
Abstract
To monitor compliance with the Comprehensive Nuclear-Test-Ban Treaty (CTBT), the International Monitoring System (IMS) is being established which will include 40 sensor systems for atmospheric xenon radioactivity. Radioactive isotopes of the noble gas xenon provide the most likely observable radioactive signatures of underground nuclear explosions. These isotopes are frequently detected by IMS noble gas systems as a result of normal operational releases from different types of nuclear facilities including nuclear power plants (NPPs), medical isotope production facilities (MIPFs), and nuclear research reactors (NRRs). Improved knowledge of the contribution of different emission sources on IMS observations strengthens the screening of radioxenon measurements to exclude observations not relevant to emissions from a nuclear explosion. The contribution of NPPs and MIPFs to the global radioxenon emission inventory is fairly well understood. NRRs have yet to be systematically assessed. This paper is the first attempt to assess the total emission inventory of NRRs expressed as annual total discharges. The results can enhance understanding of those sources most likely to impact IMS background observations and to guide future studies on contributions to IMS station background.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
1.1 Relevance of Atmospheric Radioxenon Background from Anthropogenic Sources
Xenon isotopes provide the most likely observable radioactive signatures of underground nuclear explosions. A global monitoring system for atmospheric xenon radioactivity is being established as part of the International Monitoring System to verify compliance with the Comprehensive Nuclear-Test-Ban Treaty (CTBT). The atmospheric concentrations of 135Xe, 133mXe, 133Xe and 131mXe are measured as indicators of a nuclear explosion. However, these isotopes are observed in the atmosphere as a result from normal operational releases from different types of nuclear facilities including nuclear power plants (NPPs), medical isotope production facilities (MIPFs), and nuclear research reactors (NRRs). Improving the knowledge of different emission sources leads to strengthen the interpretation on radioxenon detection results.
Due to the short half-lives of the radioxenon isotopes of interest, spent fuel reprocessing facilities are not expected to contribute to the radioxenon background significantly, as they operate on fuel that had been removed from the reactor core and cooled down for a significant amount of time, usually one two several years. The contribution of NPPs and MIPFs to the global radioxenon emission inventory is fairly well understood. The MIPFs are the main source of radioxenon in the atmosphere (Saey 2009).
Kalinowski and Tuma (2009) presented the first global radioxenon emission inventory of NPPs for a generic year. They found that 1.3 PBq of radioxenon isotopes are released by 439 NPPs per year. They used the effluent reports of several years to calculate best estimate emission data for 111 NPP sites in North America and Europe and for the remaining 84 sites for which no reported data were available they applied a typical release estimate for an individual NPP reactor unit.
In order to establish a baseline and understand the impact on the atmospheric radioxenon background at IMS stations (Gueibe 2017) compiled the best estimate of the global emission inventory for the year 2014 for the four CTBT-relevant radioxenon isotopes. This is based on peer-reviewed publications about emissions from seven MIPFs and 153 NPP sites. Their study shows fairly good agreement between estimated and observed radioxenon activity concentrations for 133Xe at IMS radioxenon systems that are located in areas with elevated background concentration levels. For other locations and for the other three radioxenon isotopes there is no agreement between the observations at IMS stations and the simulations that are based on the best estimate for the emission inventory. This applies to the average activity levels over a calendar year and even more so when comparing observations and simulations on the level of individual sample collection times. As a result, it was found necessary that further endeavours are undertaken to improve the knowledge on radioxenon emissions.
The global emission inventory of the NPPs was recently updated specifically for the year 2014 with the reported values whenever available by Kalinowski and Tatlisu (2020). The total radioxenon emission inventory of 174 NPP sites operating in 2014 and seven MIPFs are listed in Table 1.
In most studies, NRRs have been considered as a comparatively small source of radioxenon that is typically neglected for the global emission inventory for understanding the background at IMS stations. In contrast, Hoffmann/Berg (2018) found that the research reactors used for medical isotope production themselves are important contributors to the radioxenon background. Based on observations at nine IMS monitoring locations their study shows that the NRU reactor alone is typically responsible for roughly 1–5% of the inventory of emissions from the Canadian Nuclear Laboratories site, when fully operational and manufacturing medical isotopes.
Besides the four radioxenon isotopes used for nuclear explosion monitoring, there are radioxenon isotopes which are created only by activation and not by fission including 125Xe and 127Xe. The HIFR in Oak Ridge, TN (USA) is known to release these isotopes. If included at sufficiently high concentrations in air samples, these radioxenon isotopes could interfere with the signal of the CTBT-relevant isotopes and the analyst would need to make appropriate corrections for these interferences. These non-traditional radioxenon isotopes are beyond the scope of this paper and need to be addressed in future work.
This paper establishes the global emission inventory of all NRRs in order to facilitate a scientific robust decision whether to take NRR contributions into account or not with regard to studying the background at IMS stations. The main question is whether the source term of NRRs is below the level that may have an impact on the CTBT monitoring system. It was found by Bowyer (2013) that a release level below 5 GBq/day has a very low probability of being detected at the typical distance of the closest IMS station. The overall contribution of releases from NPPs to radioxenon observations at IMS stations is well known and specific detections were attributed to known emissions from certain NPPs, specifically in the form of puff releases at the time of a power shut-down and start-up (Ringbom et al. (2020). When the reactor power is ramped up or decreased or when the reactor is completely shut-down, the radioxenon concentration increases due to pressure and temperature changes that drive gas out of the fuel cladding microscopic cracks and from the water into the air. While NPPs usually keep their power constant and are shut down for revision and re-fuelling with typical periods of 16–18 months, NRRs are frequently shut-down and restarted due to many reasons such as refuelling (3–5 times per year), R&D experiments, or simply operational schedules along working days and hours. In addition, there are various other activities that may be conducted in research reactors which can cause a prompt routine release of a significant amount of radioactivity. This includes the opening of irradiation capsules, regular refuelling in on-line fuelled reactors, etc. Such routine puff emissions can vary by orders of magnitude depending on what activity is conducted. Therefore, puff releases are expected to occur more frequently from NRRs.
Besides the total activities released, an important aspect is the capability of discriminating the signatures of nuclear facility releases from nuclear explosions. It has been shown in theory by Kalinowski and Pistner (2006) and then with real observations at IMS sites by Kalinowski et al. (2010), that, even though releases from NPPs are low compared to those from MIPFs, they may be observed by the IMS and their isotopic signatures may be used to characterize them. Being in the nuclear reactor domain, the signatures cannot be confused with one from a nuclear explosion except for releases from fresh fuel elements within the first 2–3 weeks of operation. It has been shown by Tayyebi et al. (2017) that a very similar isotopic ratio trajectory may be observed in the 4-isotope plot for a research reactor, if operated in a reactor power cycle as an NPP. However, puff releases may show different isotopic signatures. For example, Saey (2009) demonstrates that the isotopic signatures of fresh releases from medical isotope production facilities are found in the nuclear explosion domain. These signatures are caused by the irradiation of production targets in research reactors for several hours up to a few days with subsequent decay periods of similar duration. This paper explores not only the detectability for releases of NRRs at IMS stations but also how they can be discriminated against nuclear explosion signals using isotopic ratios.
1.2 Literature Review on Radioxenon Emissions from NRRs
The radioxenon emissions of NRRs have not yet been comprehensively investigated. Research reactors differ from power reactors in the wide variety of designs and modes of operation, as well as a wide range of neutron applications. The primary purpose of research reactors is to provide a neutron source for research and other purposes such as testing the nuclear fuels and different materials, investigating nuclear and neutron physics, irradiating biological and medical samples, and production of radioisotopes. In terms of thermal power these facilities are smaller than the NPPs. This implies a lower fission rate and consequently a smaller radioxenon reactor inventory. On the other hand, NRRs may have a less complex containment and retention system. NPPs are often equipped with facilities that allow for an extended retention time between production and emission that allows the effluents to undergo decay before being released. NPPs are continuously operated for most time except for periods of revision and fuel reloading. NRRs have different operational working schedules from a few hours to several days that make the production and release patterns more complex. NRRs often have facilities and operational functions that expose air to neutron fluxes and as a result they may release a comparable or higher contribution of radioxenon from activation in addition to the fission source. Therefore, it is not obvious that NRRs have always significantly lower emissions of radioxenon than NPPs have.
Whereas releases from nuclear power plants are well known, there are only few publications on nuclear research reactors. In most cases the information is not available for specific isotopes but only in an aggregated way. For example, from 1993 to 1996, annual releases from two research reactors in Obninsk, Russia are reported to be in average 0.7 PBq for all noble gases (UNSCEAR 2000).
