC50 = 131.63 µM), and para derivative 2f (IC50 = 131.63 µM). The difference in observed activity might be due to the position and the number of fluorine atoms which increases or decreases the activity while para trimethyl fluoro 2j and ortho trimethyl fluoro 2k displayed almost similar potencies with IC50 values of 181.31 µM and 178.20 µM. Likewise, meta-substituted derivative 2c (IC50 = 234.68 µM) emerged as the most active followed by para-substituted derivative 2b (IC50 = 555.90 µM). The ortho-substituted 2d and ortho-para-substituted derivative 2e were inactive. Similarly, when we compared 2s (IC50 = 338.40 µM) bearing one methoxy group and 2t (IC50 = 212.20 µM), the latter showed better inhibitory potential presumably due to a greater number of methoxy groups. Lastly, all the bromo derivatives did not show any significant activity which might be attributed to the poor electron-withdrawing nature of bromine as well as its bulkiness that somehow hindered the enzyme inhibition.
Based on the above limited SAR, it can be deduced that α-amylase is mainly brought about by derivatives with higher electron withdrawing effects viz. CF3, NO2, and F while changing the position and the number of substituents resulted in variable inhibitory potencies.
Antioxidant activity evaluation
ROS accumulate in the body as a consequence of metabolic functions or due to mental and physical strain. Other external factors that cause ROS accumulation include smoking, radiation, pesticides, and organic solvents [43]. Over-accumulation of ROS has been disclosed to pivotally participate in the development of diseases that severely affect humans such as diabetes mellitus and cardiovascular diseases, [10, 44, 45]. Accordingly, the antioxidant efficacy of the synthesized compounds was evaluated using nitric Oxide (NO), Diphenyl-1-Picrylhydrazyl (DPPH), and Ferric Reducing Antioxidant Power (FRAP) assays, the results were compared to standard trolox and are depicted in Table 1 as IC50 values.
The results of NO scavenging activity revealed that most of the synthesized compounds were excellent NO scavengers better than standard trolox (IC50 > 2000 µM). Among them, derivative 2n containing bromo substituent at ortho position emerged as the most active compound with IC50 value of 44.95 µM, respectively, Changing the position of bromo substituent to meta and para positions resulted in a decrease in activity as seen in derivative 2m (IC50 = 91.66 µM) and derivative 2l (IC50 = 101.13 µM). A similar trend was observed in the nitro substituted derivatives where ortho position was favored (2g, IC50 = 128.26 µM) than meta (2p, IC50 = 142.66 µM), and para (2o, IC50 = 1226.72). Interestingly, fluoro-substituted derivatives 2h, 2g, 2f, and chloro-substituted derviatives 2d, 2c, and 2b at ortho, meta, and para positions did not follow the trend as observed above. Moreover, the substitution of the electron-withdrawing group and trifluoromethyl group at ortho and para position led to a decrease in NO scavenging activity as seen in derivatives 2k (IC50 = 256.75 µM) and 2j (IC50 = 248.34 µM) when compared to compounds 2n and 2m. Derivative 2r (IC50 = 90.29 µM) containing methyl group at meta position emerged as the second most active compound in the series. Replacement of the methyl group with methoxy group (2s and 2t) resulted in a decrease in potency. It can then be concluded that generally, electron-withdrawing substituents are detrimental to NO scavenging activity.
Enzyme kinetic studies for amylase inhibition
To evaluate the mode of inhibition of the most potent derivative in the series 2q, a time-dependent para-nitro-phenyl-β-D-glucopyranoside (pNPG) assay was used and the results are represented as Lineweaver–Burk plot (Fig. 4). The maximum velocity (Vmax) remained constant for changing inhibitor concentrations while the Michaelis–Menten constant (Km) changed denoting a competitive type of enzyme inhibition.

Lineweaver–Burk plot of the most active compound 2q against α-amylase
In-silico studies
ADME properties
At present, academics and industries are of the perspective that absorption, distribution, metabolism, and elimination (ADME) properties are equally imperative as specificity and efficacy in drug development as they are the main reason among others for clinical trial failures [46, 47]. Evaluation of ADME properties at the early stages of drug discovery increases the success rate and at the same time reduces the costs associated with research and development [48]. Herein, ADME properties of all the synthesized derivatives were computed using Qik Promodule in Schrödinger molecular modeling suite [49]. The results of ADME predictions are tabulated in Table 2.
Among the corresponding descriptors, viz. molecular weight (MW), total solvent accessible surface area (SASA), Hydrogen bond donors and (HBD) and acceptors (HBA), Octanol/water partition coefficient(QPlogPo/w), aqueous solubility (QPlogS) and binding to human serum albumin; Oral abs: human oral absorption (QPlogKhSA) were all in the accepted ranges. QPlogHERGIC50 value for the blockage of “human ether-a-go-go related gene” K+ channels (QPlogHERG) was slightly more negative than a recommended value which implies that further structural optimization may be required. Moreover, the expounding of this descriptor should be done carefully because not all hERG blockers pose a pro-TdP arrhythmic risk [50]. Additionally, synthesized derivatives showed a percentage of human oral absorption (Oral abs) ranging from 72.01 to 100 predicting substantial oral bioavailability, Lastly, all the derivatives did not violate Lipinski’s rule of five (Ro5) and Jorgensen’s rule of 3 (Ro3). Generally, it can be uttered that the synthesized derivatives possess drug-like properties.
Molecular docking and MM-GBSA calculations
To further validate the results obtained from iv vitro α-amylase studies, five promising derivatives, viz. 2q (IC50 = 96.63 µM), 2h (IC50 = 125.27 µM), 2g (IC50 = 167.10 µM), and 2k (178.20 µM) were docked at the active site of human pancreatic α-amylase enzyme (PDB ID: 2QV4) to decipher the ligand-protein interactions that explicate the observed potencies. In this analysis, the Induced Fit Docking (IFD) protocol was utilized as it seeks to optimize ligand docking with an assumption that the protein would change substantially when the ligand is docked at the active site [51]. The predictive efficiency of the docking procedure was first assessed by re-docking the co-crystallized ligand and thereafter superimposing the result pose with the crystal structure and calculating the RMSD value between them. Figure 5 illustrates the two superimposed ligands with an RMSD value of 1.8192 Å which is lower than the maximum accepted value of 2.0 Å and thus validated the accurateness of the docking protocol.

Superimposed ligand poses of co-crystallized ligand (blue) and re-docked ligand (green) with RMSD value of 1.8192 Å
The selected compounds were subsequently docked in the active site of the protein using the same docking protocol. Experimental results revealed that derivatives 2q and 2h emerged as most potent against α-amylase. These results were corroborated by their binding free energies (ΔG bind) of −54.32 kcalmol−1 and −37.21 kcalmol−1 and docking scores of −5.794 and −4.862 (Table 3), respectively. 2q-receptor binding profile revealed that carbonyl of thiazolidinone moiety afforded hydrogen bond interaction with Thr163. Another hydrogen bond was established between imine and Gln63. The binding profile was further stabilized by Pi-Pi stacking through benzyl and phenyl rings with Trp59 and Pi cation interactions with Trp59 with N attached to the benzyl ring (Figs. 6 and 7). The 2h-receptor binding profile showed that carbonyl of thiazolidinone moiety also furnished hydrogen bond interaction with Thr163 and further formed a Pi-Pi stacking interaction with Trp58.

