
Abstract
Hydrochloride of 10-{2-hydroxy-3-[N,N-bis-(2-hydroxyethyl)amino]propyl}-2-trifluoromethylphenothiazine (Flu-A) is a analogue of neuroleptic fluphenazine. Flu-A exhibits anti-multidrug resistance, antimutagenic, proapoptopic, and cancer-chemopreventive activities in screening studies. To define identity, quality, and purity of new active substance it is necessary to develop a appropriate analytical method and to establish a degradation profile. Thus, a stability-indicating reversed-phase high-performance liquid chromatography method was developed and validated for quantitative determination of Flu-A in the presence of its degradation products generated under stress conditions. The compound was subjected to oxidation, photolysis, and degradation in aqueous solutions (neutral and acidic), and solid state according to the International Council for Harmonisation Guidelines. The method was also found to be suitable for intermediate and accelerated studies and for the evaluation of kinetic mechanism of Flu-A degradation in aqueous solutions (pH 5.1–7.5, 353 K). The structures of main potential degradation products were established using high-performance liquid chromatography-Electrospray Ionization-mass spectrometry method.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer is very widespread in the modern world. According to World Health Organization in developed countries cancers are, after cardiovascular disease, the second leading cause of death. A major problem of modern oncology is the resistance of tumor cells to anticancer drugs (Klopman et al. 1997). The phenomenon of multidrug resistance (MDR) significantly impedes or even precludes effective, long-term chemotherapy in cancer. MDR frequently relates to simultaneous resistance of tumor cells to many medicinal substances, often of different chemical structure or different physicochemical properties (Klopman et al. 1997; Hendrich et al. 2003). Therefore, it seems appropriate to search inhibitors of the MDR process.
Analysis of clinical data showed that among patients with schizophrenia, treated with phenothiazines, the incidences of cancer are more seldom than in the rest of society (Fond et al. 2012). Phenothiazines, apart from their neuroleptic action, also exert different biological activities, which account for their cancer chemopreventive effect (Jaszczyszyn et al. 2012a). Unfortunately, limitations of phenothiazines use in the cancer treatment are caused by their serious side effects on the central nervous system (e.g., dyskinetic syndrome, parkinsonism, drowsiness, apathy, and depression). Hence, the structure of parent phenothiazines has been modified to obtain more hydrophilic analogues which exhibit similar antimutagenic/cancer-chemopreventive activity but fewer psychotropic effects. The modifications of neuroleptic phenothiazine structure consisted in introducing new substituents at the position 2 of the tricyclic phenothiazine ring system and changes in the length of the alkyl chain between the nitrogen atom at tricyclic skeleton and the terminal amine group in the side moiety (Gąsiorowski et al. 2003; Żyta et al. 2014).
Among the tested derivatives promising results were obtained for fluphenazine (Flu) analogue: 10-{2-hydroxy-3-[N,N-bis-(2-hydroxyethyl)amino]propyl}-2-trifluoromethylphenothiazine hydrochloride (Flu-A, Fig. 1) (Gąsiorowski et al. 2003; Żyta et al. 2014), which in screening studies exhibits antimutagenic, cancer-chemopreventive, and chemosensitizing activities. Flu-A inhibits P-glycoprotein-dependent MDR on the basis of various independent mechanisms, such as: an inhibition of protein kinase C activity, a fluidization of the structure of lipid membrane and a blocking of ligand binding sites within P-glycoprotein (Jaszczyszyn and Gąsiorowski 2006; Jaszczyszyn et al. 2012b). Moreover, Flu-A enhances apoptosis in genotoxically damaged lymphocytes 35% stronger than Flu (enhancement of apoptosis in tumor cells could be a desired mechanism of action of anticancer drugs in chemoprevention) (Gąsiorowski et al. 2003). The tested compound induces this process by stimulation of acidic sphingomyelinase and showed only a small effect on the activity of sphingomyelinase neutral (Jaszczyszyn et al. 2011). Analogue Flu-A also increased activity of caspase-3 in lympohocyte cultures genotoxically damaged in vitro with benzo[a]pyrene (Jaszczyszyn et al. 2009). The effect of Flu-A on the frequency of apoptosis was evaluated not only in cultures treated with benzo[a]pyrene, but also in undamaged cultures. However, this impact was not significant in undamaged lympohocyte what suggested that the enhancement of apoptosis could be specific, restricted to genotoxically damaged lympohocyte cultures. Simultaneously, fluphenazine derivative shows 60% lower cytotoxicity and 10% stronger antimutagenic effect than Flu (Gąsiorowski et al. 2003).