Table 2 summarizes the available values of annual activity releases of radioxenon isotopes from research reactors that are based on measurements. They are of different quality. The best quality can be expected, if the release was continuously measured and reported as annual release by the operators. In other cases, the operator reported a conservative estimate based on own experience. Some other release values are derived from a few representative measurements within the facility by converting them to a release rate and extrapolating it to the emission for a whole year. In one case, atmospheric observations are used to estimate the source term. For simplicity, we refer to all results given in Table 2 as release values and reserve the attribute “estimate” for our extrapolation to other reactors by a calculation using the Booth equations derived in this paper. The release values that are derived from measurements are ordered by the nominal reactor power ascending from top to bottom. Below we address them in the same sequence.
-
The NRU in Canada has the third largest thermal power (135 MW) of all NRRs in operation in 2014. Though no emission reports are available for the NRU, Hoffmann/Berg (2018) have inferred its release strength from remote observations of atmospheric concentrations at nine IMS monitoring locations. They found that NRU reactor alone releases about 1–5% of the total inventory of emissions from the Canadian Nuclear Laboratories site, when fully operational and manufacturing medical isotopes. This is inferred from atmospheric observations and atmospheric transport modelling to be 1.5E+16 Bq/year (Wotawa et al. 2010). Accordingly, the source strength of the NRU for 133Xe is between 1.5E+14 and 7.5E+14 Bq/year.
-
ORNL reports annual emission of noble gases from the High Flux Isotope Reactor (HFIR). The data presented in Table 2 are the median calculated from the 10 annual emission reports (2007–2016) (ORNL 2016).
-
Matuszek (1975) measured the releases of noble gases from different types of NPPs and within the pressure vessel of a heavy water moderated pressurized (HWPWR) nuclear research reactor and estimated the annually released amounts. These are estimated as being not as high as that of the studied NPPs. The results shown in Table 2 are the highest annual releases of noble gases. Matuszek et al. have measured only one sample and it is unclear whether the sample taken at the certain time of 4-week operating cycle is representative for the whole year and therefore, it is mentioned that the result for the HWPWR must be considered with caution (Matuszek et al. 1973).
-
The only source for HANARO research reactor is a report in Korean (HANARO 2006). We have used the data of Table 8, which is stated to represent a conservative estimate. The report does not specify how much higher these estimates are compared to the real release.
-
Activity concentrations that were measured in the reactor hall of the FRM II research reactor (Fontaine et al. 2021). The annual releases are included in Table 2. These are derived under the assumption that due to the fast purge of air in the reactor hall, the isotopic ratios at time of emission can be approximated by those of the concentrations measured in the reactor hall.
-
OPAL (Open Pool Australian Light-water reactor) is a research reactor located at Lucas Heights, Syndey, designed and built by Argentine company INVAP. In the reactor’s Safety Analysis Report, INVAP postulate annual releases [see table 12.2/5 in (INVAP 2004)]. We have adopted this data as a best estimate, though the releases most likely are conservative. The report makes no statement that allows to draw a conclusion on how large the difference to real emissions is.
-
The RA3 research reactor in Argentina is used to produce mainly 99Mo and 131I. Annual noble gas releases from RA3 were reported for 2005–2015 and the ranges from 7.53E+08 to 1.19E+11 for 135Xe and from 3.72E+08 to 1.75E+10 for 133Xe. The median of the emissions of all 10 years is entered in Table 2 (Quintana Domínguez 2016).
-
The noble gas releases from NETL TRIGA Mark II research reactor in the University of Texas at Austin has been measured in different locations around the reactor. Making use of measurements reported by (Johnson et al. 2017) the annual release of 133Xe is estimated and the release of the other isotopes are scaled from 133Xe using isotopic ratios for activation of air by epithermal neutrons (Klingberg 2013). The calculation is described in Sect. 2.2 and the results are presented in Table 2.
-
The radioxenon concentrations in the air above the TRIGA Mark II research reactor in Vienna have been measured by a SAUNA II system during the normal operation of the reactor (Steinhauser et al. 2012; Lechermann/Biegalski 2010). The reactor pool was covered with a plastic tent to reduce mixing with surrounding air. More detail on the measurements in that reactor is given in Sect. 2.1. The release data presented in Table 2 are estimated based on these measurements and their calculation is described in Sect. 2.2.
2 Derivation of Radioxenon Emissions for Two TRIGA Research Reactors
2.1 Vienna TRIGA Reactor
The Vienna TRIGA Mark II research reactor is in operation since March 1962 and has a continuous power output of 250 kWth. It is loaded with 81 fuel elements, 55 of which are aluminium cladded elements of type 102 (19.75% U-235 enrichment, 8.5 wt% U) and remain from the initial criticality loading. The rest are stainless steel cladded elements of type 104 (19.75% U-235 enrichment, 8.5 wt% U) and type 110 (70% U-235 enrichment, 8.5 wt% U) which had been added to the core later.
In 2009, two measurement campaigns were performed at the Vienna TRIGA research reactor (Lechermann/Biegalski 2010). The main objective of the experiment was to identify the ratios of different xenon isotopes produced in the reactor during different modes of operation. In addition, a separate study of the radioxenon signature from an irradiated highly enriched uranium (HEU) target was performed. All samples were collected and the xenon extracted with a mobile sampler of the Swedish Automatic Unit for Noble Gas Acquisition (SAUNA II) and the transport columns containing the xenon were shipped to Stockholm for measurement of the radioactive content with a SAUNA II system at the FOI laboratory.
The background samples were taken in the reactor hall after at least two full days without reactor operation. Air samples were collected on top of the reactor pool, about 7 m above the core. A plastic tent with a volume of about 10 m3 was mounted to cover the pool surface in order to reduce the exchange of air. Samples were collected at different reactor modes (reactor off; reactor at full power with primary cooling on or off).
The Vienna TRIGA reactor has a total of six ‘dry’, air-filled irradiation tubes, one central irradiation tube and five reflector irradiation tubes. The irradiation tubes were kept closed by rubber plugs during sampling from the covered volume. It is standard procedure to close off irradiation tubes that are not in use. This also allowed to distinguish the different release pathways. The sample from the irradiation tubes was a composite sample from the five reflector irradiation tubes.
During the first campaign several challenges emerged that eventually meant that only the background sample had meaningful and reliable results. High humidity as well as carbon dioxide, being released from a warming reactor pool, posed a major difficulty for the SAUNA II system that has been designed for sampling atmospheric air. The sampling method was improved for the second campaign, which included a basic humidity filter that was used for all samples. This study uses only data of the second campaign.
After the second campaign, a leaking fuel element was identified and removed from the core. Gas bubbles emerging from the fuel element as it was handled have been collected and analysed by HPGe gamma spectrometry (Steinhauser 2012).
For the purpose of this study, we will focus on the samples that were taken during full reactor operation at 250 kWth and on the sample that was taken from the irradiation tubes. Table 3 shows selected data for total radioxenon emissions based on measured radioxenon concentrations. Note, that during the time of measurement, the leaking fuel element was still present within the core.
The samples contained 133Xe activity concentrations of the order of a few up to 67 Bq/m3. The results for 131mXe ranged from 0.01 up to almost 1 Bq/m3, and the 133mXe concentrations were between 0.06 and 14 Bq/m3, where the highest value was obtained from the irradiated target. The results for 135Xe were hampered by the relatively long transport time from Vienna to Stockholm, but still, this isotope was detected in six of the samples. It should also be mentioned that the non-CTBT relevant isotope 125Xe was detected in several of the samples. This isotope is created by neutron activation of 124Xe in air.
For samples 3–7, measured activity concentrations were extrapolated to the total catchment volume of 10 m3 under the plastic cover based on the assumption of constant and homogeneous activity concentrations during sampling. These samples were taken during 3.5 h and 1 h (samples without active cooling) of reactor operation at full power.
Lastly, a composite sample from the five reflector irradiation tubes was taken during and after an irradiation time of 2 h and 8 min. Residing air was pumped out and fresh air allowed to fill the irradiation tubes before the irradiation started. This approach was chosen in order to irradiate and sample fresh atmospheric air and avoid unintended build-up. The approximate thermal neutron flux at the bottom 50 cm of the irradiation tubes was estimated to be 1.7 × 1012 cm−2 s−1. The five irradiation tubes had an estimated combined total volume of about 50 L and an active volume of 2400 cm3, which is exposed to the neutron flux. The much larger sampling volume of around 0.19 m3 is due to dead volumes in the connection to and within the air sampler. The stable xenon yield of well over 100% is likely due to the small amount of total xenon in the sample and the large relative uncertainty associated with it. The large uncertainty propagates to the total activities, which we estimate as 80%. The sampling process was preceded by completely emptying the irradiation tube system that was well-sealed to make sure not to draw in any air from outside.