3-D representation of the binding interactions of the selected compounds docked at the active site of 2QV4. Protein-ligand interactions are shown as dashed lines; hydrogen bond (yellow), pi-pi stacking (light-blue) pi-cation (green)

2-D representation of the binding interactions of the selected compounds docked at the active site of 2QV4
The reduced potencies of derivatives 2g and 2k were also validated by their ΔG bind (−35.88, −32.06, and −32.55) and docking scores (−4.051, −3.933 and −3.327) (Table 3). 2g and 2k receptor binding profiles consist of hydrogen bonds interaction between His299 and carbonyl of thiazolidinone moiety. The 2k-receptor binding profile further consisted of hydrogen bond interactions between Arg195 and the same carbonyl of thiazolidinone moiety. Further, acarbose demonstrated the highest docking score of −8.95, this is not surprising as acarbose’s higher molecular surface and its ability to interact with enzymes via several hydrophobic interactions.
Conclusion
In this research work, we have developed a simple and energy-efficient methodology for the synthesis of thiazolidinone Schiff bases (2a-2t) under neat conditions using an ultrasound irradiation technique. This procedure afforded target compounds in excellent yields ranging from 89% to 95%. The biological evaluation revealed that the synthesized compounds were stronger inhibitors of the α-amylase enzyme than the α-glucosidase enzyme with derivative 2q being established as the most active compound in the series with an IC50 value of 96.63 µM. In addition, the antioxidant evaluation revealed that derivative 2n is an excellent scavenger of NO with an IC50 value of 44.95 µM. Enzyme kinetic studies revealed a competitive mode of inhibition for compound 2q. Additionally, the docking results corroborated the biological data. It envisaged that the present compounds will serve as a template for further structural optimizations to design new potent antidiabetic compounds with dual mode of inhibition and improved antioxidant potential.
Experimental
General information
All chemicals and solvents were purchased from Sigma Aldrich and Merck and were used without any further purification. Infrared spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR spectrometer with a universal ATR sampling accessory. NMR analysis was recorded on a Bruker AVANCE III 400 MHz spectrometer (400 MHz for 1H, 100 MHz for 13 C, and 376.498 MHz for 19 F). Chemical shifts (δ) were reported in parts per million (ppm). The chemical shifts for 1H and 13 C are referenced to DMSO‑d6 at 2.50 ppm and 39.51 ppm, and CDCl3 at 7.2 ppm and 77.3 ppm, respectively. Spin multiplicities are abbreviated as follows: singlet (s), doublet (d), doublet of doublet of doublets (ddd), doublet of doublets (dd), and triplet (t). Melting points of compounds were determined in an electrothermal melting point apparatus (Electrothermal IA9100) using a sealed capillary tube and are uncorrected. High-resolution mass data were obtained using a Bruker microTQF-Q II ESI instrument operated at ambient temperatures.
Synthesis of dibenzylidenehydrazine derivatives
To a solution of ethanol (30 mL) containing corresponding benzaldehydes 10 mmol, hydrazine monohydrate 20 mmol was added, and the reaction mixture was stirred for 3–4 hours until the consumption of benzaldehydes as indicated by thin layer chromatography (TLC). The formed precipitates were filtered and recrystallized from ethanol to afford dibenzylidenehydrazine derivatives.
Synthesis of thiazolidinone derivatives
Excess thioglycolic acid 1 ml was added to the dibenzylidenehydrazine derivatives (1a-t) 1 mmol in a test tube. The mixture was then subjected to ultrasound irradiation using scientecultrasonic cleaner at 70 °C for 1–1.5 hours until dibenzylidenehydrazine derivatives have been consumed as indicated by TLC. Then, 10 ml of ethyl acetate was added and the solids crushed out were filtered in vacuo and washed with 10% washed NaHCO3 and water. The solids products were then washed with ice-cold diethyl ether to furnish the target compounds.
(E)-3-(benzylideneamino)-2-phenylthiazolidin-4-one 2a
white solid; Chemical Formula; C16H14N2OS; Molecular Weight: 281,37; Yield: 89%
FTIR νmax (cm−1): 1693.25 (C = O), 1496.82 (C = N), 1047.23 (C–N).
1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H, N = CH), 7.63 (d, 2H, Ar-H) 7.46-7.36 (m, 8H, Ar-H), 6.56 (s, 1H, thiazolidine-4-one-CH), 4.07 (d, J = 16.08 Hz, 1H, thiazolidine-4-one-CH2), 4.03 (d, J = 16.06 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.08 (C = O), 151.11 (C = N), 140.07, 134.10, 131.18, 129.51, 129.33, 128.95, 127.77, 126,47 (Aromatic), 60.90 (thiazolidine-4-one-CH), 30.52 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H14N2OS (M + Na)+: 305.0725; found 305.0731.
(E)-3-((4-chlorobenzylidene)amino)-2-(4-chlorophenyl)thiazolidin-4-one 2b
White solid; Chemical Formula: C16H12C12N2OS: Molecular Weight: 351,25; Yield: 91%
FTIR νmax (cm−1): 1683.04 (C = O), 1593.59 (C = N), 1086.27 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.22 (s, 1H, N = CH), 7.62 (d, J = 8.10, 2H, Ar-H) 7.47-7.40 (m, 6H, Ar-H), 6.49 (s, 1H, thiazolidine-4-one-CH), 4.02 (d, J = 16.08 Hz, 1H, thiazolidine-4-one-CH2), 3.83 (d, J = 16.08 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.07 (C = O), 149.95 (C = N), 139.06, 133.74, 133.01, 129.51, 129.47, 129.43, 128.50 (Aromatic), 60.41 (thiazolidine-4-one-CH), 30.46 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12C12N2OS (M + Na)+: 372.9945; found 372.9955.
(E)-3-((3-chlorobenzylidene)amino)-2-(3-chlorophenyl)thiazolidin-4-one 2c
white solid; Chemical Formula: C16H12Cl2N2OS: Molecular Weight: 351,25; Yield: 92%
FTIR νmax (cm−1): 1694.85 (C = O), 1562.31 (C = N), 1099.73 (C–N).)
1H NMR (400 MHz, DMSO-d6) δ 8.28 (s, 1H, N = CH), 7.70 (s, 1H, Ar-H), 7.62 (d, J = 7.26 Hz, 1H, Ar-H), 7.53-7.43 (m, 5H, Ar-H), 7.38 9 (d, J = 7.61 Hz, 1H, Ar-H), 6.52 (s, 1H,thiazolidine-4-one-CH), 4.13 (d, J = 15.98 Hz, 1H, thiazolidine-4-one-CH2), 3.89 (d, J = 15.98 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.20 (C = O), 149.26 (C = N), 142.46, 136.29, 134.15, 134.10, 131.44, 131.29, 130.85, 129.03, 126.98, 126.53, 125.05 (Aromatic), 60.27(thiazolidine-4-one-CH), 30.37 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12C12N2OS (M + Na)+: 372.9945; found 372.9955.
