
Abstract
The aim of the present study is to explore new selective anti-inflammatory compounds with low cardiovascular risk. Twelve thiadiazole derivatives incorporating different amino acid moieties were newly synthesized (4–15) as potential anti-inflammatory agents with low cardiovascular risks through dual COX-2/MPO inhibition. Compounds were initially screened for their anti-inflammatory effect by assay of COX-2, the most potent (4–6, 8) were further tested for COX-1 inhibition, myeloperoxidase MPO activity as well as total nitric oxide content NO in heart of irradiated rats. Cardiac toxicity potential was evaluated by assay of creatine kinase-MB (CK-MB), troponin-I (Tn-I) and lactate dehydrogenase (LDH). Celcoxcib was used as reference drug. S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino propanethioate (5) was the most potent anti-inflammatory with the least cardiotoxicity effect. It exhibited IC50 0.09 µM on COX-2 inhibition with very low activity on COX-1. Troponin I was elevated by 11% using compound 5 in non-irradiated rats. Moreover, compound (5) showed 73% reduction in MPO level. Results were supported by molecular docking into the active sites of COX-2 and MPO enzymes to have more insights about the possible dual inhibition of compound 5 of both enzymes.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disorders are the major causes of human morbidity and mortality worldwide [1]. Accumulating evidence that the inflammatory process and heart failure are inextricably linked [2]. Several studies have shown that cyclooxygenase-2 (COX-2) is easily detectable in cardiac myocytes of human suffering from heart failure [3]. Coxibs are a class of medicines that selectively inhibit COX-2 and have been developed as a safe alternative to other COX-2 inhibitors, which might cause major gastrointestinal problems as that observed with traditional NSAIDs [4]. Unfortunately, rofecoxib (Vioxx1, Merck & Co., USA), a COX-2 inhibitor, was taken off the market in 2004 because of the concerns about an increased risk for heart attack and stroke [5, 6]. The FDA, on the other hand, has decided to keep celecoxib (CEL) on the market since the benefit outweighs the potential cardiovascular risk [6]. Nevertheless, as observed in the Adenoma Prevention with Celecoxib trial, CEL may induce a considerable increase in major cardiovascular risks such as stroke, myocardial infarction or congestive heart failure, leading to the study’s early termination [6]. These serious complications may also occur in patients receiving radiotherapy for cancer treatment in the thoracic and mediastinal regions [7], reducing the therapeutic efficacy of radiotherapy and thereby, limiting the use of COX-2 inhibitors in such circumstances. Furthermore, exposure to radiation has been shown recently to be implicated in the early stages of atherosclerosis by causing vascular dysfunction, inflammation and fibrosis in the aorta [8], all of which may require the use of anti-inflammatory drugs as COX-2 inhibitors. Therefore, there is always a great effort done for developing new selective COX-2 inhibitors with a better cardiovascular profile.
Several serum cardiac markers, such as troponin I (TnI), creatine kinase-MB (CK-MB), and lactate dehydrogenase (LDH), have been studied as a potential tool for assessing patients receiving adjuvant therapy who might be at increased risk of drug-induced cardiac dysfunction or cardiotoxicity [9]. Furthermore, new research suggests that myeloperoxidase (MPO), a heme-containing peroxidase enzyme released by activated neutrophils, monocytes, and some tissue macrophages (such as those found in atherosclerotic plaques), may play a role in the progression of coronary artery disease (CAD). MPO plays a vital role in the innate immune system via producing free radicals and reactive oxygen species in the first line of defense against pathogens through a pair of chlorination-peroxidation processes. MPO produces highly toxic hypochlorous acid (HOCl) by using chloride in the presence of hydrogen peroxide, and the enzyme is transformed to an inactive peroxide form, which then undergoes two sequential reductions back to the original form of the enzyme, releasing highly reactive free radicals. Together the produced highly toxic HOCl and the reactive species destroy pathogens [10]. However, overproduction is linked to a variety of inflammatory diseases as well as oxidative damage to host tissues at inflammatory locations, such as atherosclerotic plaques [11]. MPO has been linked to atherosclerosis by pathways involving its role in inflammation [11] and nitric oxide consumption, which results in endothelial dysfunction [12].
Based on the link between MPO and the development of CAD, it was hypothesized that developing novel COX-2 inhibitors with better MPO control would be a promising approach to improve anti-inflammatory efficacy of COX-2 inhibitors as well as having higher safety margins against cardiovascular risks.
Rationale and design
Several studies had focused on designing MPO inhibitors and analyzing their binding modes in the catalytic site of MPO and this was facilitated by the availability of crystal structures of potent inhibitors. There are three main categories of MPO inhibitors: (1) Those that reversibly divert MPO from its chlorination cycle causing accumulation of the inactive peroxide form of the enzyme (e.g., indomethacin, tryptamine, 5-aminosalicylic acid, paracetamol and isoniazid). (2) Suicide or irreversible inhibitors which are oxidized by MPO leading to complete inactivation by destruction or covalent binding to heme (e.g., 4-aminobenzoic acid hydrazide (4-ABAH) and 2-thioxanthines). Inhibitors of both categories are prone to be oxidized by MPO and this is the main cause of their observed side effects, the generated reactive species cause toxic and damaging effect, besides, many have low bioavailability and are associated with gastrointestinal disorders [13]. (3) An alternate mechanism of inhibition is reversible inhibitors that compete with MPO substrates by binding into heme catalytic site (e.g., salicylhydroxamic acid (SHA)). This is an encouraging mode of inhibition that limits the oxidizing abilities of the enzyme without undesirable side effects [14].
Until now, only Verdiperstat (AZD3241) (I) is the only approved small molecule as MPO inhibitor. Verdiperstat is a selective, irreversible and orally active MPO inhibitor, with an IC50 of 630 nM for the neurodegenerative brain disorders [15].
Many efforts and SAR studies were performed in order to optimize MPO inhibition. Rivera-Antonio et al. reported new hydroxy-amino-cinnammic acid derivatives (II) as potent antioxidants and MPO inhibitors, two structural features enable this activity; phenolic group and propenoic acid side chain leading to electron delocalization upon reaction with free radicals reducing their propagation during oxidation process [16]. Aldib et al. performed structure-based design of new inhibitors and introduced potent bis-aryl propylamine derivatives (III) and showed that replacement of one aryl group with heterocyclic ring as indole led to potent MPO inhibition [17]. Santos et al. reported a series of chalcones (IV) and showed the role of terminal amino group in binding to key amino acids in MPO active site [18]. Soubhye et al. synthesized new derivatives (V) based on structure-based docking of 5-fluorotrptamine and showed that increasing the amino alkyl side chain to 4- or 5-carbons led to potent MPO inhibition, also, the unsubstituted terminal amino group is important for activity as it carries positive charge at physiological pH and forms ionic interactions with Glu102 or propenoic heme group [19]. Forbes et al. identified new substituted aromatic hydroxamates (VI) as more potent reversible MPO inhibitors than SHA. They introduced pyrimidine ring that increased stability within the active site through interaction with heme propionate group (Fig. 1A) [14].

A Some reported MPO inhibitors; B the designed target compounds
Amino acids are very important for living organisms as they are the basic units for enzymes and proteins, they are widely used as synthetic precursors in organic chemistry and drug design due to the presence of both amino and carboxylic groups [20]. Amino acid moieties are reported to be introduced to potential drugs as a strategy to maximize pharmacokinetics and pharmacodynamics [21] and due to the known antioxidant activity [22]. Etsè et al. reported a series of benzodioxole amino acid derivatives as potent anti-peroxidase, radical scavenging and non-toxic agents [23].