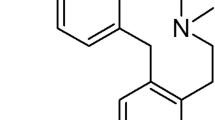
Structure of 10-{2-hydroxy-3-[N,N-bis-(2-hydroxylethyl)amino]propyl}-2-trifluoromethylphenothiazine hydrochloride (Flu-A)
Before launching a new drug, it is necessary to define its physical and chemical properties in order to facilitate the understanding of mechanism of absorption, distribution, metabolism, excretion, and toxicity of the drug substance. Another important issue in the process of testing a chemical compound, as a potential candidate for a pharmaceutical formulation, is to evaluate its stability. Stability can be defined as the capacity of the active substance or drug product to sustain its identity, strength, quality, and purity throughout the retest or expiration period. Stability testing of drug substances or products includes changes in the quality of a drug product under the influence of environmental factors such as temperature, humidity, and light (Draft Guidance for Industry Stability Testing of Drug Substances and Drug Products 1998; ICH 2003). According to the International Council for Harmonisation (ICH) Guidelines Q1A (R2) in order to conduct stability tests should be develop and validate a stability-indicating analytical procedure (ICH 2003).
Based on the above mentioned the purpose of this studies was: (1) to develop and validate stability-indicating method for fluphenazine analogue (Flu-A); (2) to determine the effect of various environmental factors on its stability.
Materials and methods
Materials and reagents
The analogue of fluphenazine: 10-{2-hydroxy-3-[N,N-bis-(2-hydroxyethyl)amino]propyl}-2-trifluoromethylphenothiazine hydrochloride (Flu-A; C20H24ClF3N2O3S; Mol. mass 464.93g/mol) was synthesized at the Department of Chemistry of Drugs, Wroclaw Medical University, Poland (Żyta et al. 2014). The same batch was use in all studies. All other chemicals and solvents were of analytical or high-performance liquid chromatographic grade.
Equipment and chromatographic conditions
The high-performance liquid chromatography (HPLC) method was developed and validated for forced degradation studies. The used HPLC system was composed of a Rheodyne Berkeley 7120 with a noose of 50 µL, an isocratic pump model liquid chromatography (LC)-61 Shimadzu and an SPD-6AV UV/VIS detector. The chromatographic conditions were as follows: an analytical column, Merck LiChrospher RP-18 (125 × 4 mm, 5 µm particle size); a mobile phase, the mixture of the solution containing 2.10 g/L of citric acid and 1.34 g/L of potassium chloride—acetonitrile (ACN) (70:30); flow rate, 1 mL/min; UV detection at 257 nm. The internal standard (I.S.) was propyl 4-hydroxybenzoate in a mixture of methanol and water (1:1) at a concentration of 0.16 mg/mL.
LC–mass spectrometry (MS) measurements were performed on a 1200 series liquid chromatograph coupled with a 6410B Triple Quad mass spectrometer (Agilent, USA). Separation was performed at 40 °C with a Poroshell 120 EC-18 column (3.0 × 75 mm, 2.7 µm, Agilent, USA). Mobile phases were: 0.1% formate buffer pH 3.2 [A] and 0.1% formic acid in acetonitrile [B]. The flow rate was 0.5 mL/min. The instrument was operated with electrospray ionization (ESI) source in the positive mode. MS conditions were: drying gas temperature (nitrogen), 300 °C; nebulizing gas flow, 8 L/min; nebulizing gas pressure, 40 psi; capillary voltage, 4 kV; fragmentor voltage, 50–250 V. The acquisition was carried out in the scan mode (m/z 50–500).
Photostability studies were carried out in a photostability chamber—the Atlas Suntest CPS + (Atlas, USA) which was equipped with temperature control system (25–100 °C), 1500 W air-cooled xenon lamps and with direct setting and control of irradiance in the wavelength range 300–800 nm (filter Solar ID65). The photostability tests were performed at 25 °C.