(E)-3-((2-chlorobenzylidene)amino)-2-(2-chlorophenyl)thiazolidin-4-one 2d
white solid; Chemical Formula: C16H12Cl2N2OS: Molecular Weight: 351,25; Yield: 90%
FTIR νmax (cm−1): 1720.20 (C = O), 1468.75 (C = N), 1042.01 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.20 (s, 1H, N = CH), 7.87 (d, J = 7.98, 1H, Ar-H), 7.58-7.56 (1H, Ar-H), 7.46-7.36 (m, 5H, Ar-H), 7.22 (s, 1H, Ar-H), 6.73 (s, 1H,thiazolidine-4-one-CH), 4.01 (d, J = 16.23 Hz, 1H, thiazolidine-4-one-CH2), 3.88 (d, J = 16.26 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.51 (C = O), 145.57 (C = N), 136.04, 134.08, 132.62, 131.72, 131.29. 130.66, 130.38, 128.64, 128.18, 127.37, 127.20 (Aromatic), 58.37 (thiazolidine-4-one-CH), 30.22 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12C12N2OS (M + Na)+: 372.9945; found 372.9956.
(E)-3-((2,4-dichlorobenzylidene)amino)-2-(2,4-dichlorophenyl)thiazolidin-4-one 2e
white solid; Chemical Formula: C16H10Cl4N2OS; Molecular Weight: 420,14; Yield: 91%
FTIR νmax (cm−1): 1682.78 (C = O), 1585.85 (C = N), 1045.14 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H, N = CH), 7.86 (d, J = 8.52 Hz, 2H, Ar-H), 7.76 (d, J = 2.13 Hz, 1H, Ar-H), 7.66 (d, J = 2.09 Hz, 1H, Ar-H), 7.50-7.44 (m, 2H, Ar-H), 7.27 (d, J = 8.42 Hz, 1H, Ar-H), 6.71 (s, 1H,thiazolidine-4-one-CH), 4.02 (dd, J = 16.13, 0.95 Hz, 1H,thiazolidine-4-one-CH2), 3.89 (d, J = 16.21 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.54 (C = O), 168.52 (C = N), 144.74, 136.38, 135.21, 134.80, 134.35, 132.77, 130.20, 129.91, 128.85, 128.65, 128.56 (Aromatic), 58.04 (thiazolidine-4-one-CH), 30.20 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H10Cl4N2OS (M + Na)+: 442.9137; found 442.9136.
(E)-3-((4-fluorobenzylidene)amino)-2-(4-fluorophenyl)thiazolidin-4-one 2f
white solid; Chemical Formula: C16H12F2N2OS; Molecular Weight: 318,34; Yield: 95%
FTIR νmax (cm−1): 1683.16 (C = O), 1600.66 (C = N), 1050.90 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H, N = CH), 7.67 (dd, 6.03, 3.09 Hz, 2H, Ar-H), 7.47 (dd, 5.48, 3.5 Hz, 2H, Ar-H), 7.26-7.19 (m, 4H, Ar-H), 6.48 (s, 1H,thiazolidine-4-one-CH), 4.03 (d, J = 16.25 Hz, 1H, thiazolidine-4-one-CH2), 3.84 (d, J = 16.27 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 197.98 (C = O), 165.18, 163.56, 162.71, 161.13 (Aromatic), 150.60 (C = N), 136.28, 136.25, 130.71, 130.78, 130.11, 130.03, 129.93, 116.56, 116.45, 116.34 (Aromatic), 60.57 (thiazolidine-4-one-CH), 30.53 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12F2N2OS (M + Na)+: 341.0544; found 341.0536.
(E)-3-((3-fluorobenzylidene)amino)-2-(3-fluorophenyl)thiazolidin-4-one 2g
white solid; Chemical Formula: C16H12F2N2OS; Molecular Weight: 318,34; Yield: 94%
FTIR νmax (cm−1): 1697.84 (C = O), 1580.99 (C = N), 1048.45 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.26 (s, 1H, N = CH), 7.47-7.39 (m, 4H Ar-H), 7.30 (m, 4H, Ar-H), 6.49 (s, 1H, thiazolidine-4-one-CH), 4.07 (d, J = 16.22 Hz, 1H, thiazolidine-4-one-CH2),3.84 (d, J = 16.28 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.07 (C = O), 164.07, 163.98, 161.63, 161.55, 149.50 (Aromatic), 149.47 (C = N), 142.93, 142.89, 136.64, 136,56, 124,30, 124,27, 122.46. 122.44, 118.11, 117.90, 116.04, 115.83, 113.64, 113.49, 113.41, 60.31 (Aromatic), 60.29 (thiazolidine-4-one-CH), 30.38 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12F2N2OS (M + Na)+: 341.0545; found 341.0536.
(E)-3-((2-fluorobenzylidene)amino)-2-(2-fluorophenyl)thiazolidin-4-one 2h
white solid; Chemical Formula: C16H12F2N2OS; Molecular Weight: 318,34; Yield: 93%
FTIR νmax (cm−1): 1700.24 (C = O), 1609.09 (C = N), 1052.87 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.36 (s, 1H, N = CH), 7.81 (ddd, 1H Ar-H), 7.51–7.20 (m, 7H, Ar-H), 6.75 (s, 1H, thiazolidine-4-one-CH), 4.00 (d, J = 16.18 Hz, 1H, thiazolidine-4-one-CH2), 3.89 (d, J = 16.20 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.2 (C = O), 162.59, 161.24, 160.10, 158,78 (Aromatic), 142,79 (C = N), 133.31, 133.22, 131.29, 131.20, 128.12, 128.09, 126.80, 126,80, 126,78, 126,61, 126,49, 125.58,125,54, 125.48, 125.45, 121.67, 121.57, 116.76, 116,65, 116.44, 55.77 (Aromatic), 55,74 (thiazolidine-4-one-CH), 30.52 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12F2N2OS (M + Na)+: 341.0544; found 341.0536.
(E)-3-((2,4-difluorobenzylidene)amino)-2-(2,4-difluorophenyl)thiazolidin-4-one 2i
White solid; Chemical Formula: C16H10F4N2OS; Molecular Weight: 354,32; Yield: 89%
FTIR νmax (cm−1): 1691.70 (C = O), 1612.08 (C = N), 1052.40 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.46 (s, 1H, N = CH), 7.88 (q, J = 14.88, 7.62, H Ar-H), 7.50 (q, J = 14.52, 7.75, 1H, Ar-H), 7.41 (q, J = 21.77, 10.83, 2H, Ar-H), 7.22-7.16 (m, 2H, Ar-H), 6.74 (s, 1H,thiazolidine-4-one-CH), 4.04 (d, J = 16.01 Hz, 1H, thiazolidine-4-one-CH2), 3.92 (d, J = 16.01 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.12 (C = O), 165.09, 163.62, 163.41, 163.33, 162.42, 161.97, 161.89, 161.13, 160.83, 160.83, 160.75, 159.47, 159.39 (Aromatic), 142.89 (C = N), 129.75, 128.58, 128.51, 123.18, 123.10, 113.29, 113.15, 112.73, 112.58, 105.48, 105.31, 105.26, 105.09, 104.92, 55.82 (thiazolidine-4-one-CH0, 30.59 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H10F4N2OS (M + Na)+: 377.0352; found 377.0348.