In previous work [24], we reported novel thiadiazole derivatives as potent anti-inflammatory agents through selective COX-2 inhibition [24]. 5-(4-Methoxy/bromobenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (2, 3) were among the most potent compounds with high selectivity towards COX-2 (IC50 = 0.24/0.19 µM), high anti-inflammatory and analgesic activities and high safety on gastric mucosa with no ulceration effect. Both compounds had similar docking scores in COX-2 active site, the 4-OCH3 group was important for binding to His90 and Arg513, the key amino acids responsible for COX-2 selectivity. Accordingly, in continuation of this work, we modified previously reported thiadiazoles and introduced amino acid moieties on thione group, with perceiving structural features to act as MPO inhibitors and protect against cardiovascular side effects reported for potent COX-2 inhibitors. Terminal NH2 group important for optimum ligand binding to MPO, α-side chain would increase the inhibitory potential by reaching the important amino acids in the heme binding site (Fig. 1B). The target compounds would be endowed with dual COX-2/MPO inhibition to act as anti-inflammatory and protective against MPO-mediated cardiovascular diseases.
Results and discussion
Chemistry
In this study, novel 12 thiadiazole derivatives bearing amino acid moieties have been synthesized as depicted in Scheme 1. The starting 5-amino-1,3,4-thiadiazole-2(3H)-thione (1) was synthesized from the reaction between thiosemicarbazide and carbon disulfide in alkaline medium as previously reported [25]. We reported compounds 5-(4-Methoxybenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (2) and 5-(4-Bromobenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (3) from most potent compounds among series of thiadiazole derivatives as selective COX-2 inhibitors [24]. They were then used in our current research as starting compounds for synthesis of more new derivatives as anti-inflammatory with low cardiovascular risk. Synthesis of target compounds 4–15 was carried by reaction of compounds 2 and / 3 with different amino acids (glycine, alanine, glutamic acid, isoleucine, phenylalanine and proline) using water as solvent in green synthesis attempt. The proposed chemical structure of these derivatives was verified by their spectral and microanalytical data, as listed in details in material and methods section. IR spectra of all new compounds showed additional strong intensity absorption bands at range 1680–1720 cm−1 ascribed for C=O of amino acid moieties introduced. IR spectra of compounds 4–15 revealed added bands for stretching NH & NH2 of introduced amino acid moieties at range 3200–3330 cm−1. 1H-NMR spectra of compounds 4–13, displayed extra upfield signal at δ 1.18–2.17 ppm attributed to NH2, exchangeable with D2O. Confirming reaction with different amino acids at specified positions. 13C spectra for all new compounds displayed signals for introduced Cs at specified positions. Where most deshielded C appears at range 182.94–194.56 ppm ascribed for introduced C=O of different amino acid moieties.

Synthetic routes for compounds 4–15
Moreover, mass spectra and microanalytical data of compounds 4–15 were in agreement with their postulated structures.
It is of added interest to mention that the newly synthesized compounds closely fulfilling Lipiniski’s rule of drug likeness.
In vitro COX-2/COX-1 inhibition
Compounds 4–15 underwent selectivity test for COX-2 compared to celecoxib, IC50 was calculated and presented in Table 1. Compounds 5 and 6 were the most potent inhibitors (IC50 = 0.09 and 0.16 µM) and even more potent than celecoxib (IC50 = 0.22 µM). Compounds 4 and 8 were of equal potency as celecoxib (IC50 = 0.24 and 0.26 µM). COX-2 inhibition for the starting materials 2 and 3 were previously reported to be 0.24 and 0.19 µM, respectively [24].
Studying the effect of para-substituting the terminal aromatic ring with either methoxy or bromine group, we can conclude that 4-methoxy phenyl substituents were more potent, this emphasize our previous finding that the methoxy group stabilized the compounds in the side pocket of COX-2 by binding to the two main amino acids responsible for selectivity (His90 and Arg513). Whereas, the bulky bromine group could lead to different orientation in the active site.
Introduction of amino acid moiety ameliorated the activity of the methoxy derivatives 5 and 6, retained similar activities as the starting material for compounds 4 and 8, but unfortunately led to decrease in the activity for the rest of compounds.
With regard to SAR of the amino acids derivatives, the alanine derivative 5 was the most active and we can conclude that a short side chain like methyl group is optimum for activity as elongation of the side chain or introduction of either carboxylic or aromatic ring decreased the activity.
In addition, the inhibitory activity of the most potent compounds (4–6, 8) were tested against COX-1 and they showed remarkable lower activity compared to that on COX-2. Accordingly, compounds 4, 5, 6 and 8 were chosen for in vivo examination because of their high selectivity to COX-2 which resembles celecoxib.
In vivo studies
Determination of acute toxicity
Acute toxicity of the most potent compounds 4, 5, 6 and 8 was evaluated by i.p injection of selected doses [100, 200, 400, 800 and 1000 mg/kg] of each compound dissolved in 5% Tween 80 (in saline). No mortality was recorded within 48 h. indicating that these compounds were practically safe in vivo (Table 2).
Cardiovascular risk for compounds 4–6, 8: assessment of CK-MB, troponin I and LDH
Annual CVD mortality is expected to reach more than 23.3 million people by 2030; thereby there is a great concern about this rising incidence of CVDs [26]. As a result, significant efforts should be made to minimize the risks of CVDs. If proper preventive measures are not in place, the increasing use of IR for cancer treatment and diagnostic purposes may result in cardiovascular damage. Many accumulating studies have shown that there is a link between cancer and inflammation where inflammation in the tumor microenvironment has a great role in promoting tumor growth [27]. Selective COX-2 inhibitors have been utilized extensively as potent anti-inflammatory medications in combination with radiotherapy in patients with COX-2-expressing malignancies, where they have been shown to improve tumor radio-sensitivity in cell and animal models [28]. Unfortunately, administration of Celecoxib, alone or combined with doxorubicin, was previously shown to cause cardiotoxicity in rats, as manifested by highly significant increase in the serum LDH, CK-MB, and Troponin level [29]. Therefore, it was of great importance to synthetize new COX-2 inhibitors with the lowest cardiovascular risk.
In the present study, the possible cardiovascular risks of the most active compounds 4, 5, 6 and 8 were evaluated using the radiation induced cardiotoxicity in rats compared to that induced by celecoxib. The response of the heart towards the tested compounds was expressed as the change in biochemical parameters obtained from sera of both irradiated and non-irradiated rats including CK-MB, Tn-I and LDH. The results revealed that celecoxib administration aggravates the increase induced by radiation in the diagnostic biomarkers of myocardial damage levels (CK-MB, Tn-I and LDH) as compared to normal control. Whereas, all the tested compounds (4, 5, 6 and 8) exhibited better cardio protective profile signified by low level of induction in these biochemical levels when compared to that induced by celecoxib either given alone or combined with radiation (Figs. 2–4).

Effect of thiadiazole derivatives and celecoxib on creatinine kinase-MB (CK-MB) (U/l) in serum of normal (A) and irradiated (B) rats (n = 8). All compounds were delivered orally in a dose of (25 mg/kg/day), in irradiated group treatment starts after one hour of irradiation (6 Gy) and treatment continued for 2 days further. Values are plotted as means ± SEM. *p ≤ 0.05 compared to normal control, †p ≤ 0.05 compared to irradiated rats
In non-irradiated rats, compounds 4, 6 and 8 caused an increase in the levels of CK-MB by 25% whereas compound 5 elevated its level by only 5% compared to a dramatic increase reaching to four folds induced by celecoxib (Fig. 2A). Moreover, TnI in non-irradiated rats was elevated by nearly 30% using compounds 4, 6 and 8 and by 11% using compound 5 whereas celecoxib increased its level by 64% (Fig. 3A). In addition, LDH in non-irradiated rats was increased after treatment with compounds 4, 6 and 8 by nearly 11% and was kept within normal range with compound 5 whereas celecoxib led to an increase by 47.7% (Fig. 4A). Fortunately, compound 5 nearly maintained these cardiac biomarkers within the normal range proofing its protective cardiovascular activity. The leakage of these cardiac enzymes into the blood stream might be due to rupture of cardiac membrane [30]. Previous studies concluded that COX-2 inhibition favors prothrombotic events by tipping the balance of prostacyclin/thromboxane in favor of thromboxane, a prethrombotic eicosanoid and thereby developing thrombotic cardiovascular events [31]. Similar findings by [32] revealed that celecoxib administration induced significant increase in the diagnostic biomarkers of myocardial damage including CK-MB, TnI and LDH levels, as compared to normal control.