The pH of the tested solutions was measured with a potentiometric Hanna Instruments 110 pH-meter using an HI 1131 combination pH electrode at the respective experimental temperatures.
Method validation
The HPLC method was validated according to the ICH Guidelines with regard to selectivity, linearity, precision, limit of detection (LOD), and limit of quantitation (LOQ).
Selectivity
The selectivity of the HPLC method was evaluated by verifying the complete separation of Flu-A from its degradation products (DP) and the I.S. For this reason, the studied compound was subjected to thermal degradation in aqueous solutions at 363 K (24 h in 0.1 M; 36 h in 1 M and 72 h in 2 M HCl; 36 h and 14 days in water), oxidation (at 298 K, 6 h in 3%; then 3 h in 1.5% H2O2) and photodegradation.
Linearity and LOD and LOQ
Linearity was established in the range from 0.02 to 0.32 mg/mL. Peak areas Pi/PI.S. (Pi and PI.S.—areas of Flu-A and I.S.) vs. Flu-A concentrations data were obtained by the least square linear regression analysis and used to calculate the calibration equations and correlation coefficients. The fourteen different concentrations were used for the linearity studies, and each sample was prepared in triplicate. The LOD and LOQ for the procedure were received directly from the calibration line using the formulas: LOD = 3.3S b /a and LOQ = 10S b /a, respectively, where S b is the standard deviation of the intercept and a is the slope of the corresponding calibration curve.
Precision
The measurement of precision was demonstrated by using the parameters of repeatability (intra-day) and intermediate precision (inter-day). In order to evaluate the repeatability of the method six replicate samples of Flu-A for three concentrations were analyzed. One of these concentrations was additionally determined (n = 6) on the second day in order to investigate the inter-day precision.
Stability studies
Flu-A was exposed to different storage conditions: degradation in aqueous solutions (neutral and acidic), photodegradation, oxidation, dry and wet heat, as defined in the ICH Guideline Q1A (R2). Alkaline degradation was not performed because Flu-A, as a salt of an organic base, is insoluble in this environment. The I.S. was added to individual tested samples before the injection of the samples on HPLC column. All solutions for Flu-A degradation time t = 0 min, used in stress, intermediate and accelerated tests, were injected on HPLC at initial Flu-A concentrations of 0.20 mg/mL. All samples were protected from the light during the degradation stage, except for sample preparation time.
Stress studies
Degradation in solutions was carried out at 363 K in water for 36 h and under acidic conditions (in 0.1 M HCl for 24 h, in 1 M HCl for 36 h and then in 2 M HCl for 72 h). After the degradation period, these solutions were neutralized (if necessary) and cooled in a mixture of ice and water.
Oxidative degradation was induced by storing the samples at room temperature (298 K) in 3 and 1.5% hydrogen peroxide for a period of 6 or 3 h, respectively.
Photostability of Flu-A compound in powder and in water solution was conducted in a quartz dish (in a layer less than 2 mm in thickness) and quartz cuvettes, respectively. All samples were inserted in a light cabinet (Atlas Suntest CSP+) and exposed to light for an overall illumination of 1.2 and then 6 million lux hours and an integrated near ultraviolet energy of 250 W/m2, at 25 °C. Control samples were protected from light with aluminum foils, placed in a light chamber and exposed concurrently.
Intermediate and accelerated stability studies
Thermal stability and sensitivity to moisture of Flu-A in a solid state were investigated during its intermediate (relative air humidity ~ 65% ± 5%, 303 K, time: initial, 7 days, 1, 2, 3, 4, 6, 9, 12 months) and accelerated (~ 75% RH ± 5% RH, 313 K, time: initial, 7 days, 1, 2, 3, 4, 5, 6 months) studies. Degradation of Flu-A at 0% relative air humidity at 393 K was carried out for 12 months at different intervals, i.e. initial, 7 days, 1, 2, 3, 6 and 12 months.
In order to determine the stability of Flu-A in dry air, the vials containing the studied substance were immersed in sand baths, in heat chambers adjusted to 393 K.
