
Abstract
Background
Gastric cancer (GC) is one of the most malignant cancers worldwide. Metabolism disorder is a critical characteristic of malignant tumors related to tumor progression and metastasis. However, the expression and molecular mechanism of malic enzyme 3 (ME3) in GC are rarely reported. In this study, we aim to investigate the molecular mechanism of ME3 in the development of GC and to explore its potential value as a prognostic and therapeutic target in GC.
Method
ME3 mRNA and protein expression were evaluated in patients with GC using RT-qPCR, WB, and immunohistochemistry, as well as their correlation with clinicopathological indicators. The effect of ME3 on proliferation and metastasis was evaluated using Cell Counting Kit-8 (CCK-8), 5-ethynyl-20-deoxyuridine (EdU) assay, transwell assay, wound healing assay, and subcutaneous injection or tail vein injection of tumor cells in mice model. The effects of ME3 knockdown on the level of metabolites and hypoxia-inducible factor-1α (HIF-1α) protein were determined in GC cells. Oxidative phosphorylation was measured to evaluate adenosine triphosphate (ATP) production.
Results
ME3 was downregulated in human GC tissues (P < 0.001). The decreased ME3 mRNA expression was associated with younger age (P = 0.02), pathological staging (P = 0.049), and lymph node metastasis (P = 0.001), while low ME3 expression was associated with tumor size (P = 0.048), tumor invasion depth (P < 0.001), lymph node metastasis (P = 0.018), TNM staging (P < 0.001), and poor prognosis (OS, P = 0.0206; PFS P = 0.0453). ME3 knockdown promoted GC cell malignancy phenotypes. Moreover, α-ketoglutarate (α-KG) and NADPH/NADP+ ratios were reduced while malate was increased in the ME3 knockdown group under normoxia. When cells were incubated under hypoxia, the NADPH/NADP+ ratio and α-KG decreased while intracellular reactive oxygen species (ROS) increased significantly. The ME3 knockdown group exhibited an increase in ATP production and while ME3 overexpression group exhibited oppositely. We discovered that ME3 and HIF-1α expression were negatively correlated in GC cells and tissues, and proposed the hypothesis: downregulation of ME3 promotes GC progression via regulating intracellular oxidative stress and HIF-1α.
Conclusion
We provide evidence that ME3 downregulation is associated with poor prognosis in GC patients and propose a hypothesis for the ME3 regulatory mechanism in GC progression. The present study is of great scientific significance and clinical value for exploring the prognostic and therapeutic targets of GC, evaluating and improving the clinical efficacy of patients, reducing recurrence and metastasis, and improving the prognosis and quality of life of patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is one of the most common aggressive malignancies and the fifth leading cause of cancer-related mortality worldwide [1]. Despite recent remarkable advances and progress in diagnostics and treatments, the five-year survival rate of GC patients remains the frustrating cause of the high recurrence and metastasis rates [2,3,4]. However, the molecular mechanism that regulates GC progression and metastasis remain poorly understood. In this study, we aim to investigate the molecular mechanism of ME3 in the development of GC and to explore its potential value as a prognostic and therapeutic target in GC.
Metabolism reprogramming is an important marker of malignant tumors [5]. The tumor cells can adjust receptors or activate related pathways through gene mutation or other ways to overcome growth-factor-dependent growth patterns, hence promoting the absorption and metabolism of nutrients or choosing new energy supply methods to provide sufficient energy for cell growth and proliferation [6,7,8]. However, metabolic remodeling affects the microenvironment of cancer cells and regulates tumor cell proliferation and distant metastasis [9, 10]. Mitochondria are an important hub for coordinating metabolism reprogramming. Mitochondria regulate energy metabolism, biosynthesis, and signal transduction pathways, allowing cancer cells to adapt to harsh survival environments and initiate invasion, metastasis, and treatment resistance [11]. Mitochondria can regulate reactive oxygen species (ROS), cell differentiation, and autophagy during metabolic remodeling. Mitochondria can also influence the malignant process of the tumor by increasing the abundance of oncogenic proteins and metabolites, such as HIF-1α and α-KG [11,12,13,14]. Studies on GC have revealed that drug treatment could cause mitochondrial ROS accumulation and promote apoptosis and autophagy. Metabolic reprogramming can sustain the survival and proliferation of GC cells when mitochondrial DNA damage occurs [15,16,17].
Cancer cells grow at an aberrantly rapid rate with the expense of overtaken oxygen, contributing to the relative hypoxia tumor microenvironment [18, 19]. The hypoxia-inducible factor-1 (HIF-1) is a key mediator of hypoxia response [20]. HIF-1, consisting of HIF-1α and HIF-1β, is a heterodimeric transcription factor. HF-1α is an oxygen concentration-sensitive subunit, while HIF-1β is a continuously expressed subunit [21, 22]. HIF-1α is hydroxylated on proline residue 402 or 564 by proline hydroxylase domain (PHD) protein under normoxia, degrading via the ubiquitin-protease pathway with a half-life of less than 1 min [23]. In contrast, PHD activity is inhibited, and the HIF-1α protein is increased under hypoxia. Then, HIF-1α and HIF-1β rapidly polymerize and regulate the downstream gene expression [24]. HIF-1 exhibits an important role in GC carcinogenesis, angiogenesis, metastasis, and chemoresistance [25,26,27,28]. However, the exact mechanisms involved in hypoxia-driven GC metastasis remain uninvestigated.