Based on above results, the radioxenon release from the reactor during normal operation is estimated. Results from samples 3 to 6 were combined to calculate the average radioxenon release rate from fission, disregarding the distinction in cooling modes. The activity of each sample is measured, and the activity concentration is calculated by dividing the activity by the air volume representative of the stable xenon detected in the sample using the abundance of xenon in atmospheric air (87 ppb). The yield relates the representative air volume to the sampling unit’s air flow meter. In case the sample was taken from underneath the tent, the total activity as reported here included the unsampled air under the tent multiplied by the activity concentration as determined in the sample.
Regarding sample 7, there is good agreement with Klingberg et al. (2013) for the ratio of 133Xe/135Xe, but the ratio of 133mXe/131mXe is drawn significantly to the domain of fission generated radioxenon. In addition, both metastable isotopes show lower than expected activities compared to 133Xe and 135Xe under the assumption of pure activation. Both observations point to the possibility of internal contamination of the irradiation tubes with uranium. Despite this and apparent discrepancies with the Texas TRIGA irradiation studies (Klingberg et al. 2013) the observations are in accord with reported and visualized results in Fig. 2 of Steinhauser et al. (2012), agreeing fully with the experimental data within the range of uncertainty. We find it quite unlikely that experiments and simulations in this peer-reviewed publication have flaws and yet happen to be in good agreement.
We further used the measured release rate from the composite sample taken from the irradiation tubes to calculate the release rate from activation in air. In this case the relevant volume is the small sample volume plus the total volume of the irradiation tubes. Since the air sampling was conducted after the irradiation had stopped, the production rate by activation is equal to the activity divided by the irradiation time. This can easily be scaled to the release of a whole year by scaling the time up to 365 days and multiplying the result with the capacity factor (see Sect. 3.2). The sum of fission and activation generated radioxenon release rates added up represent the best estimate for the reactors’ total annual radioxenon release rate. The estimated results of total annual radioxenon emissions from fission in the fuel and activation in irradiation tubes are summarized in Table 4.
2.2 Texas TRIGA Reactor
The noble gas releases from NETL TRIGA Mark II research reactor in the University of Texas at Austin has been measured by Johnson et al. (2017) in different locations around the reactor. By making use of these reported measurements, we estimate here the annual release of 133Xe. An estimate for the annual release of the other isotopes is achieved by multiplying the annual release of 133Xe with scaling factors that apply for the isotopic activity ratios after 30 min of activation of air by epithermal neutrons (Klingberg et al. 2013). The details of this calculation are as follows.
Johnson et al. (2017), have measured the 133Xe, 37Ar and 41Ar releases during different operational modes of the TRIGA reactor and various locations around the reactor. The operation mode relevant for the release estimate is full operation of the TRIGA reactor at 950 kW. We are using here the measured concentrations of 133Xe at the two stacks: 2.216 Bq/m3 for the argon purge stack and 0.508 Bq/m3 for the HVAC stack. The flow rate is 0.52 m3/s for the argon purge stack and 3.4 m3/s for the HVAC stack. With these data, the annual release R133 of 133Xe can be calculated with the following equation:
where Top [h] is operation time within one year, \(\dot{R}\) [Bq/h] is release rate during full operation at 950 kW, CF is capacity factor as a number between 0 and 1, ci [Bq/m3] is concentrations with i = 1 for argon purge stack and i = 2 for HVAC and vi [m3/h] is stack flow rate.
The result for the 133Xe release rate is 1.04E+04 Bq/h. This corresponds to a power-specific 133Xe release rate of 10.9 Bq/kWh. By considering the capacity factor of 15.4% for this reactor, the annual release can be estimated as 1.40E+07 Bq/year.
Accordingly, the scaling factors sf for the other radioxenon isotopes relative to 133Xe at the time of their generation, i.e. without decay, can be determined to be 2.21, 0.879 and 4.57 for 131mXe, 133mXe, 135Xe, respectively. Using these scaling factors, the release rates are calculated with the following equation:
where Ri [Bq/year] is annual release rate of isotope I, \(sf\)i is scaling factor according to Klingberg et al. (2013) and index i is 131 m, 133 m, 135.
The results are entered in Table 2.
3 Methodology for Extrapolating the Emission Estimates to Other Radioxenon Isotopes and to Further NRRs
3.1 Overview of the World-Wide Fleet of Research Reactors
The world-wide fleet of nuclear research reactors spans a much larger range of varieties than the nuclear power plants. This can be demonstrated specifically with the many orders of magnitude of thermal power between the smallest and the largest reactors.
Table 5 shows the number of different categories of NRRs in terms of thermal power that were in operation world-wide at different times as can be seen in the Research Reactor Database RRDB (IAEA 2019).
As of 23 January 2019, there are 227 operational research reactors in the world with wide ranges of thermal powers varying from 0 to 250 MW, of which 25 are zero power. The research reactors with more than zero power can be classified in 3 categories by thermal power: less than 1 kW (48 reactors), 1 kW–1 MW (64 reactors) and higher than 1 MW (90 reactors). The total power of the world's 227 research reactors is 2.16 GW. This is in the order of magnitude of the thermal power of one large-size NPP. For comparison, as of 23 January 2019, there are 453 nuclear power plants in operation in 30 countries with total net electrical capacity of 400 GWe (IAEA 2015). It should be noted that 49 of the research reactors in operation as of 23 January 2019 are high flux reactors, i.e. they have a neutron flux of ≥ 1014 cm−2 s−1.
There are more designs for NRRs than for NPPs. Table 6 shows all NRR types and the number of each type that was operational in 2014. The information is retrieved from IAEA (2015). A frequently used design (53 out of 160 reactors) is the pool type reactor where the core is in a large pool of water as a moderator and coolant. Tank type research reactors are similar. When counting only reactors that have a thermal power above 10 MW it is still the type with the largest number of representatives (15) followed by the 14 tank type reactors of which in total 23 were operational in 2014. The TRIGA (Training Research Isotopes General Atomics) reactor is also a wide-spread design with 35 units, however only one of them has a power larger than 10 MW. Though TRIGA reactors have an open pool of water, they are considered as a separate reactor type because they form a homogeneous sub-class. There are other designs that are moderated by heavy water or graphite. A few are homogenous, where the core is a solution of uranium salts as a liquid. There is just one high temperature gas (HTG) research reactor (in China) with 10 MW thermal power.
3.2 Operational Modes, Neutron Flux and Capacity Factors of Nuclear Research Reactors
NRRs have a large range of different operational working schedules from a few hours to continuous operation that make the production and release patterns complex. The working hours of all the research reactors can be found in the IAEA Research Reactor Database RRDB (IAEA 2019). With that database, one can calculate the capacity factor, i.e. the fraction of time that the power is up during a year.
Some research reactors work continuously 7 days a week for 24 h per day, i.e. 8760 h per year (8784 h in a leap-year). For these reactors the capacity factor is 100%. Most research reactors have a non-continuous operational mode. The following examples apply to reactors that play an important role in the study presented here:
-
Canada's National Research Universal (NRU) reactor was operated at a capacity factor of 80% (World Nuclear News 2018).
-
High flux isotope reactor (HFIR) in the USA has an operational mode of 24 h per day for 7 days a week in 24 weeks of the year, i.e. a capacity factor of 46.0%.
-
HANARO is operated continuously as well but for 28 weeks per year and has a capacity factor of 53.7%.
-
The FRM II operates 24 h for 7 days a week as well, but in 34 weeks of the year, i.e. its capacity factor is 65.2%.
-
The OPAL reactor in Australia has a capacity factor of 100%.
-
The RA-3 reactor in Argentina works 24 h per day for 5 days per week in 46 weeks per year. Its capacity factor is 63.0%.
-
The capacity factor for TRIGA II reactor at University of Texas at Austin for an operational mode of 6 h per day for 5 days per week in 45 weeks per year is 1350/8760 = 15.4%.
-
The working hours of the TRIGA II at Vienna are 7 h per day times 5 days per week for 48 weeks per year. This results in a capacity factor of 19.2%.
For those reactors for which no data on their operational mode are available, we use the median of the available data which is 1152 h per year and that is equal to a capacity factor of 13.2%.
3.3 Isotopic Ratios of Radioxenon Generation by Fission and by Activation
Fission and activation are the two production paths of radioxenon in research reactors. Both contributions need to be taken into consideration for the estimation of the radioxenon emissions from NRRs. For most reactors, one of the two production processes is the dominating source of the emissions.