The appropriate values of relative air humidity were obtained by using desiccators containing saturated solutions of suitable inorganic salts: sodium nitrite (~ 65% RH) or sodium chloride (~ 75% RH).
At individual time intervals, the vials with 2.5 mg of Flu-A were removed from the sand bath or from the desiccators placed in heat chambers, cooled to room temperature and their contents were dissolved in the mobile phase. The so-obtained solutions were quantitatively transferred into measuring flasks and diluted with a mobile phase to 10.0 mL.
Kinetic procedures
The stability of Flu-A and its DP was investigated at 353 K in aqueous buffer solutions: acetate (pH = 5.1); phosphate (pH = 6.8); borate (pH = 7.5). The ionic strength, µ = 0.5 M, was adjusted for each solution by adding a calculated amount of sodium chloride (4 M). The pH value of the buffers was measured potentiometrically. The degradation was initiated by adding a dissolved sample of Flu-A to a buffer solution of specific pH equilibrated to the required temperature in a stoppered flask.
The starting Flu-A solutions (t = 0 min) were performed in the concentration of 0.5 mg/mL. At specified time intervals 0.5 mL of the above reaction solutions was collected and either neutralized if necessary (0.5 mL) or 0.5 mL of water was added. Then the samples were instantly cooled with a mixture of water and ice. 0.25 mL of I.S. solution was added to each sample and then they were analyzed.
Identification of Flu-A products by HPLC-ESI-MS
The identification of the Flu-A degradation products was carried out by using the HPLC-ESI-MS method. The Flu-A samples were degraded under the following conditions:
-
at 363 K in buffer solutions: acetate (pH = 5.1, 357 h), borate (pH = 7.5, 92 h),
-
at 363 K in water (383 h),
-
under oxidative agent (3% H2O2, 3 h, 298 K),
-
under light (aqueous solution, dose 2700 kJ/m2, 3 h, 250 W/m2, 298 K),
-
in a solid state (RH 0%, 6 months, 393 K; RH ~ 65%, 303 K, 9 months).
Results and discussion
Method development and optimization
The main objective of the chromatographic method was to separate the generated DP from the substrate and I.S. during stability studies. DP were co-eluted by using different aqueous components of mobile phases, such as dodecyl sodium, mixture of citric acid and potassium chloride or phosphate buffer, with various pH values (3.0–7.4), and organic modifiers, including ACN and methanol. Analysis of retention time, shape and intensity of peaks, resolution between analyte and its DP, ACN consumption while using different aqueous components of mobile phases indicated that the mixture of citric acid and potassium chloride was optimal. The use of methanol in a mobile phase caused poor and long elution of all compounds, thus acetonitrile was used in further studies. The change of ratio between the organic and water components, while maintaining a constant qualitative composition, resulted in a significant change in the retention times, the shape and intensity of the peaks. At higher acetonitrile content, the retention time of substrate (Flu-A) was shorter, and the peak was narrower, more symmetric and intensive. But on the other hand resolution between the DP peaks was the worst in these conditions. To improve the quality of chromatographic separation the influence of a mobile phase pH was investigated. It was observed that with the increase of pH the tailing of Flu-A peak was longer and also time of elution was longer, therefore pH equals to about 3 was chosen as optimal.
In conclusion, the best chromatographic separation was achieved on the column LiChrospher 100 RP-18 (125 mm × 4 mm, 5 μm) using a mobile phase (ACN—the mixture of 2.10 g/L of citric acid and 1.34 g/L potassium chloride) in proportions 30:70.
Method validation
Selectivity
The results of stress tests indicated selectivity of the Flu-A quantitative method. In order to avoid the injection errors the I.S.—propyl 4-hydroxybenzoate was applied. The parent compound—Flu, was not used as a I.S. because it has a broad base as most of phenothiazines. The examples of typical chromatograms obtained for the pure Flu-A (in the presence of I.S.) and degraded samples are demonstrated in Fig. 2.