Effect of thiadiazole derivatives and celecoxib on Troponin I (ng/ml) in serum of normal (A) and irradiated (B) rats (n = 8). All compounds were delivered orally in a dose of (25 mg/kg/day), in irradiated group treatment starts after one hour of irradiation (6 Gy) and treatment continued for 2 days further. Values are plotted as means ± SEM. *p ≤ 0.05 compared to normal control, †p ≤ 0.05 compared to irradiated rats

Effect of thiadiazole derivatives and celecoxib on lactate dehydrogenase LDH activity (U/dl) in serum of normal (A) and irradiated (B) rats (n = 8). All compounds were delivered orally in a dose of (25 mg/kg/day), in irradiated group treatment starts after 1 h of irradiation (6 Gy) and treatment continued for 2 days further. Values are plotted as means ± SEM. *p ≤ 0.05 compared to normal control, †p ≤ 0.05 compared to irradiated rats
Exposure to IR caused highly significant increase in CK-MB, TnI and LDH by four folds, two folds and three folds respectively (Figs. 2B, 3B and 4B) which reflect the extent of damage in myocardial musculature. These results agree with Elkady et al. [33] who reported that whole body gamma irradiation showed a significant increase in the level of serum enzyme CK-MP, TnI and LDH due to the damage in the heart. The mechanism of radiation-induced cardiotoxicity has been reported to be through formation of superoxide anions and highly reactive and damaging hydroxyl radicals, which induces peroxidation of cell membrane lipid [34]. Radiation-induced cardiotoxicity signified by elevated level of CK-MP, TnI and LDH in irradiated rats was significantly ameliorated by compounds 4, 6, 8 and particularly compound 5 whereas they were exaggerated by celecoxib, suggesting their possible low cardiovascular risk if delivered as an adjuvant anti-inflammatory agents with radiotherapy (Figs. 2B, 3B and 4B).
Anti-inflammatory markers evaluation for compounds 4–6, 8
The anti-inflammatory potency of compounds 4, 5, 6 and 8 was investigated by measuring the MPO activity as well as total nitrite content in heart of irradiated rats.
MPO inhibitory activity for compounds 4–6, 8
MPO is a hemoperoxidase found in abundance in neutrophils, which serve as the first line of defense against microbes via phagocytosis. MPO recognizes the produced hydrogen peroxide (H2O2), generating a substrate-enzyme complex with high oxidative capability for destroying microorganisms. Under certain circumstances, however, MPO’s excessive synthesis of reactive species causes oxidation of intracellular proteins, lipids, and nucleic acids, leading to additional damage to host tissues at sites of inflammation, such as atherosclerotic plaques [35, 36]. As a result of MPO’s elevated activity during inflammation and oxidative stress in a variety of disorders, it is regarded as a highly significant biomarker [37].
Whole body exposure to IR led to an increase in MPO activity by three folds (Fig. 5). Despite Celecoxib inhibited the induced elevation in MPO activity by 42% in irradiated rats, yet it still has been associated with cardiovascular risk (Figs. 2B, 3B and 4B). Thus, it is interesting to have other possible MPO inhibitors that exhibit less cardiovascular risk than celecoxib to treat inflammatory diseases, such as the novel thiadiazole derivatives in this study, which could be used in future as a better alternative for treatment of inflammatory disorders. The tested compounds 4, 6 and 8 inhibited the increase in MPO induced by radiation by nearly 63–68% and compound 5 showed a 73% reduction in its level, thereby representing more promising anti-inflammatory agents than celecoxib with less cardiovascular risk (Fig. 5).

Effect of thiadiazole derivatives and celecoxib on myloperoxidase MPO activity (U/g tissue) in heart tissue of irradiated rats (n = 8). All compounds were delivered orally in a dose of (25 mg/kg/day) which starts after 1 h of irradiation (6 Gy) and continued for 2 days further. Values are plotted as means ± SEM. *p ≤ 0.05 compared to normal control, †p ≤ 0.05 compared to irradiated rats
NO content evaluation for compounds 4–6, 8
MPO also is capable of using nitric oxide (NO) as a physiological substrate, perhaps contributing to endothelial dysfunction. Therefore, the increase in MPO activity is associated with an increase in total nitrite content which was nearly doubled in heart tissue after exposure to radiation as shown in Fig. 6. Similarly, an increase in NO content was previously observed in mouse intestine after radiation exposure [38]. In response to pro-inflammatory stimuli, cyclooxygenase-2 (COX-2) and inducible isoform of nitric oxide synthase (iNOS) gets regulated by NF-κB that mediates subsequent inflammation process [39]. Thus agents that inhibit COX-2, iNOS expression and subsequent NO production can be a potential target for protection against radiation due to its anti-inflammatory role. In accordance, the present study showed that compounds 4, 6 and 8 inhibited the increase in total Nitric oxide content by nearly 30% whereas celecoxib caused an inhibition by 17% (Fig. 6). In particular, Compound 5 normalizes the total NO content signifying the best anti-inflammatory activity compared to the other tested compounds (Fig. 6).

Effect of thiadiazole derivatives and celecoxib on total nitric oxide content NOx (Mmol/g tissue) in heart tissue of irradiated rats (n = 8). All compounds were delivered orally in a dose of (25 mg/kg/day) which starts after 1 h of irradiation (6 Gy) and continued for 2 days further. Values are plotted as means ± SEM. *p ≤ 0.05 compared to normal control, †p ≤ 0.05 compared to irradiated rats
Docking study
In order to interpret the preliminary MPO inhibition results for the synthesized compounds, in silico evaluation using molecular docking was performed with human MPO (PDB: 1DNW), using MOE software. Docking study was performed for the most active compound (5), within the active site of MPO to have more insights about the binding mode based on the description and characterization of heme binding site reported by Fiedler et al. [40].
The methoxy phenyl group is positioned almost parallel to the plane of heme and bound with arene-cation interaction to Arg239 at the opening of the heme distal cavity. The thiadiazole ring occupies the center of the heme distal cavity and participates with two hydrogen bonds with MPO residues. One of the nitrogens of thiadiazole is bound to Arg333 while the sulfur forms an important H-bond with Thr100 which is interacted with the propionate group of heme. The amino acid chain provides the main interaction with the key residue Glu102 through the terminal NH2 group. Moreover, compound 5 is more stabilized within the active site by another H-bond with Arg424 through the carbonyl group with docking score equal to −9.32 kcal/mol (Fig. 7).

Compound 5 (magenta stick representation) bound in the active site pocket containing heme group (blue), MPO residue are shown in a stick representation (yellow). Hydrogen bonds are indicated with dashed lines
Compound (5) was also docked into the active site of COX-2 (PDB: 1CX2) in order to monitor the effect of alanine amino acid substitution on the orientation of the compound within the active site. The methoxy phenyl moiety is directed towards the primary hydrophobic pocket defined by Tyr385 and Tyr348, the thiadiazole ring is oriented towards the carboxylate site defined by Arg120 and Try355, while the alanine chain led the compound to enter the side pocket bordered with Val523 where it is involved with two hydrogen bonds with Arg513 and His90, the unique residues for COX-2 which are not present in COX-1 [41]. The docking score was equal to −12.67 kcal/mol (Fig. 8).