(E)-3-((4-(trifluoromethyl)benzylidene)amino)-2-(4-(trifluoromethyl)phenyl)thiazolidin-4-one 2j
White solid; Chemical Formula: C18H12F6N2OS; Molecular Weight: 418,36; Yield: 95%
FTIR νmax (cm−1): 1688.82 (C = O), 1619.63 (C = N), 1065.28 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.35 (s, 1H, N = CH), 7.87 (d, J = 8.16 Hz, 2H, Ar-H), 7.82 (d, J = 7.38 Hz, 4H, Ar-H), 7.66 (d, J = 8.04 Hz, 2H, Ar-H), 6.68 (s, 1H,thiazolidine-4-one-CH), 4.10 (d, J = 16.14 Hz, 1H,thiazolidine-4-one-CH2), 3.92 (d, J = 16.20 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.39 (C = O), 148.79 (C = N), 144.55, 137.95, 130.93, 130.61, 130.30, 129.53, 129.21, 128.41, 127.28, 126.54, 126.51, 126.28, 126.24, 128.80, 125.75, 123.09, 123.05 (Aromatic), 60.04 (thiazolidine-4-one-CH), 30.30 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C18H12F6N2OS (M + Na)+: 441.0479; found 441.0484.
(E)-3-((2-(trifluoromethyl)benzylidene)amino)-2-(2-(trifluoromethyl)phenyl)thiazolidin-4-one 2k
White solid; Chemical Formula: C18H12F6N2OS; Molecular Weight: 418,36; Yield: 93%
FTIR νmax (cm−1): 1720.45 (C = O), 1585.82 (C = N), 1030.51 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.06 (d, J = 7.86 Hz, 1H, Ar-H), 8.01 (s, 1H, N = CH), 7.85 (d, J = 7.86 Hz, 1H, Ar-H), 7.74-7.68 (m, 3H, Ar-H), 7.62-7.56 (m, 2H, Ar-H), 7.43 (d, J = 7.86, 1H, Ar-H),), 6.62 (s, 1H,thiazolidine-4-one-CH), 4.07 (d, J = 16.14 Hz, 1H,thiazolidine-4-one-CH2), 3.90 (d, J = 16.20 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.72 (C = O), 143.62 (C = N), 137.61, 134.44, 133.40, 131.72, 131.19, 129.72, 127.79, 127.49, 127.41, 127.20, 127.15, 127.09, 127.04, 126.49, 126.42, 126.36, 126.31, 125.97, 125.72, 125.42, 125.12, 123.25, 123.25, 122.70 (Aromatic), 56.86 (thiazolidine-4-one-CH), 29.93 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C18H12F6N2OS (M + Na)+: 441.0479; found 441.0484.
(E)-3-((4-bromobenzylidene)amino)-2-(4-bromophenyl)thiazolidin-4-one 2l
White solid; Chemical Formula: C16H12Br2N2OS; Molecular Weight: 440,15; Yield: 93%
FTIR νmax (cm−1): 1686.90 (C = O), 1590.38 (C = N), 1068.55 (C–N).
1H NMR (400 MHz, DMSO-d6) δ 8.21 (s, 1H, N = CH), 7.62–7.53 (m, 6H, Ar-H), 7.35 (d, J = 8.46, 2H, Ar-H), 6.47 (s, 1H,thiazolidine-4-one-CH), 4.02 (dd, J = 16.25, 0.89 Hz, 1H,thiazolidine-4-one-CH2),3.84 (d, J = 16.25 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (101 MHz, DMSO-d6) δ 168.10 (C = O), 149.85 (C = N), 139.46, 133.33, 132.44, 132.40, 129.63, 128.76, 124.59, 122.05 (Aromatic), 60.35 (thiazolidine-4-one-CH), 30.42 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12Br2N2OS (M + Na)+: 462.8910; found 462.8914.
(E)-3-((3-bromobenzylidene)amino)-2-(3-bromophenyl)thiazolidin-4-one 2m
White solid; Chemical Formula: C16H12Br2N2OS; Molecular Weight: 440,15; Yield: 94%
FTIR νmax (cm−1): 1693.71 (C = O), 1570.95 (C = N), 1069.02 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.20 (s, 1H, N = CH), 7.80 (s, 1H, Ar-H), 7.62 (t, J = 7.18 Hz, 3H, Ar-H), 7.52 (d, J = 6.18 Hz, 1H, Ar-H), 7.38–7.33 (m, 3H, Ar-H), 6.46 (s, 1H,thiazolidine-4-one-CH), 4.07 (d, J = 16.27 Hz, 1H, thiazolidine-4-one-CH2), 3.83 (d, J = 16.38 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.21 (C = O), 149.09 (C = N), 142.62, 136.50, 133.74, 131.71, 131.56, 129.88, 129.37, 126.91, 125.39, 122.62 (Aromatic), 60.12 (thiazolidine-4-one-CH), 30.35 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12Br2N2OS (M + Na)+: 462.8925; found 462.8914.
(E)-3-((2-bromobenzylidene)amino)-2-(2-bromophenyl)thiazolidin-4-one 2n
white yield; Chemical Formula: C16H12Br2N2OS; Molecular Weight: 440,15; Yield: 89%
FTIR νmax (cm−1): 1719.34 (C = O), 1585.09 (C = N), 1020.22 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.07 (s, 1H, N = CH), 7.86 (dd, J = 1.65, 7.80 Hz, 1H, Ar-H), 7.74(d, J = 7.80 Hz, 1H, Ar-H), 7.62 (d, J = 7.80 Hz, 1H, Ar-H), 7.45–7.23 (m, 4H), 7.16 (d, J = 7.59 Hz, Ar-H), 6.62 (s, 1H,thiazolidine-4-one-CH), 4.00 (d, J = 16.35 Hz, 1H, thiazolidine-4-one-CH2), 3.87 (d, J = 16.31 Hz,1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.58 (C = O), 147.88 (C = N), 137.37, 133.96, 133.60, 132.85, 132.75, 130.88, 129.22, 128.70, 127.76, 124.42, 121.89 (Aromatic), 60.82 (thiazolidine-4-one-CH), 30.12 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12Br2N2OS (M + Na)+: 462.8920; found 462.8914.
(E)-3-((4-nitrobenzylidene)amino)-2-(4-nitrophenyl)thiazolidin-4-one 2o
Brown solid; Chemical Formula: C16H12N4O5S; Molecular Weight: 372,36; Yield: 91%
FTIR νmax (cm−1): 1691.56 (C = O), 1596.46 (C = N), 1055.35 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.03 (s, 1H, N = CH), 8.25 (d, J = 8.57 Hz, 4H, Ar-H), 7.87 (d, J = 8.39 Hz, 2H, Ar-H), 7.68 (d, J = 8.52 Hz, 4H, Ar-H), 6.68 (s, 1H,thiazolidine-4-one-CH), 4.08 (d, J = 16.28 Hz, 1H, thiazolidine-4-one-CH2), 3.90 (d, J = 16.30 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.43 (C = O), 148.78 (Aromatic), 147.91 (C = N), 147.88, 147.13, 140.12, 128.82, 127.77, 124.54 (Aromatic), 59.85 (thiazolidine-4-one-CH), 30.29 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12N4O5S (M + Na)+: 395.0434 found 395.0433.