HPLC chromatograms of Flu-A degradation samples: a in water, 363 K, t = 0; b in water, 363 K, t = 14 days; c in HCl (2 M), 363 K, t = 72 h; d in H2O2 (1.5%), 298 K, t = 60 min.; e in solution, 298 K (55 046 lux h), t = 60 min.; where I.S.—internal standard, P—degradation products, Flu-A—10-{2-hydroxy-3-[N,N-bis-(2-hydroxylethyl)amino]propyl}-2-trifluoromethylphenothiazine hydrochloride
Linearity and LOD and LOQ
The calibration curve for aqueous solutions of Flu-A was linear over the tested concentration range and the obtained correlation coefficient was 0.999 (n = 14) which shows an excellent correlation between the peak area and the analyte concentration. The equation of regression was as follows: y = (20.06 ± 0.48) · x. The value b = 0.091 calculated from y = ax + b was statistically insignificant, because the value t b = b/S b was lower than the critical one t 0.05(12) (estimated for f = n−2 degrees of freedom (n = 14) at p = 0.05). The LOD and LOQ were 0.07 × 10−1 mg/mL and 0.02 mg/mL, respectively.
Precision
The method had adequate repeatability because the relative standard deviation (RSD) for six repeated assays of samples was less than 2.0% for three different concentrations of Flu-A: 0.58% for c = 0.10 mg/mL, 0.85% for c = 0.20 mg/mL, 1.63% for c = 0.30 mg/mL. The RSD for inter-day precision was 1.10% for c = 0.20 mg/mL (n = 12).
Stability studies
Stress tests of active substances or pharmaceutical products can help to identify the likely DP and to establish the decomposition pathways, which can be useful at the stage of development or modification of their chemical structure, manufacturing process, as well as their formulation or determining the storage conditions or validation of the stability-indicating analytical method. Moreover long-term, intermediate and accelerated stability studies allow evaluating the re-test period at quality monitoring or a shelf life of a drug product (Draft Guidance for Industry Stability Testing of Drug Substances and Drug Products 1998; ICH 2003).
Stress, intermediate and accelerated stability studies
Flu-A was very stable in acidic solutions (HCl, 363 K) whereas in water degradation was observed (Table 1). In above cases color changes of tested solutions were observed (bright yellow or orange yellow color). The appearance of the color may indicate the presence of oxidation product—a radical cation. This is probable because in the presence of oxygen (from the air) in aqueous and buffered solutions of some phenothiazine derivatives, besides hydrolytic degradation the oxidation can occur (Pawełczyk et al. 1975; Underberg 1978). The thermal degradation of aqueous solutions of phenothiazines depends, first of all, on the kind of substituent at C2 and N10 positions (Pawełczyk et al. 1975).
Significant degradation of fluphenazine derivative was observed at 298 K under the oxidizing agents (6 h in 3% H2O2—total degradation; 3 h in 1.5% H2O2—sufficient decomposition) which classifies the compound as very unstable under an oxidative factor (Table 1).
Flu-A underwent photodegradation in aqueous solutions and was stable in the solid state on exposure to light (Table 1).
The significant degradation of Flu-A was observed at 0% relative air humidity at 393 K and ~ 65% RH at 303 K (intermediate studies). But during accelerated testing (~ 75% RH, at 313 K) the fluphenazine analogue was stable (Table 1). The phenomena of lower Flu-A stability under milder conditions of air humidity and temperature may be related to the oxidative properties of sodium nitrite, which was applied to obtain the suitable value of humidity. Thus, a different kind of salt should be used for stability studies at 65% RH.
Kinetics studies
Based on the results of stress tests, the kinetic mechanism of Flu-A compound degradation was evaluated in a range of pH 5.1–7.5 at 353 K (at higher pH Flu-A compound underwent precipitation, and at lower pH its degradation time was significantly elongated).
The degradation of Flu-A in buffer solutions is a reversible first-order reaction relative to substrate concentration (Fig. 3).