Compound 5 (purple stick representation) bound in COX-2 active site, COX-2 residue are shown in yellow and hydrogen bonds are indicated with dashed lines
Conclusion
In Conclusion, our interest in developing novel COX-2 inhibitors arises from the high risk of cardiovascular toxicity associated with the use of this class. Patients undergoing radiotherapy or exposed to hazards sources of radiation are at great risk of normal tissue toxicity and they were using the anti-inflammatory agents especially COX-2 inhibitors on a large scale. Therefore, efforts have been undertaken to develop novel synthesized agents that protect normal tissues with the lowest possible cardiovascular toxicity. In this work, we performed structural modification of previously reported selective COX-2 inhibitors, different amino acids were introduced on thione group of thiadiazole leading to more potent COX-2 inhibition and low cardiovascular risk than that induced by celecoxib or radiation with nearly the same anti-inflammatory activity. The alanine derivative 5 in particular, showed the most promising anti-inflammatory activity with the lowest cardiovascular risk. The obtained activity was supported with docking of compound (5) into COX-2 and MPO active sites suggesting possible dual inhibition of both enzymes.
Materials and method
Chemistry
A Stuart melting point apparatus (Stuart Scientific, Redhill, UK) was used for recording of uncorrected melting points and were carried in open capillary tubes. FTIR Shimadzu spectrometer (Shimadzu, Tokyo, Japan) was used for recording Infrared (IR) spectra of the compounds. A Bruker (400 MHz for 1H-NMR and 100 MHz for 13C-NMR) spectrometer (Bruker Bioscience, MA, USA) was used for recording 1H NMR and 13C NMR spectra, using TMS as an internal Standard and DMSO-d6 as solvent. Mass spectra were run on HP Model MS-5988 (Hewlett Packard, Palo, Alto, California, USA). A Carlo Erba 1108 Elemental Analyzer (Heraeus, Hanau, Germany), was used for obtaining microanalyses values. Pre-coated SiO2 gel (HF254, 200 mesh) aluminum plates (Merk, Darmstadt, Germany) were used as TLC for checking of reactions’ Completion, where a developing solvent system of chloroform/methanol (7:3) was used and the spots were visualized under UV light. IR, 1H-NMR, 13C-NMR, Mass and elemental analysis were consistent with the assigned structures. All reagents used were purchased from Sigma (St. Louis, MO) of analytical grade.
General procedure for the preparation of 5-(4-substituted-benzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (2,3)
A mixture of 5-Amino-3H-[1,3,4]thiadiazole-2-thione 1 (0.53 g, 0.004 mol) and the appropriate aldehydes (0.004 mol) was refluxed in ethanol containing (1 ml) glacial acetic acid for 5 h. The reaction mixture was cooled, poured onto ice water and the precipitated solid was collected by filtration, dried, and crystallized from methanol to give 2 and 3, respectively (as reported).
5-(4-Methoxybenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (2) as reported [24].
5-(4-Bromobenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (3) as reported [24].
General procedure for the preparation of compounds (4–15)
In a 250-ml round bottom flask, Na2CO3 (0.01 mol) was dissolved in 10 ml water and then different L-amino acids was added; glycine, alanine, glutamic acid, isoleucine, phenylalanine and proline (0.02 mol). To this mixture, addition of 5-(4-Methoxybenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (2) (0.02 mol) and/OR 5-(4-Bromobenzylideneamino)-1,3,4-thiadiazole-2(3H)-thione (3) (0.02 mol) with continuous stirring was done for half an hour. Then the mixture was stirred at reflux for 6–9 h. After the completion of the reaction (checked by TLC), a precipitate was formed, filtered while hot. The product obtsained, washed with water, dried and crystallized from ethanol to give the assigned products 4–15, respectively.
S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino ethanethioate (4)
Yield, 86%, mp 200–202 °C. IR (KBr, cm−1): 3320, 3230, 3200 (NH, NH2), 2939, 2870 (CH aliph.), 3048 (CH arom.), 1720 (C=O), 1626 (C=N). 1H-NMR (DMSO-d6, ppm): 1.62 (s, 2H, NH2, exchangeable with D2O), 3.76 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 4.98 (s, 1H, CH-thiadiazole), 6.96 (d, 2H, J = 7.5 Hz, Ar-H), 7.27 (d, 2H, J = 7.5 Hz, Ar-H), 7.70 (s, 1H, N=CH), 9.44 (s, 1H, NH, exchangeable with D2O).13C-NMR (DMSO- d6, ppm): 48.67 (CH2), 56.04 (OCH3), 104.05 (CH-thiadiazole), 114.49, 128.31, 132.32 (5C-aromatic), 164.06 (C-OCH3), 165.84 (C-thiadiazole), 168.15(C=N), 182.94 (C=O). MS m/z: 310 [M+]. Analysis calculated for C12H14N4O2S2: C, 46.43; H, 4.55; N, 18.05, found: C, 46.45; H, 4.57; N, 18.02.
S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino propanethioate (5)
Yield, 82%, mp 220–222 °C. IR (KBr, cm−1): 3322, 3231, 3202 (NH, NH2), 2937, 2872 (CH aliph.), 3046 (CH arom.), 1680 (C=O), 1626 (C=N). 1H-NMR (DMSO-d6, ppm): 1.45 (s, 2H, NH2, exchangeable with D2O), 1.37 (d, 3H, CH3, J = 6.5 Hz), 3.59 (q, 1H, CH, J = 6.1 Hz), 3.71 (s, 3H, OCH3), 4.88 (s, 1H, CH-thiadiazole), 6.88 (d, 2H, J = 7.5 Hz, Ar-H), 7.16 (d, 2H, J = 7.5 Hz, Ar-H), 7.80 (s, 1H, N=CH), 9.76 (s, 1H, NH, exchangeable with D2O).13C-NMR (DMSO-d6, ppm): 22.59 (CH3), 56.14 (OCH3), 57.71 (CH), 104.35 (CH-thiadiazole), 114.79, 128.51, 132.22 (5C-aromatic), 164.26 (C-OCH3), 165.94 (C-thiadiazole), 168.35 (C=N), 190.61 (C=O). MS m/z: 324 [M+]. Analysis calculated for C13H16N4O2S2: C, 48.13; H, 4.97; N, 17.27, found: C, 48.33; H, 4.77; N, 17.47.
4-Amino-5-((5-((4-methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl) thio)-5-oxopentanoic acid (6)
Yield, 89%, mp 245–247 °C. IR (KBr, cm−1): 3420 (OH), 3319, 3232, 3202 (NH, NH2), 2939, 2870 (CH aliph.), 3047 (CH arom.), 1720 (C=O), 1629 (C=N). 1H-NMR (DMSO-d6, ppm): 1.93 (s, 2H, NH2, exchangeable with D2O), 2.16 (td, 2H, CH2, J = 8.1, 0.6 Hz), 2.31 (t, 2H, CH2, J = 7.8 Hz), 3.53 (t, 1H, CH, J = 7.4 Hz), 3.88 (s, 3H, OCH3), 4.92 (s, 1H, CH-thiadiazole), 6.82 (s, 1H, NH, exchangeable with D2O), 7.09 (d, 2H, J = 7.5 Hz, Ar-H), 7.14 (d, 2H, J = 7.5 Hz, Ar-H), 7.38 (s, 1H, N=CH), 9.50 (s, 1H, OH, exchangeable with D2O).13C-NMR (DMSO-d6, ppm): 30.95 (2CH2), 56.24 (OCH3), 59.77 (CH), 104.25 (CH-thiadiazole), 114.59, 128.41, 132.39 (5C-aromatic), 164.46 (C-OCH3), 165.74 (C-thiadiazole), 168.15 (C=N), 176.74 (COOH), 194.56 (C=O). MS m/z: 382 [M+]. Analysis calculated for C15H18N4O4S2: C, 47.11; H, 4.74; N, 14.65, found: C, 47.31; H, 4.64; N, 14.45.