(E)-3-((3-nitrobenzylidene)amino)-2-(3-nitrophenyl)thiazolidin-4-one 2p
Yellow solid; Chemical Formula: C16H12N4O5S; Molecular Weight: 372,36; Yield: 93%
FTIR νmax (cm−1): 1706.06 (C = O), 1512.27 (C = N), 1054.22 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.42 (s, 1H, Ar-H), 8.35 (s, 1H, N = CH), 8.27 (s, 1H, Ar-H) 8.24 (d, J = 7.98 Hz, 1H, Ar-H), 8.18 (d, J = 6.36 Hz, 1H, Ar-H), 8.05 (d, J = 7.50 Hz, 1H, Ar-H), 7.82 (d, J = 7.56, 1H, Ar-H), 7.71–7.68 (m, 2H, Ar-H), 6.65 (s, 1H,thiazolidine-4-one-CH), 4.09 (d, J = 16.14 Hz, 1H, thiazolidine-4-one-CH2), 3.87 (d, J = 16.20 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.45 (C = O), 148.64, 148.61 (Aromatic), 148.34 (C = N) 142.03, 135.83, 133.78, 132.78, 131.29, 130.99, 125.43, 123.97, 122.07, 121.60 (Aromatic), 59.90 (thiazolidine-4-one-CH), 30.24 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12N4O5S (M + Na)+: 395.0434 found 395.0433.
(E)-3-((2-nitrobenzylidene)amino)-2-(2-nitrophenyl)thiazolidin-4-one 2q
Yellow solid; Chemical Formula: C16H12N4O5S; Molecular Weight: 372,36; Yield: 95%
FTIR νmax (cm−1): 1712.27 (C = O), 1513.40 (C = N), 1054.10 (C–N).
1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 8.14 Hz, 1H,Ar-H), 8.22 (s, 1H, N = CH), 7.99 (d, J = 8.18 Hz, 1H, Ar-H,) 7.92 (d, J = 7.65 Hz, 1H, Ar-H), 7.84 (t, J = 8.01, 1H, Ar-H) 7.80 (t, J = 7.65, 1H, Ar-H), 7.67–7.64 (m, 2H, Ar-H) 7.30 (d, J = 7.53 Hz,1H, Ar-H), 6.78 (s, 1H, thiazolidine-4-one-CH), 3.98 (d, J = 16.44 Hz, 1H, thiazolidine-4-one-CH2), 3.82 (d, J = 16.40 Hz, 1H, thiazolidine-4-one-CH2).
13C NMR (151 MHz, DMSO-d6) δ 168.91 (C = O), 148.77, 146.44 (Aromatic), 146.01 (C = N), 135.84, 134.68, 134.32, 131.70, 130.35, 128.94, 128.65. 126.74, 126.44, 125.11 (Aromatic), 57.18 (thiazolidine-4-one-CH), 29.54 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C16H12N4O5S (M + Na)+: 95.0431 found 95.0433.
(E)-3-((3-methoxybenzylidene)amino)-2-(3-methoxyphenyl)thiazolidin-4-one 2r
White solid; Chemical Formula: C18H18N2O3S; Molecular Weight: 342,41; Yield: 89%
FTIR νmax (cm−1): 1713.24 (C = O), 1573.05 (C = N), 1041.79 (C–N).
1H NMR (400 MHz, DMSO-d6) δ 8.16 (s, 1H, N = CH), 7.34 (q, J = 7.84, 2H, Ar-H), 7.1.6 (s, 2H, Ar-H), 7.00 (d, J = 8.20, 1H, Ar-H), 9.93–6.87 (m, 3 h, Ar-H), 6.46 (s, 1H, thiazolidine-4-one-CH), 4.03 (d, J = 16.04 Hz, 1H, thiazolidine-4-one-CH2), 3.82 (d, J = 16.12 Hz, 1H, thiazolidine-4-one-CH2), 3.75 (s, 3H, CH3), 3.74 (s, 3H, CH3).
13C NMR (101 MHz, DMSO-d6) δ 168.09 (C = O), 160.12, 159.96 (Aromatic), 150.77 (C = N), 141.66, 135.55, 130.74, 130.46, 120.69, 118.29, 117.25, 114.21, 112.21, 112.02 (Aromatic), 60.70 (thiazolidine-4-one-CH), 55.64 (OCH3), 55.62 (OCH3), 30.48 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C18H18N2O3S (M + Na)+: 365.0939; found 365.0942.
(E)-3-((3-methylbenzylidene)amino)-2-(m-tolyl)thiazolidin-4-one 2s
White solid; Chemical Formula: C18H18N2OS; Molecular Weight: 310,41; Yield: 91%
FTIR νmax (cm−1): 1698.66 (C = O), 1580.15 (C = N), 1060.37 (C–N).
1H NMR (400 MHz, DMSO-d6) δ 8.11 (s, 1H, N = CH), 7.41 (s, 1H, Ar-H), 7.38 (d, J = 7.36 Hz, 1H, Ar-H), 7.30–7.11 (m, 6H, Ar-H), 6.46 (s, 1H, thiazolidine-4-one-CH), 4.02 (d, J = 16.30 Hz, 1H, thiazolidine-4-one-CH2), 3.82 (d, J = 16.22 Hz, 1H, thiazolidine-4-one-CH2), 2.29 (s, 6H, 2CH3)
13C NMR (101 MHz, DMSO-d6) δ 168.07 (C = O), 150.84 (C =n), 140.06, 138.61, 134.08, 131.84, 129.65, 129.39, 129.21, 128.02, 126.71, 125.23, 123.46 (Aromatic), 60.69 (thiazolidine-4-one-CH), 30.74 (CH2), 21.46 (CH3), 24.24 (CH3).
HRMS: (ESI+-MS, m/z) calcd for C18H18N2OS (M + Na)+: 333.1043; found 333.1038.
(E)-3-((2,3,4-trimethoxybenzylidene)amino)-2-(2,3,4-trimethoxyphenyl)thiazolidin-4-one 2t
White solid; Chemical Formula: C22H26N2O7S; Molecular Weight: 462,52; Yield: 89%
FTIR νmax (cm−1): 1689.96 (C = O), 1578.01 (C = N), 1119.01 (C–N).
1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H, N = CH), 6.95 (s, 2H, Ar-H), 6.69 (s, 2H, Ar-H), 6.36 (s, 1H, thiazolidine-4-one-CH), 4.08 (d, J = 15.96 Hz, 1H, thiazolidine-4-one-CH2), 3.77 (13H, thiazolidine-4-one-CH2, 4OCH3,), 3.67 (s, 3H, OCH3), 3.64 (s, 3H, OCH3).