The isotopic ratios of annual average releases are plotted in Fig. 1 for the HANARO, HFIR, TRIGA II Vienna and FRM II reactors (see Sect. 1.2; Table 2). In addition, the operational isotopic ratio trajectories are marked for an NPP (Kalinowski et al. 2010) and for the TRR (Tayyebi 2017). Finally, the isotopic ratios resulting from activating atmospheric air by thermal and by epithermal neutrons for 30 min, as found in Klingberg et al. (2013) are marked for different times of decay after 30 min of irradiation. The highest datum represents no decay. For thermal neutrons, the lower markers represent decay of 17, 41, and 65 h. For epithermal neutrons, the lower markers represent decay of 6, 53, and 82 h. These are found at a clear distance from the equilibrium points in the trajectories shown for the NPP and the TRR. The results of the different Booth equations for pool, TRIGA and other reactors (see Sect. 3.5) are marked as well. The plot shows that the isotopic compositions resulting from fission and activation can in most cases be clearly distinguished. The isotopic ratios by fission according to the Booth plot and activation by epithermal neutrons according to Klingberg et al. (2013) are compared in Table 7. With a different neutron spectrum, the isotopic ratios from activation may be different.

4-isotope activity ratio plot
Specifically, for the Vienna TRIGA II, the activation products are found closely to the fission products on the four-isotope plot in Fig. 1. This is in disagreement with the radioxenon ratios reported by Klingberg et al. (2013). The study shows perfect agreement between simulations and measurements of irradiated air samples. According to Steinhauser et al. (2012) simulation for irradiating air at the Vienna TRIGA reactor is in full agreement with measured samples irradiated at that reactor. However, contrary to the expectations, the results of these two papers are not in agreement to each other. Surprisingly, the Vienna TRIGA data for radioxenon from activation in the sealed-off irradiation tubes show isotopic ratios like those of radioxenon from fission.
An important difference was accounted for in the simulations of Klingberg et al. (2013) and subsequently in the use of the data in this paper. Most activation in the Texas TRIGA reactor occurs by irradiation of air dissolved in the moderator water Fay (2014), whereas the major source of activation products in the Vienna TRIGA reactor takes place in the irradiation tubes., Specifically for the two metastable isotopes of Fig. 1, there is a remarkable difference that is not yet understood.
It should be noted that the main origin of the 133Xe and 37Ar released at the Texas TRIGA reactor at the time of the study undertaken by Klingberg et al. (2013) was concluded to be activation Biegalski et al. (2018). Based on the radioargon activity concentrations, the expected activated radioxenon activity concentration was validated. The numbers for calculated and measured 133Xe match up within the expected range of uncertainties. Monthly surveys on the pool water of the Texas TRIGA reactor did not reveal any fission products. This provides additional confirmation for activation being the dominant source, in this instance.
This confirms that under favorable conditions both production mechanisms can be distinguished by their respective isotopic ratios. The exception to this rule is when the isotopic ratios from activation fall into the same domain as the fission products. Therefore, for a reactor that has an isotopic composition in the main cluster of activation around 133mXe/131mXe = 0.1 can be deduced to have a release that is dominated by activation generation. The entries of the isotopic ratios for the HFIR reactor are close to the activation ratios, whereas the other three research reactors are close to the equilibrium point of the NPP and the TRR. This confirms that the emissions from some NRRs are dominated by activation and for others dominated by fission.
Of course, these observations apply only for the releases from the stack. As soon as the release enters the atmosphere it gets mixed with radioxenon background from other sources. It was already shown by Kalinowski and Pistner (2006) that the mixing of multiple emissions moves the resulting isotopic ratios towards the left, i.e. from the fission domain into the activation dominated area of the four-isotope plot. The marker for accumulated activation demonstrates this effect. It indicates where isotopic ratios can be found when allowing for accumulation of continuously generated radioxenon over extended time while decay applies (see Table 10). This may give a clue at the possible reason for HFIR entries being shifted towards the left from the simulated entries.
Even though it cannot be expected that the clear pattern as shown in Fig. 1 persist, the conclusions that can be drawn have still relevance for interpreting observations of the IMS noble gas system. Specifically, reactors that have emissions dominated by activation-generated radioxenon may also release other activation products like 127Xe at activity levels that could have an impact on IMS spectra.
The Vienna TRIGA II reactor shows a mix of fission and activation production. This results in its entry in Fig. 1 being shifted a bit away from the fission-generated entry (the marker for the sum of fission and activation releases is almost hidden behind the fission marker, appearing like a shadow of the fission marker). Fortunately, for this reactor separate estimates can be made for releases from fission and from activation production (see Sect. 2.2). The isotopic ratios for each of these specific sources are marked in the 4-isotope activity ratio plot.
There are no release reports for radioxenon emissions from the NRU other than for 133Xe. Therefore, it cannot be included in the four-isotope plot. Instead, we refer to Fig. 8 in Hoffman and Berg (2018). The environmental measurements made at Ottawa while the medical isotope production was shut down were shown to be dominated by emissions from the NRU. The isotopic composition of these observations is clustered around the Hanaro NRR isotopic ratios with extremes getting close to the activation ratios one side and touching the nuclear explosion domain on the other.
An additional entry is made to Fig. 1 for the Molten Salt Reactor (MSR). This entry is based on the MSR Experiment (MSRE) conducted at Oak Ridge National Laboratory (ORNL) from 1965 to 1969. The estimated daily emissions of all four radioxenon isotopes are reconstructed based on a timetable of the MSRE reactor power (Burnett 2019). In the four-isotope plot the MSR is found as the highest reactor ratio for 135Xe to 133Xe and in the nuclear test domain but very close to the separation line. This shows that special reactor types may be found to have isotopic ratio characteristics unlike all other known reactor types. MSRs could have a larger source term than current reactors with sealed fuel because they do not have the barrier of a fuel cladding.
Since there is evidence that radioxenon releases from some nuclear research reactors are dominated by activation and most others are dominated by fission, the question arises whether any technical features can be used as indicators whether fission or activation might be dominating. Tight fuel cladding is an important factor for minimizing fission gas releases and the intensity of irradiation activities (high neutron flux, large irradiation tube volume, allowing ambient air rather than inert gases to flow through irradiation tubes) has an impact on enlarged production and release of activation products. Most of these parameters are hard to get information about. The neutron flux is available from the Research Reactor Data Base (IAEA 2019). In Fig. 2, for each nuclear research reactor in operation in 2014, the neutron flux is plotted against the capacity factor multiplied by thermal power, which defines the fission rate. The fission gas production rate is linear with the fission rate whereas the activation rate scales with the neutron flux and other parameters that are not sufficiently known. The plot shows that reactors with the same fission rate can reach a range of neutron fluxes that spans several orders of magnitude. The HFIR is not only the reactor with the second highest neutron flux but also is found at the upper end of neutron fluxes that reactors with a similar fission rate achieve. A similar situation applies to the FRM II and its radioxenon releases might also be dominated by activation. However, Table 1 shows that this is not the case and this example demonstrates that no simple conclusion can be drawn. The two TRIGA reactors are not at the top of the neutron flux range and still the radioxenon releases of one of them (Texas TRIGA II) appear to be dominated by activation.

Scatter plot of neutron flux versus capacity factor multiplied to thermal power for each nuclear research reactor in operation in 2014
In the following sections, the method for estimating emissions from fission and activation are described.
3.4 Scaling the Radioxenon Fission Production Rate by Thermal Power
A common expression used for fission birth rate in terms of generation of activity inventory per time interval for a certain fission product is proportional to the fission rate F [see, for example, Bonka (1982)]:
where Bf [Bq/year] is fission birth rate, λi [1/s] is decay constant of the radioisotope of interest indicated by I, F [1/s] is fission rate, Yi is cumulative fission yield of the radioisotope of interest and Tsy is 3.1536E+07—number of seconds per year.
The fission rate F depends on the power of the reactor and its capacity factor. As a first linear approximation, the fission birth rate can be calculated from the following equation:
where CF is capacity factor as a number between 0 and 1, P [MW] is thermal power of the reactor, Ef [MeV] is typical fission energy release deposited in the reactor per fission event, it is about 200 meV, and 1.6E−19 is the factor that converts MeV to MJ.
The fission yields of the radioxenon isotopes from fission of 235U are given in Table 8. We apply the cumulative yield in the calculations to consider all the radioxenon isotopes produced through the decay chain.
3.5 Scaling Radioactive Noble Gas Emissions from Fission Production Rate
Due to the sparse data of radioxenon emissions from NRRs (Table 2), we explore a half-life dependent approach to determine a best estimate for the release of all four CTBT-relevant radioxenon isotopes. Due to the dependence of fission gas releases from reactor designs, we apply this approach to TRIGA and pool reactors. For any reactor of these two types, the annual emissions are estimated by extrapolations from empirical data and a theoretical understanding. Obviously, the higher the production rate, the larger the inventory that can potentially be released. The emission rate depends on the containment effectiveness of the different compartments: from the fuel through cladding into the water, further into the reactor hall and out through the stack. The longer the residence times are, the more activity has decayed before the release takes place. As a result, short-lived radionuclides have a smaller release rate than longer-lived isotopes.