S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino-3-methylpentanethioate (7)
Yield, 79%, mp 210–212 °C. IR (KBr, cm−1): 3327, 3235, 3250 (NH, NH2), 2939, 2880 (CH aliph.), 3045 (CH arom.), 1691 (C=O), 1628 (C=N). 1H-NMR (DMSO-d6, ppm): 0.94–1.09 (m, 6H, 2CH3), 1.22–1.24 (m, 2H, CH2), 1.61–1.64 (m, 1H, CH), 2.13 (s, 2H, NH2, exchangeable with D2O), 2.99 (d, 1H, CH, J = 6.5 Hz), 3.86 (s, 3H, OCH3), 4.68 (s, 1H, CH-thiadiazole), 7.01 (d, 2H, J = 7.5 Hz, Ar-H), 7.35 (d, 2H, J = 7.5 Hz, Ar-H), 7.44 (s, 1H, N=CH), 8.04 (s, 1H, NH, exchangeable with D2O). 13C-NMR (DMSO- d6, ppm): 11.56 (CH3), 16.19(CH3), 24.33 (CH2), 39.13 (CH), 56.04 (OCH3), 63.74 (CH), 104.08 (CH-thiadiazole), 114.49, 128.21, 132.22 (5C-aromatic), 164.06 (C-OCH3), 165.74 (C-thiadiazole), 168.15 (C=N), 192.23 (C=O). MS m/z: 366 [M+]. Analysis calculated for C16H22N4O2S2: C, 52.43; H, 6.05; N, 15.29, found: C, 52.53; H, 6.25; N, 15.19.
S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino-3-phenylpropanethioate (8)
Yield, 80%, mp 230–232 °C. IR (KBr, cm−1): 3324, 3232, 3202 (NH, NH2), 2940, 2870 (CH aliph.), 3045 (CH arom.), 1717 (C=O), 1622 (C=N). 1H-NMR (DMSO-d6, ppm): 1.35 (s, 2H, NH2, exchangeable with D2O), 3.25 ((m, 2H, CH2), 3.79 (s, 3H, OCH3), 4.51 (t, 1H, CH, J = 7.1 Hz), 4.78 (s, 1H, CH-thiadiazole), 7.10–7.38 (m, 10H, 5H phenyl, 4H Ar, NH, exchangeable with D2O), 7.76 (s, 1H, N=CH).13C-NMR (DMSO-d6, ppm): 39.97 (CH2), 56.08 (OCH3), 62.09 (CH), 104.55 (CH-thiadiazole), 114.58, 128.21, 132.39 (5C-aromatic), 126.98, 129.06, 129.17, 138.03 (6C-phenyl), 163.46 (C-OCH3), 166.84 (C-thiadiazole), 168.35 (C=N), 193.36 (C=O). MS m/z: 400 [M+]. Analysis calculated for C19H20N4O2S2: C, 56.98; H, 5.03; N, 13.99, found: C, 56.88; H, 5.13; N, 13.79.
S-(5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino ethanethioate (9)
Yield, 83%, mp 240–242 °C. IR (KBr, cm−1): 3330, 3230, 3201 (NH, NH2), 2949, 2880 (CH aliph.), 3048 (CH arom.), 1683 (C=O), 1629 (C=N). 1H-NMR (DMSO-d6, ppm): 1.39 (s, 2H, NH2, exchangeable with D2O), 3.76 (s, 2H, CH2), 4.99 (s, 1H, CH-thiadiazole), 6.89 (s, 1H, NH, exchangeable with D2O), 7.13 (d, 2H, J = 7.5 Hz, Ar-H), 7.59 (d, 2H, J = 7.5 Hz, Ar-H), 7.70 (s, 1H, N=CH).13C-NMR (DMSO-d6, ppm): 48.67 (CH2), 104.05 (CH-thiadiazole), 128.85 (C-Br), 131.29, 132.30, 133.87 (5C-aromatic), 165.84 (C-thiadiazole), 168.15 (C=N), 182.94 (C=O). MS m/z: 359 [M+], 361 [M + 2]. Analysis calculated for C11H11BrN4OS2: C, 36.77; H, 3.09; N, 15.59, found: C, 36.87; H, 3.29; N, 15.49.
S-(5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino propanethioate (10)
Yield, 78%, mp 261–262 °C. IR (KBr, cm−1): 3322, 3233, 3205 (NH, NH2), 2939, 2875 (CH aliph.), 3049 (CH arom.), 1716 (C=O), 1628 (C=N). 1H-NMR (DMSO-d6, ppm): 1.18 (s, 2H, NH2, exchangeable with D2O), 1.41 (d, 3H, CH3, J = 6.5 Hz), 3.59 (q, 1H, CH, J = 6.1 Hz), 4.8 (s, 1H, CH-thiadiazole), 7.11 (d, 2H, J = 7.5 Hz, Ar-H), 7.65 (d, 2H, J = 7.5 Hz, Ar-H), 7.63 (s, 1H, N=CH), 8.18 (s, 1H, NH, exchangeable with D2O). 13C-NMR (DMSO-d6, ppm): 22.59 (CH3), 57.71 (CH), 104.35 (CH-thiadiazole), 128.95 (C-Br), 131.39, 132.40, 134.87 (5C-aromatic), 166.84 (C-thiadiazole), 168.95 (C=N), 190.61 (C=O). MS m/z: 373 [M+], 375 [M + 2]. Analysis calculated for C12H13BrN4OS2: C, 38.61; H, 3.51; N, 15.01, found: C, 38.71; H, 3.41; N, 15.21.
4-amino-5-((5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)thio)-5-oxopentanoic acid (11)
Yield, 86%, mp 245–247 °C. IR (KBr, cm−1): 3420 (OH), 3321, 3236, 3203 (NH, NH2), 2941, 2873 (CH aliph.), 3045 (CH arom.), 1694 (C=O), 1623 (C=N). 1H-NMR (DMSO-d6, ppm): 1.42 (s, 2H, NH2, exchangeable with D2O), 2.16 (td, 2H, CH2, J = 8.1, 0.6 Hz), 2.37 (t, 2H, CH2, J = 7.5 Hz), 3.76 (t, 1H, CH, J = 6.6 Hz), 4.48 (s, 1H, CH-thiadiazole), 6.87 (s, 1H, NH, exchangeable with D2O), 7.45 (d, 2H, J = 7.5 Hz, Ar-H), 7.57 (d, 2H, J = 7.5 Hz, Ar-H), 7.95 (s, 1H, N=CH), 9.55 (s, 1H, OH, exchangeable with D2O). 13C-NMR (DMSO-d6, ppm): 30.95 (2CH2), 59.77 (CH), 104.65 (CH-thiadiazole), 129.85 (C-Br), 131.69, 132.30, 133.77 (5C-aromatic), 164.84 (C-thiadiazole), 168.35 (C=N), 176.74 (COOH), 194.56 (C=O). MS m/z: 431 [M+], 433 [M + 2]. Analysis calculated for C14H15BrN4O3S2: C, 38.98; H, 3.51; N, 12.99, found: C, 38.88; H, 3.61; N, 12.79.
S-(5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino-3-methylpentanethioate (12)
Yield, 85%, mp 267–269 °C. IR (KBr, cm−1): 3332, 3223, 3210 (NH, NH2), 2949, 2881 (CH aliph.), 3046 (CH arom.), 1722 (C=O), 1622 (C=N). 1H-NMR (DMSO-d6, ppm): 0.89–1.09 (m, 6H,2 CH3), 1.32–1.42 (m, 2H, CH2), 1.79–1.87 (m, 1H, CH), 2.17 (s, 2H, NH2, exchangeable with D2O), 2.99 (d, 1H, CH, J = 6.5 Hz), 4.98 (s, 1H, CH-thiadiazole), 7.13 (d, 2H, J = 7.5 Hz, Ar-H), 7.62 (d, 2H, J = 7.5 Hz, Ar-H), 7.33 (s, 1H, N=CH), 8.25 (s, 1H, NH, exchangeable with D2O). 13C-NMR (DMSO-d6, ppm): 11.56 (CH3), 16.19(CH3), 24.33 (CH2), 39.13 (CH), 63.74 (CH), 104.05 (CH-thiadiazole), 128.75 (C-Br), 131.29, 132.50, 133.87 (5C-aromatic), 165.64 (C-thiadiazole), 168.35 (C=N), 192.23 (C=O). MS m/z: 415 [M+], 417 [M + 2]. Analysis calculated for C15H19BrN4OS2: C, 43.37; H, 4.61; N, 13.49, found: C, 43.27; H, 4.71; N, 13.59.