Plots of: a lnP = f(t) for reactions of Flu-A degradation and product A formation in acetate buffer, c = 0.10 M, (pH ~ 5.1, temp. 353 K); b ln(P t −P ∞) = f(t) for Flu-A degradation reaction and ln(P ∞−P t ) = f(t) for product A formation in acetate buffer (c = 0.10 M, pH ~ 5.1, temp. 353 K)
During this study one main degradation product was analyzed. The concentration of product A in time interval from t 0 to t ∞ increased from 0 to P ∞ (no further degradation of product A shows that the process is not a subsequent reaction) (Fig. 3a). Both processes: Flu-A degradation and product A formation can be described by the following equations:
where: P 0, P t , P ∞—the ratio of the peak area of Flu-A or product A to the peak area of I.S. at time zero, t and t ∞, respectively; k obs—the observed first-order reaction rate constants; t—time (Fig. 3b). The values of reaction rate constants of Flu-A degradation and product A formation were compared using a parallelism test (Table 2). The results indicated that there are no statistically significant differences between them, which may suggest that product A is produced from Flu-A. But it does not exclude formation of product A by radicals. In this case rate constants of Flu-A and radicals degradation would be equal to rate constant of product A formation.
The catalytic effect was determined by measuring the rate of degradation of Flu-A at a constant pH (in all buffers), ionic strength (μ = 0.5 M) and temperature (353 K) (Table 3). Only the buffer concentration at a specific pH was different. The results obtained show (NaBO2, H3BO3) at concentrations applied demonstrate a catalytic effect. In acetate and phosphate buffers the rate constant (k obs) for the degradation of Flu-A is independent of their concentrations. It means that components like CH3COOH, CH3COONa, KH2PO4, Na2HPO4 do not catalyze the degradation reaction. This data can be useful at the selection of pharmaceutical excipients during formulation process.
MS analysis
An LC-MS study was carried out to determine the molecular weight and structure of major Flu-A degradation products formed under oxidation, photodegradation and degradation both in solution (acidic and neutral medium) and a solid state. Flu-A degradation in all studied samples was incomplete, hence the peak at t R = 11.3 min corresponding to Flu-A occurred at all obtained chromatograms. In the MS analysis at low fragmentor voltage (FV 50 V) of Flu-A a pseudo-molecular ion [M + H]+ at m/z 429 (Fig. 4a) and ammonium [M + NH4]+ and sodium [M + Na]+ adducts at m/z 445 and m/z 451, respectively were found. Moreover one or two major Flu-A degradation products were observed at chromatograms (DP I at 10.7 min; DP II at 10.5 min) (Fig. 4b,c). In all the above degradation conditions the most intensive product peak was observed at 10.7 min (DP I). Its mass spectrum exhibited a protonated molecular ion at m/z 445 (Fig. 4b) and ammonium (m/z 461) and sodium (m/z 467) adducts. Moreover, a pseudo-molecular ion [M + H]+ at m/z 405 (DP II) was found during photodegradation (Fig. 4c).

MS fragmentaion spectrum of substrate (Flu-A) a and its photodegradation products DP I b and DP II c (FV 250 V)
As it is known from literature that phenothiazine drugs undergo degradation under various factors (Nałęcz-Jawecki et al. 2008; Puzanowska-Tarasiewicz et al. 2009; Trautwein and Kümmerer 2012). They are easily oxidized by different chemical, photochemical, enzymatic (Puzanowska-Tarasiewicz et al. 2009), and electrochemical (Sobczak et al. 2015) agents often with a color change. The process of oxidation can proceed in several one-electron stages forming colored intermediate products—cationic radicals. Further oxidation leads to a generation of colorless sulphoxide. The first step of converting a substrate to radicals is relatively fast process opposite to the last stage of sulfoxide formation. Due to structure and lability of the radicals, they are not always recorded by every analytical methods. Stability of cationic radicals depends on a presence and a structure of substituents in positions 2 and 10, the nature of an oxidant applied, pH of solutions, the type and concentration of acid or buffer used, temperature and time of reaction (Borg and Cotziaz 1962; El-Gindy et al. 2002; Puzanowska-Tarasiewicz et al. 2005; Nałęcz-Jawecki et al. 2008; Puzanowska-Tarasiewicz et al. 2009). The hydrolysis of phenothiazines was also described in the literature (Pawełczyk and Marciniec 1974; Pawełczyk et al. 1975; Egorov 1998; Nałęcz-Jawecki et al. 2008).