Figure 3 shows the dependency of the release to birth rate ratios (R/B) for different noble gas isotopes as a function of the decay constant λ for normal reactor operation during steady state conditions (Lewis 1989). This graphical representation is known as the Booth plot because it goes back to the original idea of (Booth 1957). It has the advantage of providing an easy approximation of release rates without the need for a full understanding of the exact retention and transport mechanisms through the various compartments. The following equation can be fitted to the data in the Booth plot and describes the total transfer from the location of its generation in fuel to the gaseous effluents (Bonka 1982).
where R [Bq/year] is release rate, B [Bq/year] is birth rate, k is emission rate constant, λ [1/s] is decay constant of the radioisotopes of interest and α is exponential.

The Booth plot of Bonka (1982) that inspired our approach used empirical release data of NPP that were measured in 1969. It had a slope of α = 0.636. In the meantime, the containment and retention mechanisms have been enhanced significantly. The underlying theoretical approach of the diffusion equation is still valid. Therefore, the results need to be adjusted to recent empirical data.
Figure 3 provides a comparison between the old NPP releases and more recent NPP emissions (Gueibe 2017). The upper part shows data of Bonka (1982) that were measured at a BWR in 1969 (Bonka 1982). In the lower part of the plot, the data of show the average R/B for a typical NPP in 2014. The best estimates for release rates of NPPs in 2014 are given by Gueibe et al. (2017) and for the birth rate we are using the average thermal power of all 389 NPPs that were in operation in 2014 (2.90E+06 kW) approximated by multiplying by a factor of three the average electric power as given by IAEA (2015).
The emission data have strongly reduced, about four orders of magnitude, and the slope with increasing decay constant has become steeper which implies longer retention times (Gueibe 2017).
There are few reported experimental data on radioxenon releases from NRRs (Quintana Domínguez 2016; Fontaine et al. 2021; INVAP 2004; Lechermann/Biegalski 2010; HANARO 2006; Matuszek et al. 1975; Hoffman/Berg 2018). To estimate the global contribution of all operational NRRs, the Booth plot might be applied but the result is not reliable because only very few empirical data are available and there are many different reactor designs. Instead we apply the Booth plot to visualize the wide spread of results and to identify opportunities for getting estimates for certain groups of reactors that show a relatively small spread.
If applicable to a group of reactors, the Booth equation is calculated as best linear fitting curve of the sparse data for the release to birth rate ratio, R/B, as a function of the decay constant λ of each isotope. With the birth rate calculated for each reactor of reactors belonging to the same group based on its thermal power and capacity factor (see Sect. 3.4), the resulting Booth equation parameters can be used to calculate an estimate for the release rate R for each of the four radioxenon isotopes of interest.
All NRR emission data used for this purpose are listed in Table 2 and the release to birth rate ratio, R/B, are visualised in Fig. 4 The value of B is calculated according to Eq. (5) for each reactor from its power and capacity factor as described in Sect. 3.2. In the bottom part of this set of plots, the data for all nine reactors are entered. The birth rates of the HFIR and Texas TRIGA reactor should be calculated for activation since this is the dominant production path. Since the birth rates are unknown, the fission product generation rate is used instead to allow for a comparison under the assumption that the release from fission would be equal to the release from activation. A huge spread of up to seven orders of magnitude can be observed for the longer-lived isotopes 131mXe and 133Xe. The spread gets smaller for shorter half-lives but is still about three orders of magnitude for 135Xe. This trend is due to facilities with larger emissions having a steeper slope of the Booth line. Having smaller release to birth ratios R/B for shorter half-lives may indicate a longer residence time.

Booth plot for NRRs, the numbers in the legend shows multiplication of thermal power in kW multiplied by capacity factor, the solid lines show the Booth equations. The left upper plot shows pool type reactors, the right upper plot is for TRIGA reactors. The lower plot contains all available data irrespective of reactor type
This huge spread among only a few reactors discourages from the attempt to create a global radioxenon emission inventory for the global NRR fleet from this sparse data set. In order to account for the large variety of different NRR designs, a separate Booth equation should be determined for each type of NRRs in combination with an indication of the uncertainty range as determined. This can only be done for an NRR type, if a minimum number of representatives of that reactor type are available. The nine reactors for which we have release estimates from publications represent four pool reactors (FRM II, HANARO, OPAL and RA3) and two TRIGA reactors (TRIGA Texas and TRIGA Mark II Vienna). The two heavy water reactors are too different from each other to create a group. As can be seen in the upper two sub-plots of Fig. 4 each group has representatives that still span two or three orders of magnitude. This approach is a first approximation and can be expanded as more data become available.
The Booth equation for a whole group is derived in two steps. First, a linear fit is done for each individual reactor representing this group for which release data are available. Next, the parameter α for the group is calculated as the arithmetic mean of the α values of all reactors representing this group. The parameter k is the log average of k for this parameter in each individual booth equation. The resulting fits of the Booth plot for these three groups of NRRs are shown in Fig. 4 located in the middle of the spread of individual booth lines for the reactors of the same group. The resulting Booth equations for reactor groups are described by the following equations. For pool reactors:
For the pool reactors this average Booth equation provides a best estimate with a lower and upper range for the spread of the four reactor examples for which we have release data. For two of these reactors (HANARO, OPAL), we are using conservative estimate (see Sect. 1.2) and therefore their fitting curves can be considered more conservative rather than realistic. As result, the best estimate and upper ranges for pool reactors is conservative.
For TRIGA reactors:
This equation is less reliable than the one for pool reactors since it is based on two rather than four example reactors. The two TRIGA reactor examples indicate a minimum spread of R/B ratios that may be expected to be found at TRIGA type reactors. The Booth line determined as log-average of the two individual TRIGA reactor booth lines can be used as a best estimate with an uncertainty range indicated by the upper case of the Texas TRIGA reactor and the lower case of the Vienna TRIGA reactor.
We create a third Booth equation from the data of all nine NRRs in order to create a baseline for a global radioxenon emission inventory and with the understanding that this is not reliable. For this reason, we refer to it as hypothetical radioxenon emissions. Future studies presenting more reliable results can use this baseline for comparison.
The Booth equation can be used to estimate the emission of each reactor and each type for which the equation is available by solving it for R and using Eq. (4) for B.
Solving the equation for R/(P \(\times\) CF) and converting the unit y into h with 1 y = 365.25 × 24 h, one can calculate the energy specific release rate E′ in Bq/kWh. The result for 133Xe is entered as last column into Table 9.
Table 9 summarizes all the parameters of all Booth equations determined in this study. In general, those values of k and alpha that are based on estimated emissions for the respective reactor are of high quality and reproduce with low uncertainty the known emission values. This is not true for HFIR and Texas TRIGA MARK II. The quality of their Booth equation is low because their emissions are dominated by radioxenon from activation. Therefore, the birth rate that is based on fission yields cannot be applied to estimate the releases in case they are dominated by an activation source. Specifically, the experimentally determined value for 133Xe is found much lower than the Booth equation at the λ value of this isotope. For HFIR, it is about two orders of magnitude lower, for the Texas TRIGA MARK II, it is a factor of five lower. A different approach needs to be used to estimate releases dominated by activation products. This is described in Sect. 3.6. The quality of the Booth equations for the groups of pool and TRIGA reactors is medium with large uncertainties due to the spread of input data over more than one order of magnitude. The uncertainties of release estimates are discussed in Sect. 4.1.
3.6 Scaling the Radioxenon Release Rate Originating from Activation
To determine the radioxenon production rate resulting from activation, we have empirical data available from the TRIGA reactor at University of Texas at Austin, from the high flux isotope reactor, HFIR at Oak Ridge National Laboratory, and from the Vienna TRIGA Mark II reactor. There are indications that the most radioxenon produced in HFIR is from neutron activation of air in the beam tubes. For the Texas TRIGA reactor no evidence was found that any release of fission products from the reactor core can be observed and the releases are dominated by activation occurring on gas dissolved in the moderator water (Biegalski et al. 2018). The isotopic activity ratios of the HFIR in Fig. 1 gives evidence that activation is the dominant production mechanism as well and according to Klingberg et al. (2013) it is cause by irradiation with epithermal neutron energies. However, the radioxenon releases from the Vienna TRIGA Mark II reactor are dominated by fission generation and the contribution of activation is smaller for every isotope. For 133Xe it is smaller by one order of magnitude (see Table 4). Therefore, it is not included here in the assessment of radioxenon releases from reactors that have activation as the dominating source.