S-(5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)2-amino-3-phenylpropanethioate (13)
Yield, 81%, mp 270–272 °C. IR (KBr, cm−1): 3329, 3235, 3209 (NH, NH2), 2939, 2870 (CH aliph.), 3043 (CH arom.), 1690 (C=O), 1625 (C=N). 1H-NMR (DMSO-d6, ppm): 1.57 (s, 2H, NH2, exchangeable with D2O), 3.03 (m, 2H, CH2), 3.97 (t, 1H, CH, J = 7.6 Hz), 4.88 (s, 1H, CH-thiadiazole), 7.15–7.26 (m, 6H, 5H phenyl, NH, exchangeable with D2O), 7.36 (s, 1H, N=CH), 7.37 (d, 2H, J = 7.5 Hz, Ar-H), 7.63 (d, 2H, J = 7.5 Hz, Ar-H). 13C-NMR (DMSO-d6, ppm): 39.97 (CH2), 62.09 (CH), 104.05 (CH-thiadiazole), 126.98, 129.06, 129.17, 138.03 (6C-phenyl), 128.25 (C-Br), 131.99, 132.40, 133.97 (5C-aromatic), 165.24 (C-thiadiazole), 168.15 (C=N), 193.36 (C=O). MS m/z: 449 [M+], 451 [M + 2]. Analysis calculated for C18H17BrN4OS2: C, 48.11; H, 3.81; N, 12.47, found: C, 48.31; H, 3.71; N, 12.57.
S-(5-((4-Methoxybenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)-pyrrolidine-2-carbothioate (14)
Yield, 79%, mp 262–264 °C. IR (KBr, cm−1): 3231, 3205 (NH), 2941, 2880 (CH aliph.), 3049 (CH arom.), 1721 (C=O), 1629 (C=N). 1H-NMR (DMSO-d6, ppm): 1.86 (s, 1H, NH, exchangeable with D2O), 1.73–2.09 (m, 4H, 2CH2-pyrrolidine), 3.32–3.41(m, 2H, CH2-pyrrolidine), 3.68 (t, 1H, CH-pyrrolidine, J = 8.5 Hz), 3.80 (s, 3H, OCH3), 4.98 (s, 1H, CH-thiadiazole), 6.96 (d, 2H, J = 7.5 Hz, Ar-H), 7.15 (d, 2H, J = 7.5 Hz, Ar-H), 7.69 (s, 1H, N=CH), 9.46 (s, 1H, NH, exchangeable with D2O). 13C-NMR (DMSO-d6, ppm): 24.68, 33.34, 46.90 (3CH2-pyrrolidine), 56.04 (OCH3), 67.49 (CH-pyrrolidine), 104.05 (CH-thiadiazole), 114.49, 128.61, 132.72 (5C-aromatic), 164.76 (C-OCH3), 165.84 (C-thiadiazole), 168.35 (C=N), 193.36 (C=O). MS m/z: 350 [M+]. Analysis calculated for C15H18N4O2S2: C, 51.41; H, 5.18; N, 15.99, found: C, 51.51; H, 5.38; N, 15.79.
S-(5-((4-Bromobenzylidene)amino)-2,3-dihydro-1,3,4-thiadiazol-2-yl)pyrrolidine-2-carbothioate (15)
Yield, 82%, mp 232–234 °C. IR (KBr, cm−1): 3240, 3209 (NH), 2937, 2870 (CH aliph.), 3044 (CH arom.), 1687 (C=O), 1628 (C=N). 1H-NMR (DMSO-d6, ppm): 1.15 (s, 1H, NH, exchangeable with D2O), 1.71–2.01 (m, 4H, 2CH2-pyrrolidine), 2.70–2.73 (m, 2H, CH2-pyrrolidine), 4.04 (t, 1H, CH-pyrrolidine, J = 8.6 Hz), 4.98 (s, 1H, CH-thiadiazole), 7.10 (d, 2H, J = 7.5 Hz, Ar-H), 7.55 (d, 2H, J = 7.5 Hz, Ar-H), 7.62 (s, 1H, N=CH), 7.71 (s, 1H, NH, exchangeable with D2O). 13C-NMR (DMSO-d6, ppm): 23.88, 33.64, 47.70 (3CH2-pyrrolidine), 66.69 (CH-pyrrolidine), 104.85 (CH-thiadiazole), 128.85 (C-Br), 131.29, 132.70, 133.97 (5C-aromatic), 165.94 (C-thiadiazole), 168.25 (C=N), 192.36 (C=O). MS m/z: 399 [M+], 401 [M + 2]. Analysis calculated for C14H15BrN4OS2: C, 42.11; H, 3.79; N, 14.03, found: C, 42.21; H, 3.89; N, 14.23.
In vitro COX-2/COX-1 enzyme inhibition assay
The in vitro ability of the 12 synthesized compounds and celecoxib to inhibit the COX-2/COX-1 isozymes was carried out using Cayman colorimetric COX (ovine) inhibitor screening assay kit (kit catalog number 560101, Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions. Percent inhibition was calculated by the comparison of compound treated to various control incubations. The concentration of the test compound causing 50% inhibition (IC50, μM) was calculated from the concentration-inhibition response curve following triplicate determinations.
In vivo studies
Animals
Male Wistar rats (150–180 g) and mice (25 ± 5 g) purchased from the animal breeding unit of the National Research Centre, Giza, Egypt, and acclimatized in the animal facility of the National Centre for Radiation Research and Technology (NCRRT)-Atomic Energy Authority, Cairo, Egypt for 1 week before being used. Animals were housed at a temperature of 25 ± 5 °C, humidity of 60 ± 5% and 12/12-h light-dark cycle. They were fed standard pellet diet obtained from the National Research Centre, Dokki, Cairo and enabled free access to water ad libitum. All animal experiments comply with the ARRIVE guidelines. The study was conducted U.K. Animals (Scientific Procedures) Act, 1986, and associated guidelines set by EU Directive (2010/63/EU) for animal experiments.
Irradiation
Animals were exposed to whole body irradiation at an acute dose level of 6 Gy at the NCRRT using the Gamma Cell-40 biological irradiator with a Caesium137 source (Atomic Energy of Canada Ltd; Sheridan Science and Technology Park, Mississauga, Ontario, Canada). The radiation dose level was 6 Gy with a dose rate of 0.643 Gy/min.
Determination of acute toxicity
The most potent compounds (4, 5, 6 and 8) were chosen after in vitro COX-2 inhibition assay for in vivo studies. The approximate 50% lethal dose (ALD50) of the compounds (4, 5, 6 and 8) was determined. Adult male Swiss albino mice (3–4 months) weighing 25–35 g were obtained from the animal breeding unit of the National Research Centre, Giza, Egypt. A total of 160 mice were randomly divided into four main groups (corresponding to compounds 4, 5, 6 and 8), each eight mice received I.p. one acute dose of each compound as follows: 100, 200, 400, 800 and 1000 mg/kg.b.wt. All mice were observed for 48 h during which any mortality in each group was recorded. After drug administration, all the animals had free access to food and water. The LD50 was calculated from the data obtained [42].