13C NMR (151 MHz, DMSO-d6) δ 167.90 (C = O), 153.89, 153.43 (Aromatic), 152.75, 140.76, 138.39, 134.46, 129.04, 105.11, 103.12 (Aromatic), 63.74 (thiazolidine-4-one-CH), 60.94 (OCH3), 60.86 (OCH3), 56.31 (OCH3), 56.25 (OCH3), 31.34 (CH2).
HRMS: (ESI+-MS, m/z) calcd for C22H26N2O7S (M + Na)+: 442.9137; found 442.9136.
Pharmacological assays
Nitric Oxide (NO) activity
The antioxidant capacity of the compounds to mop up nitric oxide radicals was determined with the modified protocol utilized by Kurian et al. [52]. Briefly, 250 μL of sodium nitroprusside (10 mM) prepared in Sodium phosphate buffer (pH 7.4) was added to 500 μL of the compounds (250 - 2000 mM) solution or distilled water (control). The resulting solution was incubated at 37 °C for 2 h. Then, 250 μL of Griess reagent was added to the reaction mixture before the absorbance was measured at 540 nm. The % nitric oxide scavenging activity of the compounds was calculated with the formula:
2,2′-diphenyl-1-picrylhydrazyl (DPPH) activity
The compounds were evaluated on their ability to scavenge stable DPPH radicals by adopting the method of Turkoglu et al. [53] with slight modifications. 2 mL of varied concentrations (250 - 2000 mM) of the compounds or Trolox was added to 2 mL 0.3 mM DPPH prepared in methanol. After thorough mixing, the solution was kept in the dark chamber at room temperature (25 °C) for 30 min. Then, the absorbance was measured at 517 nm, and the DPPH radical mopping activity was calculated as follows:
Ferric reducing antioxidant (FRAP)power
The ferric-reducing antioxidant power of the chemical compounds was evaluated using the modified method by Oyaizu [54]. 500 μL of varying concentrations of the compounds or Trolox (250 - 2000 mM) was added to 250 μL distilled water, 100 μL of 200 mM phosphate buffer (pH = 6.6), and 100 μL of 1% potassium ferricyanide [K3Fe (CN)6]. The mixture was incubated at 50 °C for 20 min, followed by acidification with 100 μL trichloroacetic acid (10%). After centrifugation at 3500 rpm for 10 min, 200 μL of the supernatant was transferred into another test tube containing 200 μL distilled water and 0.8 mL of FeCl3 (0.1%). Finally, the absorbance was read at 700 nm, and the total reductive antioxidant power was calculated thus:
α-glucosidase inhibitory activity
The α- glucosidase inhibitory activity was evaluated with the previous method of Ademiluyi and Oboh [55] with slight modifications. Briefly, 100 µL aliquot) of each compound or acarbose (75–600 μM) was added to α-glucosidase (1.0 U/mL) solution in 100 mM sodium phosphate buffer (pH 6.8). The reaction was incubated at 37 °C for 15 min before adding 50 µL of para nitrophenyl β-D-glucopyranoside solution (5 mM). After further incubation at 37 °C for another 30 min, the absorbance of the resulting solution was measured at 405 nm. The compounds’ inhibitory activity was calculated as the percentage of the control sample using the expression below:
α-amylase inhibitory activity
α-Amylase inhibitory activity was determined according to Ibitoye et al. [56] with slight modifications. Briefly, 200 μL of the compounds or acarbose (75–600 μM) was incubated at 25 °C for 10 min with 200 μL solution of porcine pancreatic amylase (0.5 mg/mL) prepared in 200 mM sodium phosphate buffer (pH 6.8). Then, 500 μL of 1% starch solution was added before further incubation at 25 °C for 15 min. The reaction in the mixture was terminated with a 1 mL dinitrosalicylate reagent before boiling for 10 min. The cooled mixture was diluted with 5 mL of distilled water, and the absorbance was read at 540 nm. The inhibitory activity was calculated using the formula below:
Molecular modeling protocols
The Schrödinger molecular modeling suite (release 2021-3) and OPLS4 force were used for all computations.
Ligand preparation
The structures of compounds 2q, 2h, 2g, 2k and acarbose were saved as chemdraw files and imported onto the software, the 3D models with low energy conformation were then generated with LigPrep [57] while their protonation states were assigned using Epik [58] at pH 7 ± 2.0.
Protein preparation
The three-dimensional structure (3D) of α-amylase was retrieved from Protein Data Bank and the structure was prepared using Protein Preparation Wizard [59] using default workflow.
Induced-fit docking and MM-GBSA binding free energy
The prepared ligands were docked at the active site of the human pancreatic α-amylase enzyme (PDB ID: 2QV4) by employing the induced-fit docking (IFD) [60] procedure following standard protocol. The usual protocol parameters included defining the receptor’s grid box as the centroid of the native ligand of ligands size of ≤20 Å with a conformational sampling at a 2.5 kcal mol−1 energy window. The initial docking stage with glide constituted generating poses that can be docked on the receptor and followed by Prime refinement to an RMSD of 1.8 Å. Subsequently, structures with an energy of 30 kcal.mol−1 were submitted for glide redocking with extra precision to account for fall positives. Thereafter, the best receptor-ligand complex was redocked to improve the final pose. Complexes presenting the best poses were selected based on docking score, glide emodel and glide energy and then used as input structures to calculate ligand binding free energy using Prime Molecular Mechanics-Generalized Borne Surface Area (MM-GBSA) [61].
References
Sroor FM, Abbas SY, Basyouni WM, El-Bayouki K, El-Mansy MF, Aly HF, et al. Synthesis, structural characterization and in vivo anti-diabetic evaluation of some new sulfonylurea derivatives in normal and silicate coated nanoparticle forms as anti-hyperglycemic agents. Bioorg Chem. 2019;92:103290.
Sameeh MY, Khowdiary MM, Nassar HS, Abdelall MM, Amer HH, Hamed A, et al. Thiazolidinedione derivatives: in silico, in vitro, in vivo, antioxidant and anti-diabetic evaluation. Molecules. 2022;27:830.
Missioui M, Mortada S, Guerrab W, Serdaroğlu G, Kaya S, Mague JT, et al. Novel antioxidant quinoxaline derivative: Synthesis, crystal structure, theoretical studies, antidiabetic activity and molecular docking study. J Mol Struct. 2021;1239:130484.
Rezaei M, Rabizadeh S, Mirahmad M, Hajmiri MS, Nakhjavani M, Hemmatabadi M. et al. The association between advanced glycation end products (AGEs) and ABC (hemoglobin A1C, blood pressure, and low-density lipoprotein cholesterol) control parameters among patients with type 2 diabetes mellitus. Diabetol Metab Syndr. 2022;14:1–10.
Mehmood R, Mughal EU, Elkaeed EB, Obaid RJ, Nazir Y, Al-Ghulikah HA, et al. Synthesis of novel 2, 3-dihydro-1, 5-benzothiazepines as α-glucosidase inhibitors: In vitro, in vivo, kinetic, SAR, molecular docking, and QSAR studies. ACS Omega. 2022;7:30215–32.