In order to estimate the release rate, we estimate the power-specific release rate of 133Xe and then we derive the rates of the other isotopes from isotopic ratios established for activation. For the Texas TRIGA reactor, the power-specific release rate of 133Xe can be estimated as 1.09E+01 Bq/kWh (see Sect. 2.2). By using the isotopic ratio for epithermal neutron energies as reported in Klingberg et al. (2013), the power-specific scaling factors SF for the other radioxenon isotopes at the time of their generation, i.e. without decay, can be determined to be 4.98E+01, 9.60E+00, 2.41E+01 Bq/kWh for 135Xe, 133mXe, 131mXe respectively.
For HFIR, the median of 10-year annual releases of 133Xe is 1.92E+11 Bq/year. In view of the capacity factor or 46% and power of 85 MW, the power-specific release rate of 133Xe from HFIR is 5.59E+02 Bq/kWh. For the other isotopes the same calculation is applied. The power-specific release rate from HFIR is 1.51E+04, 2.18E+03, and 3.13E+03 Bq/kWh for 131mXe, 133mXe, and 135Xe respectively. For each isotope, the logarithmic average of the pair of values from TRIGA and HFIR are considered as scaling factor to estimate the radioxenon release from activation from any reactor. The scaling factors for 131mXe, 133Xe, 133mXe, and 135Xe are 6.03E+02, 7.81E+01, 1.45E+02, and 3.95E+02 Bq/kWh, respectively.
As stated above for the Booth equation for fission using data from all reactors, the results cannot be reliably applied to any specific reactor. The same applies here. For comparison, the resulting isotopic ratios are shown in Fig. 1 with a marker for “Activation, best estimate”.
4 Results for Radioxenon Emissions from NRRs
4.1 Distribution of Release Estimates for NRRs
Since the power of the NRRs is spreading over about 8 orders of magnitude from 1 MW to more than 100 MW and the capacity factor add further variations, the radioxenon emissions estimated as a linear function of the power multiplied by capacity factor is spreading over 10 orders of magnitude. This spread is shown in Fig. 5 which presents the distribution of hypothetical annual 133Xe releases from fission and activation by reactors ordered with increasing release. The 220 reactors did not all operate at the same time.

Distribution of the estimated 133Xe release as calculated with the Booth equation
The real uncertainty of these release estimates is not known. Taking the variability of radioxenon releases from NPPs into account that was demonstrated by Kalinowski/Tatlisu (2020), it can be assumed to range over several orders of magnitude. As an indication of the minimum uncertainty, we refer to the spread of the reported releases according to Table 2 that can be observed in the set of reactors representing the whole group. It propagates into the uncertainty budget for the release estimate. This effect is demonstrated in Fig. 6. For each reactor for which release information is available, this is marked in the distribution of releases calculated for each reactor. The markers used in Fig. 6 for upper and lower bounds are resembling error bars and are derived from the difference between estimated and reported annual releases. The upper bound for TRIGA reactors is not given by the Texas TRIGA reactor but by the Booth estimate. The reason for this can be seen in Fig. 4 where the orange marker for 133Xe release from the Texas TRIGA reactor is lying lower than the blue line of the booth equation for the TRIGA reactor type. This isotope is found at significantly lower activities than the other three isotope markers due to the specific isotopic ratios of activation. Therefore, no upper bound is marked by an error bar marker.

Distribution of 133Xe release by group of reactor types and within the group ordered by increasing release estimate
In Fig. 6, the distribution of 133Xe release estimates is repeated but now shown in three groups. Fairly reliable are the estimates for the annual radioxenon releases of pool and TRIGA reactors based on separate Booth equations in combination with an indication of the uncertainty range as determined in Sect. 3.5.
The third group applies to all other NRRs. It is a hypothetical release estimate based on the available data of all nine reactors. It is shown here for comparison purposes and no upper and lower ranges are indicated because no information is available about the uncertainties. The only available indication about the uncertainty range is given by the NRU and the HWPWR reactor. Both have relatively high R/B ratios compared to the other reactor types, and the NRU has a ratio that is about three orders of magnitude higher than the HWPWR. In summary, the extraordinary high release estimate for the NRU in comparison to all other NRRs can be explained by the combination of three factors and the NRU being at the upper end of the range of all three factors:
-
1.
The fission rate determines the production rate of fission products. The NRU is the third largest of all NRRs regarding the thermal power. With a capacity factor of 80%, it has one of the largest production rates of radioxenon among all NRRs.
-
2.
When comparing the average R/B ratios of reactors of certain types for which release estimates are available from the literature, the heavy water reactors to which the NRU belongs are at the higher end, the pool reactors in the middle range and the TRIGA reactors at the lower end.
-
3.
Within each reactor type, the individual representatives of that group cover a wide range of release rates over several orders of magnitude. Among the two representatives of heavy water reactors, the NRU reactor happens to be at the upper end.
In Fig. 7, a different representation is used to demonstrate the difference between fission and activation generated radioxenon. It shows the distribution of hypothetical annual 133Xe releases ordered by increased release per year. For most reactors, the release assumed to come from activation is slightly lower than that from fission. In the intermediate range of annual release levels, the release from activation are significantly higher. Since the hypothetical releases from activation use the same equation for all reactors, this difference is due to the specific equations for fission-based releases from pool and TRIGA reactors. This shows that the release would be higher than the best estimate for fission, if activation dominates the release.

Distribution of hypothetical annual 133Xe releases either from fission (green asterisk) or by activation (magenta plus) by all reactors ordered by increased release per year
In order to aggregate this distribution of estimated emissions for all NRRs, Fig. 8 shows the box and whisker plot for all 4 isotopes by fission and activation. The TRIGA reactors have in general a narrow range of release estimates at a comparatively low level. For the pool reactors, the upper quartile of 133Xe emissions from fission is slightly higher than for activation. However, for all other isotopes, emissions from activation are always higher for the same reactor power times capacity factor. If activation dominates, the activity release is slightly smaller than the estimate for 133Xe but for all other isotopes it is by two to three orders of magnitude higher. The usual experience is that 133Xe is the lead nuclide and most regularly observed in samples that contain only one isotope. It is rarely missing in samples that contain other isotopes, except for the longest-lived isotope, 131mXe. It is important to note that this sequence is reversed when activation is the dominating source. In that case, 133Xe is the isotope with the lowest activity, 131mXe is the lead isotope and 135Xe follows with the second highest activity release.

Box and Whisker Plot for all 4 isotopes by fission (in colour) and activation (in black and white)
4.2 Assessing the Maximum Spike Releases
In the case of NPPs, it is a well-known fact that the batch releases are much more likely to cause detections at IMS stations above the normal background than continuous emissions from this source type. See for example (Ringbom et al. 2020). The same is most likely the case for NRRs. Therefore, it is important to get an idea about the possible characteristics of the release maxima and their pattern over time (typical duration of a pulse release, frequency of pulse releases).
For an assumed continuous release of radioxenon, the maximum spike release can be assessed with a simply approach as described in the following.
We can estimate upper limits for a pulse release of xenon activation products, if we assume that the reactor containment is 100% tight and instead of a continuous release all radioactivity is held back for a certain retention period and then released in one pulse. The pulse will release the cumulative radioactivity according to the continuous release, i.e. the integral over the release rate with decay correction applied. For this assessment, we assume retention times of T = 1 day, 1 week, 1 month and 1 year. The accumulated activity A is calculated based on the daily release rate Rd as
Table 10 summarizes scaling factors s(T) for each isotope and retention time T that can be applied to each release rate given in Bq/day rather than Bq/year. It shows that one will never get a one-time release for 133Xe that is higher than 7.56 times the cumulative daily release, irrespective how long one allows the activity to accumulate. For 133mXe, an increase of a total daily release by an order of magnitude is never possible through accumulation followed by a puff release of all accumulated activity. For 135Xe, accumulated activity releases would always be lower than continuous releases due to the short half-live. 131mXe has the largest potential for increasing release rates due to internal accumulation.
For illustration purposes, Table 11 provides the value of the maximum one-time releases after accumulation from all nine research reactors for that an annual release estimate is listed in Table 2. The isotopic ratios resulting from activation products being accumulated over longer time is shown as an icon in Fig. 1. Its location is shifted down and left from the initial isotopic activity ratios of freshly generated activation products.
The estimate of a maximum release made above assumes a constant release rate under normal operational conditions being withheld for a certain time and allows the activity to accumulate.