Experimental design
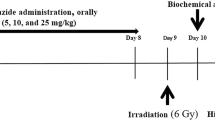
Rats were blindly allocated into 12 groups, consisting of 8 rats each (n = 8). Group 1: served as control non-irradiated animals, Group 2: rats were irradiated at 6 Gy [43, 44] but left untreated, Group 3: irradiated rats were given celecoxib (25 mg/kg) as a reference drug [45], Group 4, 5, 6, 7: irradiated rats were given the newly synthesized compounds 4, 5, 6 and 8 respectively in a dose of (25 mg/kg) similar to the dose of celecoxib. All compounds were dissolved in normal saline and delivered orally after 1 h of irradiation and treatment continued for 2 days further. Group 8: non-irradiated rats were given celecoxib (25 mg/kg). Group 9, 10, 11 and 12: non-irradiated rats were given the newly synthesized compounds 4, 5, 6 and 8 respectively in a dose of (25 mg/kg). On the third day, the rats were sacrificed by decapitation under deep ether anesthesia. Blood samples were collected in non-heparinised tubes, allowed to clot and centrifuged for 15 min at 1000 × g using Hettich Mikro 22R centrifuge (Germany). The separated serum of irradiated and non-irradiated rats was taken for the assay of LDH, Creatinin kinase MB (CK-MB) and TnI. The heart tissues of irradiated rats were rinsed with ice cold saline and rapidly excised. The excised tissue was weighed and homogenized in ice cold normal saline (20% w/v homogenate) using Glass-Col1 homogenizer (Terre Haute, Indiana, USA). The homogenate was immediately stored in −80 °C in order to prepare later homogenates in appropriate media according to the inflammatory parameters to be measured as Myloperoxidase and Nitric oxide.
Cardiac damage marker
(A) Creatinine kinase MB (CK-MB)
Measurement of CK-MB is a specific test for detection of cardiac muscle damage and is used to monitor myocardial infarction. The CK-MB activity is measured using a kit “manufactured by Egyptian Company for Biotechnology (Cairo, Egypt)” according to the recommendation of the International Federation of Clinical Chemistry (IFCC) as shown in the manufacturer instructions. Values are expressed as U/l.
(B) Troponin I (Tn I)
TnI is used as a specific marker for cardiac muscle damage. Its concentration was measured by ELISA using a commercially available kit according to the manufacturer’s instructions from Life Diagnostics, Inc., Cat. No. CTN-1-HIS (West Chester, USA). An ELISA plate reader (Dynatech® MR5000, Guernsey, Channel Islands, UK) was used to measure the absorbance of each sample and was set at 450 nm. Values were expressed as ng/ml.
(C) Lactate dehydrogenase activity (LDH)
Sera of rats were used for the determination of LDH activity LDH in serum was determined according to the method of [46] using kinetic ultraviolet (UV) kit from BioSystems S.A (Barcelona, Spain), and the assay was carried out according to the manufacturer’s instructions. LDH catalyzes the reduction of pyruvate by NADH to form lactate and NAD. The catalytic activity was determined from the rate of decrease of NADH, measured at 340 nm. Values were expressed as U/dl.
Inflammatory markers
(A) Myeloperoxidase (MPO) assay
MPO activity was determined in heart tissue homogenates prepared in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (1:1 ratio) The assay method was that described by [47]. Values were expressed as U/g tissue.
(B) Nitric oxide assay
Total nitrate/nitrite (NOx) was measured according to the method described by [48] using vanadium tri-chloride (VCl3), sulfanilamide N-(1-napthyl) ethylenediamine dihydrochloride (NEDD). Pink color chromophore of the end product was measured at 540 nm using double beam spectrophotometer (Unicam 8625 UV/Vis, Cambridge, UK). Values were expressed as Mmol/g tissue.
Docking study
Molecular docking study was performed utilizing Molecular Operating Environment (MOE) software, version 2014.090. Structure of compound 5 was drawn on MOE. Hamiltonian‐Force Field‐MMFF94x was used to minimize compound’s energy. The forcefield partial charges were calculated. Default settings were utilized for the analysis of the conformational stochastic of the compound. X‐ray crystal structure of MPO in complex with isothiocyanate (PDB ID: 1DNW) and that of COX-2 in complex with SC-558 (PDB ID: 1CX2) were downloaded from http://www.rscb.org/pdb. The protein–ligand complex obtained from the Protein Data Bank (pdb) was prepared for docking, the enzyme was protonated three‐dimensionally and then the system was optimized. Repeated chains of the protein and co-crystallized water molecules were removed. Determination and isolation of the binding pocket were performed and then the backbone was hidden. MOEDOCK was used to determine flexible docking of the ligand‐rigid receptor of the most stable conformers. Scoring was performed using the alpha triangle placement method and London dG as afunction. Forcefield refinement was performed on the obtained poses using the same scoring function. Retention of 50 of the most stable docking models of the ligand with the best‐scored conformation was performed.
References
Ritter O, Neyses L. The molecular basis of myocardial hypertrophy and heart failure. Trends Mol Med. 2003;9:313–21. https://doi.org/10.1016/s1471-4914(03)00114-x.
Velten M, Duerr GD, Pessies T, Schild J, Lohner R, Mersmann J, et al. Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc Res. 2012;96:422–32. https://doi.org/10.1093/cvr/cvs280.
Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–3. https://doi.org/10.1161/01.cir.98.2.100.
Simon LS, Lanza FL, Lipsky PE, Hubbard RC, Talwalker S, Schwartz BD. et al. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor: efficacy and safety in two placebo-controlled trials in osteoarthritis and rheumatoid arthritis, and studies of gastrointestinal and platelet effects. Arthritis Rheum. 1998;41:1591–602. https://doi.org/10.1002/1529-0131(199809)41:9<1591::aid-art9>3.0.co;2-j.
McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–44. https://doi.org/10.1001/jama.296.13.jrv60011.
Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–80. https://doi.org/10.1056/NEJMoa050405.
Taylor CW, Kirby AM. Cardiac side-effects from breast cancer radiotherapy. Clin Oncol. 2015;27:621–9. https://doi.org/10.1016/j.clon.2015.06.007.
Hamada N, Kawano KI, Nomura T, Furukawa K, Yusoff FM, Maruhashi T, et al. Vascular damage in the aorta of wild-type mice exposed to ionizing radiation: sparing and enhancing effects of dose protraction. Cancers. 2021;13. https://doi.org/10.3390/cancers13215344.
Christenson ES, James T, Agrawal V, Park BH. Use of biomarkers for the assessment of chemotherapy-induced cardiac toxicity. Clin Biochem. 2015;48:223–35. https://doi.org/10.1016/j.clinbiochem.2014.10.013.
Malle E, Furtmüller PG, Sattler W, Obinger C. Myeloperoxidase: a target for new drug development? Br J Pharm. 2007;152:838–54. https://doi.org/10.1038/sj.bjp.0707358.
Kullo IJ, Gau GT, Tajik AJ. Novel risk factors for atherosclerosis. Mayo Clin Proc. 2000;75:369–80. https://doi.org/10.4065/75.4.369.
Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–32. https://doi.org/10.1074/jbc.275.48.37524.
Cabrera Pérez LC, Gutiérrez Sánchez M, Mendieta Wejebe JE, Hernández Rgodríguez M, Fragoso Vázquez MJ, Salazar JR, et al. Novel 5-aminosalicylic derivatives as anti-inflammatories and myeloperoxidase inhibitors evaluated in silico, in vitro and ex vivo. Arab J Chem. 2019;12:5278–91. https://doi.org/10.1016/j.arabjc.2016.12.026.
Forbes LV, Sjögren T, Auchère F, Jenkins DW, Thong B, Laughton D, et al. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J Biol Chem. 2013;288:36636–47. https://doi.org/10.1074/jbc.M113.507756.
Patnaik A, Axford L, Deng L, Cohick E, Ren X, Loi S, et al. Discovery of a novel indole pharmacophore for the irreversible inhibition of myeloperoxidase (MPO). Bioorg Med Chem. 2020;28:115548. https://doi.org/10.1016/j.bmc.2020.115548.
Rivera-Antonio A, Rosales-Hernández MC, Balbuena-Rebolledo I, Santiago-Quintana JM, Mendieta-Wejebe JE, Correa-Basurto J, et al. Myeloperoxidase inhibitory and antioxidant activities of (E)-2-hydroxy-α-aminocinnamic acids obtained through microwave-assisted synthesis. Pharmaceuticals. 2021;14:513. https://doi.org/10.3390/ph14060513.