Kanwugu ON, Glukhareva TV, Danilova IG, Kovaleva EG. Natural antioxidants in diabetes treatment and management: prospects of astaxanthin. Crit Rev Food Sci Nutr. 2021;62:1–24.
Ezati M, Ghavamipour F, Adibi H, Pouraghajan K, Arab SS, Sajedi RH, et al. Design, synthesis, spectroscopic characterizations, antidiabetic, in silico and kinetic evaluation of novel curcumin-fused aldohexoses. Spectrochim Acta A Mol Biomol Spectrosc. 2023;285:121806.
Aroua LM, Almuhaylan HR, Alminderej FM, Messaoudi S, Chigurupati S, Al-Mahmoud S, et al. A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorg Chem. 2021;114:105073.
Gummidi L, Kerru N, Ebenezer O, Awolade P, Sanni O, Islam MS, et al. Multicomponent reaction for the synthesis of new 1,3,4-thiadiazole-thiazolidine-4-one molecular hybrids as promising antidiabetic agents through α-glucosidase and α-amylase inhibition. Bioorg Chem. 2021;115:105210.
Khirallah SM, Ramadan H, Aladl H, Ayaz NO, Kurdi L, Jaremko M, et al. Antidiabetic potential of novel 1,3,5-trisubstituted-2-thioxoimidazloidin-4-one analogues: insights into α-glucosidase, α-amylase, and antioxidant activities. Pharmaceuticals. 2022;15:1576.
Venditti P, Di Meo S. The role of reactive oxygen species in the life cycle of the mitochondrion. Int J Mol Sci. 2020;21:2173.
Luo Y, Peng B, Wei W, Tian X, Wu Z. Antioxidant and anti-diabetic activities of polysaccharides from guava leaves. Molecules. 2019;24:1343.
Gupta A, Kumar R, Pandey AK. Antioxidant and antidiabetic activities of Terminalia bellirica fruit in alloxan induced diabetic rats. South Afr J Bot. 2020;130:308–15.
Xie L, Shen M, Wen P, Hong Y, Liu X, Xie J. Preparation, characterization, antioxidant activity and protective effect against cellular oxidative stress of phosphorylated polysaccharide from Cyclocarya paliurus. Food Chem Toxicol. 2020;145:111754.
Ibrahim SA, Salem MM, Elsalam HAA, Noser AA. Design, synthesis, in-silico and biological evaluation of novel 2-Amino-1,3,4-thiadiazole based hydrides as B-cell lymphoma-2 inhibitors with potential anticancer effects. J Mol Struct. 2022;1268:133673.
Bar M, Skóra B, Tabęcka-Łonczyńska A, Holota S, Khyluk D, Roman O, et al. New 4-thiazolidinone-based molecules Les-2769 and Les-3266 as possible PPARγ modulators. Bioorg Chem. 2022;128:106075.
Jampilek J. Heterocycles in medicinal chemistry. Molecules. 2019;24:3839.
Pradhan T, Gupta O, Kumar V, Sristi, Chawla G. A comprehensive review on the antidiabetic attributes of thiazolidine‐4‐ones: Synthetic strategies and structure–activity relationships. Arch Pharm. 2023;356:2200452.
Mech D, Kurowska A, Trotsko N. The bioactivity of thiazolidin-4-ones: a short review of the most recent studies. Int J Mol Sci. 2021;22:11533.
Lebovitz HE. Thiazolidinediones: the forgotten diabetes medications. Curr Diab Rep. 2019;19:151.
bin Ahmad Kamar AKD, Ju Yin L, Tze Liang C, Tjin Fung G, Avupati VR. Rhodanine scaffold: a review of antidiabetic potential and structure–activity relationships (SAR). Med Drug Discov. 2022;15:100131.
Zhang J, Liu R, Kuang HY, Gao XY, Liu HL. Protective treatments and their target retinal ganglion cells in diabetic retinopathy. Brain Res Bull. 2017;132:53–60.
Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opin Drug Saf. 2010;9:347–54.
Ullah H, Uddin I, Rahim F, Khan F, Sobia, Taha M, et al. In vitro α-glucosidase and α-amylase inhibitory potential and molecular docking studies of benzohydrazide based imines and thiazolidine-4-one derivatives. J Mol Struct. 2022;1251:132058.
Kaur R, Kumar R, Dogra N, Yadav AK. Design, synthesis, biological evaluations and in silico studies of sulfonate ester derivatives of 2-(2-benzylidenehydrazono)thiazolidin-4-one as potential α-glucosidase inhibitors. J Mol Struct. 2022;1247:131266.
Singh R, Kumar P, Sindhu J, Devi M, Kumar A, Lal S, et al. Parsing structural fragments of thiazolidin-4-one based α-amylase inhibitors: a combined approach employing in vitro colorimetric screening and GA-MLR based QSAR modelling supported by molecular docking, molecular dynamics simulation and ADMET studies. Comput Biol Med. 2023;157:106776.
Ibrahim M, Latif A, Ahmad M, Ahmad S, Ali A, Siddique AB, et al. Sulfonylbis(acylhydrazones) as anticholinesterase inhibitors: Synthesis, in vitro biological evaluation and computational studies. J Mol Struct. 2022;1252:132215.
Bushra S, Shamim S, Khan KM, Ullah N, Mahdavi M, Faramarzi MA, et al. Synthesis, in vitro, and in silico evaluation of Indazole Schiff bases as potential α-glucosidase inhibitors. J Mol Struct. 2021;1242:130826.
Taha M, Hayat S, Rahim F, Uddin N, Wadood A, Nawaz M, et al. Exploring thiazole-based Schiff base analogs as potent α-glucosidase and α-amylase inhibitor: their synthesis and in-silico study. J Mol Struct. 2023;1287:135672.
Adalat B, Rahim F, Taha M, Hayat S, Iqbal N, Ali Z, et al. Synthesis of Benzofuran–based Schiff bases as anti-diabetic compounds and their molecular docking studies. J Mol Struct. 2022;1265:133287.
Kerru N, Gummidi L, Bhaskaruni SVHS, Maddila SN, Jonnalagadda SB. Ultrasound-assisted synthesis and antibacterial activity of novel 1,3,4-thiadiazole-1H-pyrazol-4-yl-thiazolidin-4-one derivatives. Monatshefte Chem. 2020;151:981–90.
Rezki N, Al-Sodies SA, Aouad MR, Bardaweel S, Messali M, El Ashry el SH. An eco-friendly ultrasound-assisted synthesis of novel fluorinated pyridinium salts-based hydrazones and antimicrobial and antitumor screening. Int J Mol Sci. 2016;17:766.
Mahmood S, Khan SG, Rasul A, Christensen JB, Abourehab M. Ultrasound assisted synthesis and in silico modelling of 1,2,4-triazole coupled acetamide derivatives of 2-(4-Isobutyl phenyl)propanoic acid as potential anticancer agents. Molecules. 2022;27:7984.
Kohl B, Granitzka V, Singh A, Quintas P, Xiromeriti E, Mörtel F, et al. Comparative α-glucosidase and α-amylase inhibition studies of rhodanine–pyrazole conjugates and their simple rhodanine analogues. Med Chem Res. 2019;28:143–59.