There may be other causes of pulse emissions. The experience from NPPs is that radioxenon generated in the fuel from fission may accumulate in tiny gas bubbles in cracks of the fuel cladding. Fast temperature and pressure changes could lead to their release and cause a higher than usual fraction of radioxenon being released from the fuel through the water and into the plenum gas or reactor hall. This has the potential of giving rise to a pulse release with quite high activity. Besides of this effect, NRRs have additional processes that may lead to puff releases. For example, irradiation of atmospheric air in a target capsule placed into the reactor core exposing it to high neutron flux instead of filling the target container with gas that undergoes low activation. Upon opening the target capsule after irradiation is finished, the activated air may be released.
4.3 Comparison of NRR Emission Estimates with Other Source Types
The major sources of radioxenon are medical isotope production facilities (MIPFs) of which about 10 are in operation worldwide. All NPPs together have a global annual emission inventory that is in the same order of magnitude or even less than the annual emission of each of the seven largest MIPFs.
The global emission inventory of NPP relative to their combined total power generation is 97 Bq/kWh for 133Xe when an average capacity factor of 70% is applied. The nine NRRs listed in Table 2 have specific release rates ranging from 0.43 Bq/kWh (Vienna TRIGA) to 7.93E+05 Bq/kWh (NRU). At the lower range it is more than two orders of magnitude less than for an average NPP, at the upper end it is more than three orders of magnitude higher. The Booth equation for TRIGA reactors indicates a 20 times lower value, for pool reactors it lies at the same level as for NPPs (see Table 9). This comparison of power specific emission rates can be interpreted as NRRs having a significant overlap with NPPs but with an extreme upper end represented by the NRU reactor. Given that the power of NRRs is in general much smaller than of NPPs, it remains to be seen how the absolute release estimates of NRRs compare to NPPs.
Irrespective of assuming fission or activation as the dominating generation process, this study reveals that the NRRs with the highest emissions are not negligible when compared to typical NPPs. Table 12 shows the typical daily emissions for three classes of nuclear facilities in comparison to the activity of radioxenon generated by a very small nuclear explosion and the assumed 10% prompt release if it is conducted underground. This confirms that for 133Xe, the NRR with the largest release (NRU) is three orders of magnitude higher than for a typical NPP if fission is assumed to be the dominating source (~ 1 GBq/day). The second largest fission source among the nine reactors listed in Table 2 is as strong as a typical NPP. If activation is the major production mechanism, the largest NRR considered here (HFIR) has a release of the same order of magnitude than a typical NPP. However, for 131mXe and with activation being the major production mechanism, the release of the HFIR is two orders of magnitude above the typical release of an NPP. For 133mXe it is one order of magnitude higher and for 135Xe it is of the same order of magnitude. In both cases of NRU and HFIR, the NRR available release estimates are—depending on the radioisotope and on fission or activation—are much smaller than the signal of a nuclear explosion. However, for certain cases they are not significantly different. The 133Xe release of the NRU is only two orders of magnitude below and the 131mXe release of the HFIR only one order of magnitude below the 10% prompt release scenario for a 1 kt TNT equivalent underground nuclear explosion.
Figure 9 compares daily 133Xe releases of the nine NRRs in Table 2 (blue horizontal lines) with two types of data for the European NPPs, an estimate for a generic year (blue histogram bars) and the real release amounts as reported for 2014 (red bars). This depicts in more detail what has been explained above. Two NRRs have a release higher than from a typical NPP (~ 1 GBq/day), OPAL has a slightly higher one (1.4 GBq/day) but the release rate of the NRU reactor is three orders of magnitude higher (2.1 TBq/day) and even one order or magnitude higher than for any NPP shown in this figure. The HFIR has a release rate slightly below 1 GBq/day and this is of special interest since activation is assumed to be the source. The two TRIGA reactors have release rates that are lower than estimates and reports for any NPP included in this figure. The other four NRRs are in the middle range and lower than almost all NPP estimates but higher than the reported real release rates of some NPPs.

The tremendous difference between the estimated and real release rates for the NPPs highlights the extremely large uncertainties of all estimates. It can be expected that a similar uncertainty applies to the NRRs. This implies that their actual release rate might occasionally be higher by one or more orders of magnitude. When comparing the Booth equations for NPPs and NRRs, one finds that the slope α is steeper for NPPs. This indicates that NRRs have shorter retention times as compared to NPPs. This may imply that the containment of NRRs is not as tight as for NPPs.
Bowyer et al. (2013) determine 5 GBq/day as an emissions threshold for 133Xe below which a detection at IMS stations would have a very low probability when using a minimum detectable concentration (MDC) of 0.1 mBq/m3 and assuming the existing sites of medical isotope production facilities and NPPs as release locations. Eight NRRs stay below this value if a continuous release is assumed. However, the release of the NRU lies almost three orders of magnitude higher (2.1 TBq/day) than the above-mentioned emission threshold. It should also be noted that the NRR locations were not considered in above mentioned study.
To what extend the IMS noble gas systems may be affected by an NRR release depends not only on the source strength but also on the geographical proximity between the NRR and the closest IMS noble gas system, on the normal level of background concentrations at the IMS location and the simultaneous influence of other sources, and on the meteorological conditions. It is beyond the scope of this paper to provide a systematic analysis of these conditions on the possible impact of NRRs on the IMS sensors.
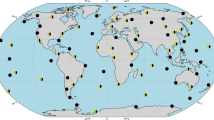
In order to facilitate an overview of geographic locations of NRRs and the IMS noble gas systems. Figure 10 shows a global map with all IMS noble gas systems and the 50 NRR that have the highest estimated hypothetical radioxenon source strength. The source strength scales with the product of capacity factor and thermal power. The cut-off used here for this parameter is 1.03 MW. All 50 NRRs that have this or a larger value are included on the map. If more than one NRR is co-located on the same site, this is indicated by a different colour.

Map showing all IMS noble gas systems and the location of the 50 NRRs with the highest estimated hypothetical radioxenon source strength
5 Conclusions
Only for seven NRRs reports are available about their annual radioxenon emissions. For two other NRRs their emissions are determined in this paper based on published experimental data. Though this data base is insufficient for the attempt to establish of a global emission inventory of the global fleet of NRRs, several conclusions can be drawn from the existing data.
The isotopic ratios provide evidence that most NRRs are emitting radioxenon that originates mainly from nuclear fission and for some reactors neutron activation is the dominant production mechanism. According to Fig. 1, activation-based releases are clearly discriminated from nuclear explosion domain and except for one case (Vienna TRIGA reactor) also distinct from fission-based reactor releases. Entries for the latter are grouped close to the discrimination line but still in the reactor domain. Special reactor types may be found in the nuclear explosion domain as the rare example of a molten salt reactor demonstrates.
In this paper, release values of nine NRRs for which experimental data are available are discussed in comparison to other relevant sources. For two reactor types (TRIGA and pool), experimental data of several reactors and multiple radioxenon isotopes are available. This enables the derivation of a Booth equation which can be used to calculate release estimates of all four CTBT-relevant radioxenon isotopes for any reactor of the same type.
Irrespective of assuming fission or activation as the dominating generation process, this study reveals that the NRRs with the highest emissions are not negligible when compared to typical NPPs. The distribution of source strengths for the nine NRRs and NPPs are overlapping. As expected, most NRRs studied here have low release rates compared to NPPs, and two of them have lower releases than any European NPP in 2014. However, at the upper end, there are two NRRs with fission as the dominating source that have a daily release estimate that is larger than the release rate of a typical NPP (~ 1 GBq/day). Another relevant reference value is a release rate of 5 GBq/day, which was identified to be a “reasonable radioxenon release” per single source according to Bowyer et al. (2013). Releases below this value were found to have no significant impact on IMS sensors. The NRR with the highest 133Xe release rate (NRU) has a release that is a factor of 400 larger than 5 GBq/day and two orders of magnitude higher than any European NPP had in 2014. The 133Xe release of the NRU is only two orders of magnitude below the 10% prompt release scenario for a 1 kt TNT equivalent underground nuclear explosion.
Both 133Xe and 135Xe releases of the HFIR with activation being the major production mechanism are of the same order of magnitude as for a typical NPP. However, for 131mXe, the release for the HFIR is two orders of magnitude above the typical release of an NPP, for 133mXe it is one order of magnitude higher. The 131mXe release of the HFIR is only one order of magnitude below the 10% prompt release scenario for a 1 kt TNT equivalent underground nuclear explosion.
When discussing all these numbers, it needs to be taken into consideration that they are subject to high uncertainties and high variability.