The most frequently reported modifications of the molecule concerned S/N-oxidation, wherein N-oxides are formed mainly by oxidation of the nitrogen atoms present at the side chain at the 10-position. Oxidation at the nitrogen atom in phenothiazine ring is less likely to occur because substitution of an alkyl group at the 10-position causes oxidation at ring to be more difficult. Moreover transformation of the trifluoromethyl to a carboxylic group, elimination of amine side chains or dimerization can be also observed (Pawełczyk et al. 1975; Heyes 1982; Egorov 1998; Nałęcz-Jawecki et al. 2008; Trautwein and Kümmerer 2012).
Based on the above literature data connected with degradation of different phenothiazine derivatives, the molecular formula of DP I at m/z 445 could be defined as C20H23F3N2O4S and the corresponding structure could be attributed to any oxides of Flu-A: sulfoxides or N-oxides. For unambiguous identification of DP I higher FV (250 V) was used caused in-source fragmentation of compound. Fragmentation ions of the phenothiazine core combined with oxygen at m/z 296 and 340, which indicates oxidation at a sulfur atom in a phenothiazine ring, were observed. Furthermore, MS fragments at m/z 162 and 118 exclude oxidation at an aliphatic amine side chain. On this basis the product at m/z 445 could be attributed to a sulfoxide of Flu-A (DP I) (Fig. 4b).
Under light DP II formation were observed. MS fragmentation of DP II led to fragment ions m/z 224 and 256, which may indicate a transformation of the trifluoromethyl moiety to a carboxylic group (Fig. 4c).
DP I and II eluted at earlier retention time than their parent compound, which indicates their higher polarity. This observation may confirm the hypothesis that DP I (logPcalc. = 1.90; the value calculated by ChemDraw program) is a sulfoxide of Flu-A (for Flu-A logPcalc. = 2.93) and DP II is a carboxylic derivative of Flu-A (logPcalc. = 1.57).
Conclusions
In this paper stability of new fluphenazine analogue (Flu-A), which was designed to counteract the resistance of cancer cells to cytostatics, was investigated. Based on the results we obtained, it was demonstrates that Flu-A in solutions is sensitive to light and oxidative agents, so the substance should be protected before these factors in the future studies (preformulation process, testing on animal models etc.). Flu-A susceptibility to oxidation and photodegradation was confirmed by HPLC-ESI-MS and degradation profile obtained.
Abbreviations
- Flu:
-
fluphenazine
- Flu-A:
-
analogue of fluphenazine
- ICH:
-
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- I.S.:
-
internal standard
- LOD:
-
limit of detection
- LOQ:
-
limit of quantitation
- MDR:
-
multidrug resistance
- RH:
-
relative air humidity
- RSD:
-
relative standard deviation
References
Borg DC, Cotziaz GC (1962) Interaction of trace metals with phenothiazine drug derivatives, III. Theoretical part. Biochemistry 48:643–652
Food and Drug Administration (1998) Draft guidance for industry stability testing of drug substances and drug products. FDA, Rockville
Egorov AP (1998) [The detection of 10-acyl derivatives of phenothiazine by staining reactions and a spectrophotometric method for their hydrolysis products]. Sud Med Ekspert 41:29–31
El-Gindy A, El-Zeany B, Awad T, Shabana MM (2002) Derivative spectrophotometric, thin layer chromatographic-densitometric and high performance liquid chromatographic determination of trifluoperazine hydrochloride in presence of its hydrogen peroxide induced-degradation product. J Pharm Biomed Anal 27:9–18
Fond G, Macgregor A, Attal J, Larue A, Brittner M, Ducasse D, Capdevielle D (2012) Antipsychotic drugs: pro-cancer or anti-cancer? A systematic review. Med Hypotheses 79:38–44
Gąsiorowski K, Malinka W, Świątek P, Jaszczyszyn A (2003) Antimutagenic activity of new analogues of fluphenazine. Cell Mol Biol Lett 8:927–942