Cele N, Awolade P, Seboletswe P, Olofinsan K, Islam MS, Singh P. α-Glucosidase and α-amylase inhibitory potentials of quinoline–1, 3, 4-oxadiazole conjugates bearing 1, 2, 3-triazole with antioxidant activity, kinetic studies, and computational validation. Pharmaceuticals. 2022;15:1035.
Almasirad A, Ghadimi M, Mirahmadi S, Ahmadian Kodakan P, Jahani R, Nazari M, et al. Design, synthesis, and preliminary pharmacological evaluation of novel thiazolidinone derivatives as potential benzodiazepine agonists. Mol Divers. 2022;26:769–80.
El‐Bordany EA, Abdel Aziz A, Abou‐Elmagd WSI, Hashem AI. Synthesis and spectroscopic characterization of some novel pyrazoloquinoline, pyrazolyltetrazine, and thiazolidinone derivatives. J Heterocycl Chem. 2018;55:291–6.
Desai NC, Jadeja KA, Jadeja DJ, Khedkar VM, Jha PC. Design, synthesis, antimicrobial evaluation, and molecular docking study of some 4-thiazolidinone derivatives containing pyridine and quinazoline moiety. Synth Commun. 2021;51:952–63.
Shrivastava B, Sharma O, Sharma P, Singh J. Synthesis, characterization and antimicrobial evaluation of novel azole based (Benzoic Acid) derivatives. Asian J Pharm Pharm. 2019;5:368–72.
Altowyan MS, Barakat A, Al-Majid AM, Al-Ghulikah HA. Spiroindolone analogues as potential hypoglycemic with dual inhibitory activity on α-amylase and α-glucosidase. Molecules. 2019;24:2342.
Rafique R, Khan KM, Arshia, Kanwal, Chigurupati S, Wadood A, et al. Synthesis of new indazole based dual inhibitors of α-glucosidase and α-amylase enzymes, their in vitro, in silico and kinetics studies. Bioorg Chem. 2020;94:103195.
Khan Y, Khan S, Hussain R, Maalik A, Rehman W, Attwa MW, et al. The synthesis, in vitro bio-evaluation, and in silico molecular docking studies of pyrazoline–thiazole hybrid analogues as promising anti-α-glucosidase and anti-urease agents. Pharmaceuticals. 2023;16:1650.
Aytac S, Gundogdu O, Bingol Z, Gulcin İ. Synthesis of Schiff bases containing phenol rings and investigation of their antioxidant capacity, anticholinesterase, butyrylcholinesterase, and carbonic anhydrase inhibition properties. Pharmaceutics. 2023;15:779.
Bouali N, Hammouda MB, Ahmad I, Ghannay S, Thouri A, Dbeibia A, et al. Multifunctional derivatives of spiropyrrolidine tethered indeno-quinoxaline heterocyclic hybrids as potent antimicrobial, antioxidant and antidiabetic agents: design, synthesis, in vitro and in silico approaches. Molecules. 2022;27:7248.
Tenuta MC, Deguin B, Loizzo MR, Dugay A, Acquaviva R, Malfa GA, et al. Contribution of flavonoids and iridoids to the hypoglycaemic, antioxidant, and nitric oxide (NO) inhibitory activities of arbutus unedo L. Antioxidants. 2020;9:184.
Amine Khodja I, Boulebd H, Bensouici C, Belfaitah A. Design, synthesis, biological evaluation, molecular docking, DFT calculations and in silico ADME analysis of (benz)imidazole-hydrazone derivatives as promising antioxidant, antifungal, and anti-acetylcholinesterase agents. J Mol Struct. 2020;1218:128527.
Shen J, Cheng F, Xu Y, Li W, Tang Y. Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model. 2010;50:1034–41.
Butina D, Segall MD, Frankcombe K. Predicting ADME properties in silico: methods and models. Drug Discov Today. 2002;7:S83–8.
Schrödinger Release 2022-1: QikProp, S.N.Y., NY: LLC, 2022).
Awolade P, Cele N, Kerru N, Singh P. Synthesis, antimicrobial evaluation, and in silico studies of quinoline—1H-1,2,3-triazole molecular hybrids. Mol Divers. 2021;25:2201–18.
Chike-Ekwughe A, Adegboyega AE, Johnson TO, Adebayo AH, Ogunlana OO. In vitro and in-silico inhibitory validation of Tapinanthus cordifolius leaf extract on alpha-amylase in the management of type 2 diabetes. Inform Med Unlocked. 2023;36:101148.
Kurian GA, Suryanarayanan S, Raman A, Padikkala J. Antioxidant effects of ethyl acetate extract of Desmodium gangeticum root on myocardial ischemia reperfusion injury in rat hearts. Chin Med. 2010;5:1–7.
Turkoglu A, Duru ME, Mercan N, Kivrak I, Gezer K. Antioxidant and antimicrobial activities of Laetiporus sulphureus (Bull.) Murrill. Food Chem. 2007;101:267–73.
Oyaizu M. Studies on products of browning reaction antioxidative activities of products of browning reaction prepared from glucosamine. Jpn J Nutr Dietetics. 1986;44:307–15.
Ademiluyi AO, Oboh G. Soybean phenolic-rich extracts inhibit key-enzymes linked to type 2 diabetes (α-amylase and α-glucosidase) and hypertension (angiotensin I converting enzyme) in vitro. Exp Toxicol Pathol. 2013;65:305–9.
Ibitoye OB, Olofinsan KA, Teralı K, Ghali UM, Ajiboye TO. Bioactivity‐guided isolation of antidiabetic principles from the methanolic leaf extract of Bryophyllum pinnatum. J Food Biochem. 2018;42:e12627.
Schrödinger. Release 2021-2: LigPrep, S.; LLC: New York, NY, USA, 2021.
Schrödinger. Release 2021-2: Epik, S.; LLC: New York, NY, USA, 2021.
Schrödinger Release 2021-2: Protein Preparation Wizard; Epik, Schrödinger (New York, NY: LLC, 2016); Impact, Schrödinger (New York, NY: LLC, 2016); Prime, Schrödinger (New York, NY: LLC, 2021).
Schrödinger. Release 2021-2: Induced Fit Docking Protocol, Glide, S.; LLC: New York, NY, USA, 2021.
Schrödinger. Release 2021-2: Prime MM-GBSA, Schrödinger, LLC, New York, NY. 2021, 202.
Acknowledgements
PS gratefully acknowledges the National Research Foundation (South Africa) for a competitive grant for rated researchers (Grant Number: SRUG2204092857), and the Center for High-Performance Computing (CHPC), Cape Town for computational resources.
Funding
Open access funding provided by University of KwaZulu-Natal.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seboletswe, P., Kumar, G., Kubone, L. et al. Ultrasound-assisted synthesis of 4-thiazolidinone Schiff bases and their antioxidant, α-glucosidase, α-amylase inhibition, mode of inhibition and computational studies. Med Chem Res (2024). https://doi.org/10.1007/s00044-024-03268-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00044-024-03268-2