As a conclusion, NRRs cannot be ignored as having neglectable impact on the IMS. Some NRRs may have an observable impact on IMS noble gas detectors, if they have comparatively large radioxenon emissions and are operating at geographic locations that IMS sensors may occasionally have a high source–receptor sensitivity for. Therefore, this source type needs to be taken into consideration and further studies are required to get a better understanding of the following questions:
-
What mechanisms of radioxenon formation and release can explain the variability of releases from NRRs and what are the maximum release amounts on 1 day that can be expected?
-
Identify which individual NRRs may have an impact on which specific regional IMS stations.
References
Biegalski, S., Edward, A., Peter, S. (2018), Research reactor contributions to radioxenon atmospheric inventories. Presentation at the workshop on signatures of man-made isotope production (WOSMIP) VII in Sydney, Australia, 3–7 December 2018; complemented by private communication by email from Steven Biegalski to MK dated 12 November 2020.
Bonka, H. (1982). Strahlenexposition durch radioaktive Emissionen aus kerntechnischen Anlagen im Normalbetrieb. . TÜV Rheinland.
Booth, A. H. (1957). A suggested method for calculating the diffusion of radioactive rare gas fission products from UO* sub 2/fuel elements and a discussion of proposed in-reactor experiments that may be used to test its validity (No DCI-27). . Atomic Energy of Canada Ltd. Chalk River Project.
Bowyer, T. W., Kephart, R., Eslinger, P. W., Friese, J. I., Miley, H. S., & Saey, P. R. J. (2013). Maximum reasonable radioxenon releases from medical isotope production facilities and their effect on monitoring nuclear explosions. Journal of Environmental Radioactivity, 115, 192–200.
Burnett Jonathan, L., & Milbrath, B. (2019), Radionuclide signatures of molten salt reactors. Poster T2.1-P19 presented at the CTBT: Science and Technology 2019 Conference SnT2019 in Vienna.
CD (1995), Conference on disarmament, working group 1-verification international monitoring system expert group, conference on disarmament. Ad Hoc Committee on a Nuclear Test Ban. Paper number CD/NTB/WP.224.
ENDF (2018). Evaluated nuclear data library ENDF/B-VIII.0. https://www.nndc.bnl.gov/endf/b8.0/.
Fay, A. (2014). Characterization of sources of radioargon in a RESEARCH REACTOR, the University of Texas at Austin.
Fontaine et al. (2021) Emissions of radioactive noble gases from nuclear facilities in Europe with a focus on 37Ar. To be published in Journal of Environmental Radioactivity.
Gueibe, C., Martin, B. K., Jonathan, B., Abdelhakim, G., Monika, K., & Jolanta, K.-M. (2017). Setting the baseline for estimated background observations at IMS systems of four radioxenon isotopes in 2014. Journal of Environmental Radioactivity, 178–179, 297–314.
HANARO (2006), Byung Chul Lee, gaseous source term of environmental release during HANARO normal operation, KAERI/TR-3180/2006.
Hoffman, I., & Berg, R. (2018). Medical isotope production, research reactors and their contribution to the global xenon background. Journal of Radioanalytical and Nuclear Chemistry, 318(1), 165–173.
IAEA-International Atomic Energy Agency (2015). Operating experience with nuclear power stations in member states in 2014. Power Reactor Information System. https://pris.iaea.org/PRIS/home.aspx.
IAEA-International Atomic Energy Agency (2019), Research Reactor Data Base (RRDB). https://nucleus.iaea.org/RRDB/RR/ReactorSearch.aspx?rf=1.
INVAP (2004). Safety analysis report, chapter 12: Operational radiological safety, Document Number: RRRP-7225-EBEAN-002-REV0-CHAPTER-12.
Johnson, C., Biegalski, S. R., Artnak, E. J., Moll, E., Haas, D. A., Lowrey, J. D., Aalseth, C. E., Seifert, A., Mace, E. K., Woods, V. T., & Humble, P. (2017). Production and release rate of 37Ar from the UT TRIGA Mark-II research reactor. Journal of Environmental Radioactivity, 167, 249–253.
Jorge, A. Q. D. (2016). RA3 research reactor, base of the reactor radioisotope production in Argentina, WOSMIP VI, PNNL-26793.
Kalinowski, M. (2011). Characterization of prompt and delayed atmospheric radioactivity releases from underground nuclear tests at Nevada as a function of release time. Journal Environmental Radioactivity, 102, 824–836.
Kalinowski, M., Anders, A., Marc, B., Xavier, B., Theodore, W. B., Guy, B., Simon, H., Justin, I. M., Jana, P., Christoph, P., Maria, R., Anders, R., Saey, P. R. J., Schlosser, C., Stocki, T. J., Thomas Taffary, R., & Kurt, U. (2010). Discrimination of nuclear explosions against civilian sources based on atmospheric xenon isotopic activity ratios. Pure and Applied Geophysics, 167, 517–539.
Kalinowski, M., & Christoph, P. (2006). isotopic signature of atmospheric xenon released from light water reactors. Journal of Environmental Radioactivity, 88, 215–235.
Kalinowski, M., & Halit, T. (2020). Global radioxenon emission inventory for 2014 by normal operational releases from nuclear power plants and medical isotope production facilities. Published in this issue of Pure and Applied Geophysics. https://doi.org/10.1007/s00024-020-02579-w
Kalinowski, M., & Tuma, M. P. (2009). Global radioxenon emission inventory based on nuclear power reactor reports. Journal of Environmental Radioactivity, 100, 58–70.
Klingberg, F. J., Biegalski, S. R., & Fay, A. G. (2013). Radioxenon signatures from activation of environmental xenon. Journal of Radioanalytical and Nuclear Chemistry, 296, 117–123.
Lechermann, M. S. B. (2010). The effect of research reactors on the global radioxenon background, 2010.
Lewis B. J., et al. (1989). A model to describe defected HJEL behavior for the EIJ5SIM fuel performance code. In: Second international conference on CANDU fuel.
Matuszek, J. M., Carl, J. P., & Charles, O. K. (1973). Characterization of stack effluents from certain nuclear facilities. . Health Research, and Inc New York State Department of Health Albany.
Matuszek, J. M., Paperiello, C. J., & Kunz, C. O. (1975). Reactor contributions to atmospheric noble gas radioactivity levels. Noble Gases, 20, 360–364.
Oak Ridge Reservation Annual Site Environmental Report, ORNL. (2016).
Ringbom A., Axelsson, A., Björnham, O., Brännström, N., Fritioff, T., Grahn, H., Olsson, M.,& Hennigor, S. (2020). High time-resolution observations of radioxenon releases from BWRs compared to stack data and reactor operation parameters. Submitted to the topical issue of PAGEOPH on Nuclear Explosion Monitoring and Verification: Scientific and Technological Advances.
Saey, P. R. J. (2009). the influence of radiopharmaceutical isotope production on the global radioxenon background. Journal of Environmental Radioactivity, 100, 396–406.
Steinhauser, G., Michael, L., Anders, A., Helmuth, B., Anders, R., Paul, R. J. S., Clemens, S., & Mario, V. (2012). Research reactors as sources of atmospheric radioxenon. Journal of Radioanalytical and Nuclear Chemistry. https://doi.org/10.1007/s10967-012-1949-x.
Tayyebi, P., Fereydoun, A. D., Mohsen, T., Marzieh, E., & Hossein, A. (2017). Computational investigation of isotopic signature of radioxenon released from Tehran research reactor. Journal of Radioanalytical and Nuclear Chemistry, 311(1), 571–576.
UNSCEAR. (2000). United Nations Scientific Committee on the Effects of Atomic Radiation report to the general assembly, Sources and effects of ionizing radiation, Vol. 1.
World Nuclear News (2018). Canadian isotope reactor enters retirement. https://www.world-nuclear-news.org/RS-Canadian-isotope-reactor-enters-retirement-0308185.html. Accessed on 16 Mar 2021.
Wotawa, G., Becker, A., Kalinowski, M., Saey, P., Tuma, M., & Zahringer, M. (2010). Computation and analysis of the global distribution of the radioxenon isotope Xe-133 based on emissions from nuclear power Plants and radioisotope production facilities and its relevance for the verification of the nuclear-test-ban treaty. Pure and Applied Geophysics, 167, 541–557.
Acknowledgements
The original work was carried out while P. Tayyebi was as an intern at CTBTO/SA/SM under supervision of Martin B. Kalinowski. She would like to thank him for such an interesting research. The authors thank the anonymous reviewers for their very instructive comments and suggestions that motivated us to do a thorough revision.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kalinowski, M.B., Tayyebi, P., Lechermann, M. et al. Global Radioxenon Emission Inventory from Nuclear Research Reactors. Pure Appl. Geophys. 178, 2711–2739 (2021). https://doi.org/10.1007/s00024-021-02719-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00024-021-02719-w