Hendrich AB, Wesołowska O, Motohashi N, Molnar J, Michalak K (2003) New phenothiazine-type multidrug resistance modifiers: anti-MDR activity versus membrane perturbing potency. Biochem Biophys Res Commun 304:260–265
Heyes WF (1982) Studies on the oxidative degradation of fluphenazine decanoate in oily solution. Doctoral thesis, Liverpool John Moores University 17-23; 53-54
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2003). ICH Harmonised Tripartite Guideline: Stability Testing of New Drug Substances and Products Q1A (R2), Step 5. ICH, Geneva
Jaszczyszyn A, Gąsiorowski K (2006) Mechanisms of chemopreventive activity of newly synthesized fluphenazine analoques (Polish). Borgis® Wydawnictwo Medyczne, Warsaw
Jaszczyszyn A, Gąsiorowski K, Świątek P, Malinka W (2009) The impact of the newly synthesized fluphenazine analogues on apoptosis and necrosis in lymphocyte cultures. Polish Oncol 12:143–149
Jaszczyszyn A, Gąsiorowski K, Świątek P, Malinka W (2011) Proapoptotic activity of fluphenazine analogues after blocking sphinogomyelinases in cultures of human lymphocytes. Polish Oncol 14:43–53
Jaszczyszyn A, Gąsiorowski K, Świątek P, Malinka W, Cieślik-Boczula K, Petrus J, Czarnik-Matusewicz B (2012a) Chemical structure of phenothiazines and their biological activity. Pharmacol Rep 64:16–23
Jaszczyszyn A, Gąsiorowski K, Świątek P, Malinka W, Cieślik-Boczula K, Petrus J, Czarnik-Matusewicz B (2012b) New fluphenazine analogues as inhibitors of P-glycoprotein in human lymphocyte cultures. Contemp Oncol 16:332–337
Klopman G, Shi LM, Ramu A (1997) Quantitative structure-activity relationship of multidrug resistance reversal agents. Mol Pharmacol 52:323–334
Nałęcz-Jawecki G, Hajnas A, Sawicki J (2008) Photodegradation and phototoxicity of thioridazine and chlorpromazine evaluated with chemical analysis and aquatic organisms. Ecotoxicology 17:13–20
Pawełczyk E, Marciniec B (1974) Kinetics of drug decomposition, Part XXIX, Autoxidation and hydrolysis of thioridazine hydrochloride in aqueous solutions. Pol J Pharmacol Pharm 26:465–472
Pawełczyk E, Marciniec B, Matlak B (1975) Kinetics of drug decomposition, Part XXXVI, Stability of 10-(1’-methyl-4’-piperazinylpropyl)phenothiazine derivatives on the grounds of kinetics of thermal degradation and Hammett equation. Pol J Pharmacol Pharm 27:317–325
Puzanowska-Tarasiewicz H, Kuźmicka L, Karpińska J, Mielech-Łukasiewicz K (2005) Efficient oxidizing agents for determination of 2,10-disubstituted phenothiazines. Anal Sci 21:1149–1153
Puzanowska-Tarasiewicz H, Karpińska J, Kuźmicka L (2009) Analytical applications of reactions of iron(III) and hexacyanoferrate(III) with 2,10-disubstituted phenothiazines. Int J Anal Chem 2009:1–8
Sobczak A, Rębiś T, Milczarek G (2015) Electrocatalysis of NADH oxidation using electrochemically activated fluphenazine on carbon nanotube electrode. Bioelectrochemistry 106:308–315
Trautwein C, Kümmerer K (2012) Ready biodegradability of trifluoromethylated phenothiazine drugs, structural elucidation of their aquatic transformation products, and identification of environmental risks studied by LC-MSn and QSAR. Environ Sci Pollut Res Int 19:3162–3177
Underberg WJM (1978) Oxidative degradation of pharmaceutically important phenothiazines III: Kinetics and mechanism of promethazine oxidation. J Pharm Sci 67:1133–1338
Żyta J, Jaszczyszyn A, Świątek P, Gąsiorowski K, Malinka W (2014) Synthesis, pro-apoptotic activity and 2D-QSAR studies of new analogues of fluphenazine. Acta Pol Pharm 71:49–58
Acknowledgements
This study was partly supported by Poznan University of Medical Sciences grant 502-14-03305411-08304 and 502-01-03305411-04981.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sobczak, A., Teżyk, A., Szyndlarewicz, J. et al. New fluphenazine analogue with antimutagenic and anti-multidrug resistance activity—degradation profile and stability-indicating method. Med Chem Res 26, 2443–2451 (2017). https://doi.org/10.1007/s00044-017-1944-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1944-4