Aldib I, Gelbcke M, Soubhye J, Prévost M, Furtmüller PG, Obinger C, et al. Novel bis-arylalkylamines as myeloperoxidase inhibitors: design, synthesis, and structure-activity relationship study. Eur J Med Chem. 2016;123:746–62. https://doi.org/10.1016/j.ejmech.2016.07.053.
Santos MBD, Carvalho Marques B, Miranda Ayusso G, Rocha Garcia MA, Chiquetto Paracatu L, Pauli I, et al. Chalcones and their B-aryl analogues as myeloperoxidase inhibitors: in silico, in vitro and ex vivo investigations. Bioorg Chem. 2021;110:104773. https://doi.org/10.1016/j.bioorg.2021.104773.
Soubhye J, Prévost M, Van Antwerpen P, Zouaoui Boudjeltia K, Rousseau A, Furtmüller PG, et al. Structure-based design, synthesis, and pharmacological evaluation of 3-(aminoalkyl)-5-fluoroindoles as myeloperoxidase inhibitors. J Med Chem. 2010;53:8747–59. https://doi.org/10.1021/jm1009988.
Luo SH, Yang K, Lin JY, Gao JJ, Wu XY, Wang ZY. Synthesis of amino acid derivatives of 5-alkoxy-3,4-dihalo-2(5H)-furanones and their preliminary bioactivity investigation as linkers. Org Biomol Chem. 2019;17:5138–47. https://doi.org/10.1039/c9ob00736a.
Zaher NH, Salem AA, Ismail AF. Novel amino acid derivatives bearing thieno[2,3-d]pyrimidine moiety down regulate NF-κB in γ-irradiation mediated rat liver injury. J Photochem Photobio B. 2016;165:328–39. https://doi.org/10.1016/j.jphotobiol.2016.10.029.
Hwang H-S, Winkler-Moser JK, Doll KM, Gadgil M, Liu SX. Factors affecting antioxidant activity of amino acids in soybean oil at frying temperatures. Eur J Lipid Sci Technol. 2019;121:1900091. https://doi.org/10.1002/ejlt.201900091.
Etsè KS, Demonceau A, Zaragoza G, Serteyn D, Mouithys-Mickalad A. Design, synthesis and biochemical evaluation of novel 2-amino-3-(7-methoxybenzo[d][1,3]dioxol-5-yl)propanoic acid using Horseradish peroxidase (HRP) activity, cellular ROS inhibition and molecular docking study. J Mol Struct. 2022;1250:131668. https://doi.org/10.1016/j.molstruc.2021.131668.
Ragab FA, Heiba HI, El-Gazzar MG, Abou-Seri SM, El-Sabbagh WA, El-Hazek RM. Synthesis of novel thiadiazole derivatives as selective COX-2 inhibitors. MedChemComm. 2016;7:2309–27. https://doi.org/10.1039/c6md00367b.
Cho NS, Kim GN, Párkányi C. Synthesis of 5-aroylamino-3H-1,3,4-thiadiazole-2-thiones and their tautomerism. J Heterocycl Chem. 1993;30:397–401. https://doi.org/10.1002/jhet.5570300219.
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. https://doi.org/10.1371/journal.pmed.0030442.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. https://doi.org/10.1038/nature07205.
Petersen C, Petersen S, Milas L, Lang FF, Tofilon PJ. Enhancement of intrinsic tumor cell radiosensitivity induced by a selective cyclooxygenase-2 inhibitor. Clin Cancer Res. 2000;6:2513–20.
Ahmad S, Panda BP, Kohli K, Fahim M, Dubey K. Folic acid ameliorates celecoxib cardiotoxicity in a doxorubicin heart failure rat model. Pharm Biol. 2017;55:1295–303. https://doi.org/10.1080/13880209.2017.1299768.
Sabeena Farvin KH, Anandan R, Kumar SH, Shiny KS, Sankar TV, Thankappan TK. Effect of squalene on tissue defense system in isoproterenol-induced myocardial infarction in rats. Pharm Res. 2004;50:231–6. https://doi.org/10.1016/j.phrs.2004.03.004.
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–9. https://doi.org/10.1001/jama.286.8.954.
Abdelazeem AH, Safi El-Din AG, Abdel-Fattah MM, Amin NH, El-Moghazy SM, El-Saadi MT. Discovery of novel urea-diarylpyrazole hybrids as dual COX-2/sEH inhibitors with improved anti-inflammatory activity and highly reduced cardiovascular risks. Eur J Med Chem. 2020;205:112662. https://doi.org/10.1016/j.ejmech.2020.112662.
Elkady AA, Mohamed ET. Possible role of Withania somnifera against gamma radiation induced cardiotoxicity in male albino rats. Pak J Zool. 2016;48:539–45.
Hemnani T, Parihar MS. Reactive oxygen species and oxidative DNA damage. Indian J Physiol Pharm. 1998;42:440–52.
van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11:2899–937. https://doi.org/10.1089/ars.2009.2538.
Koyani CN, Flemmig J, Malle E, Arnhold J. Myeloperoxidase scavenges peroxynitrite: a novel anti-inflammatory action of the heme enzyme. Arch Biochem Biophys. 2015;571:1–9. https://doi.org/10.1016/j.abb.2015.02.028.
Loria V, Dato I, Graziani F, Biasucci LM. Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008;2008:135625. https://doi.org/10.1155/2008/135625.
Freeman SL, MacNaughton WK. Ionizing radiation induces iNOS-mediated epithelial dysfunction in the absence of an inflammatory response. Am J Physiol Gastrointest Liver Physiol. 2000;278:G243–50. https://doi.org/10.1152/ajpgi.2000.278.2.G243.
Chiu FL, Lin JK. Tomatidine inhibits iNOS and COX-2 through suppression of NF-kappaB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008;582:2407–12. https://doi.org/10.1016/j.febslet.2008.05.049.
Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. J Biol Chem. 2000;275:11964–71. https://doi.org/10.1074/jbc.275.16.11964.
Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Disco. 2003;2:179–91. https://doi.org/10.1038/nrd1034.
Smith QE. Pharmacological screening test progress in medicinal chemistry. London: Butterworths; 1960.
de Freitas RB, Boligon AA, Rovani BT, Piana M, de Brum TF, da Silva Jesus R, et al. Effect of black grape juice against heart damage from acute gamma TBI in rats. Molecules. 2013;18:12154–67. https://doi.org/10.3390/molecules181012154.
E SD, Ismail IA, El-Hallous EI, W FA. Protective role of Juniperus phoenicea L. leaves extract against gamma-irradiation-induced oxidative stress. Pak J Biol Sci. 2020;23:922–30. https://doi.org/10.3923/pjbs.2020.922.930.
El-Hazek RMM, El-Sabbagh WA, El-Hazek RM, El-Gazzar MG. Anti-inflammatory and analgesic effect of LD-RT and some novel thiadiazole derivatives through COX-2 inhibition. Arch Pharm. 2020;353:e2000094. https://doi.org/10.1002/ardp.202000094.
Gay RJ, McComb RB, Bowers GN Jr. Optimum reaction conditions for human lactate dehydrogenase isoenzymes as they affect total lactate dehydrogenase activity. Clin Chem. 1968;14:740–53.
Bradley PP, Christensen RD, Rothstein G. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood. 1982;60:618–22.
Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 2001;5:62–71. https://doi.org/10.1006/niox.2000.0319.
Acknowledgements
The authors kindly appreciate the staff members of gamma irradiation unit at the National Center for Radiation Research and Technology (NCRRT), for their cooperation in carrying out the irradiation experiment.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaher, N.H., El-Sheikh, M.M., El-Hazek, R.M. et al. Potential effect of novel thiadiazole derivatives against radiation induced inflammation with low cardiovascular risk in rats. Med Chem Res 31, 1875–1888 (2022). https://doi.org/10.1007/s00044-022-02948-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02948-1