Malic enzymes (MEs) are essential decarboxylases in the tricarboxylic acid cycle (TCA), catalyzing the conversion of malate to pyruvate [29, 30]. In human cells, malic enzymes are encoded by three homologous genes: ME1 is located in the cytoplasm, and its enzymatic activity depends on NADP+; ME2 and ME3 are both located in mitochondria, and their enzymatic activity depends on NAD+ and NADP+, respectively [31, 32]. According to studies, ME1 plays an important role in various diseases. ME1 is upregulated to maintain redox homeostasis [33], while ME2 is frequently hemizygously co-deleted with SMAD4 in GC. Another study reveals a positive feedback mechanism between TP53 and MEs (ME1 and ME2) that modulates the metabolism reprogramming and senescence. Inhibiting ME1 and ME2 expression induces cellular senescence (rather than apoptosis) by activating p53 feed-forwardly via the Mdm2 and AMPK pathways, respectively, whereas ME1 and ME2 overexpression inhibit cellular senescence [32]. The co-deletion effect of ME2 and SMAD4 could be replenished by ME3 in pancreatic cancers, implying that ME3 inhibitors might be a promising target of ME2-null tumors [34]. However, little research has been conducted on the role of ME3 in cancers, especially GC. Mitochondrial malic enzymes’ altered expression or activity affects other TCA cycle intermediate metabolites, including citric acid and α-KG. Increased α-KG facilitated modification of HIF-1α by PHDs and further recognition by Von Hippel Lindau protein (pVHL) for degradation via the proteasomal pathway [35]. Therefore, we attempt to investigate whether the ME3 downregulation could promote GC growth and metastasis by regulating HIF-1α expression and to provide a mechanistic understanding of hypoxia and tumor metabolism pathways.
According to proteomic sequencing of GC samples from our center, ME3 is differentially expressed in GC patients with high or low metastatic tendency. However, there must be more research on the ME3 expression in GC and its impact on the biological behavior of GC cells, including tumorigenesis, progression, and potential regulatory mechanisms. Tumor cells adapt to this change by metabolic reprogramming because they have a higher demand for material and energy supplements. Increasing intracellular ROS and the onset of hypoxia accompany this process. Malic enzymes are involved in intracellular substance metabolism, and their abnormal expression and function in a hypoxic environment might disrupt intracellular redox homeostasis. This signaling pathway activation can be controlled by regulating α-KG production, affecting the HIF-1α protein degradation. Therefore, we investigate the effects of abnormal ME3 expression on the HIF-1α signaling and its potential regulatory mechanisms in promoting GC growth and metastasis. We hope that this study would provide new prospectives and scientific evidence for exploring the prognostic and therapeutic targets of GC, evaluating and improving the clinical efficacy of patients, reducing recurrence and metastasis, and improving the prognosis and quality of life of patients.
Methods
Patients and clinical specimens
GC and para-cancerous tissue samples were collected randomly from patients undergoing gastrectomy between 2015 and 2020 at the First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang Province, China. All the participants have signed “written informed consent” to take part in this study. All the samples were pathologically diagnosed as GC, and all patients had not received radiotherapy or chemotherapy before gastrectomy, and follow-up information was available. Patients were informed that their specimens were stored by the hospital and potentially used for scientific research. The study was conducted under the Declaration of Helsinki and its amendments and approved by the the Medical Ethics Committee of the First Affiliated Hospital of Zhejiang University (Approval Number: 2021 − 869).
Cell culture and transfection
The human immortalized normal gastric mucosal cell line GES-1 was obtained from the China Center for Type Culture Collection (CCTCC). The GC cell lines (AGS, HGC-27, BGC823, MGC803, SGC7901, SNU-1, NCI-N87, and MKN45) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). GC cell lines were cultured in RPMI (Roswell Park Memorial Institute)-1640 medium (supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin), while GES-1 was cultured in DMEM (dulbecco’s modified eagle medium) medium (supplemented with 10% fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin) at 37℃ with 5% CO2 considered as a normoxic condition. A hypoxic culture chamber (ELECTROTEK, England) was employed to evaluate the effects of hypoxic conditions (37℃, 1% O2, and 5% CO2).
The specific small interfering RNA (siRNA) targeting ME3 and a negative control siRNA were obtained from GenePharma (Shanghai, China). Plasmids expressing ME3 shRNA or control shRNA were chemically synthesized by Generay Biotech (Shanghai, China). The cells were cultured in a six-well plate and transfected with siRNA or shRNA using lipofectamine 3000 (Invitrogen, USA), according to the manufacturer’s instructions. The cells were harvested 48 h after transfection for further experiments.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from tissues and cells using Trizol (Invitrogen, Carlsbad, CA, USA) followed by RT-PCR. The qRT-PCR was performed using TB Green® Premix EX Taq™ II (Takara, Osaka, Japan) via StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative expression analysis was performed using the comparative CT method (2−ΔΔCT), and gene mRNA level was normalized to GAPDH expression. qPCR primers were purchased from Tsingke (Beijing, China).
Western blot (WB) analysis
Total protein was extracted from GC cells and tissues, and Pierce™ Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific) was used to measure protein concentrations. An equal quantity of protein samples was separated using SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membranes were probed with primary antibodies overnight and incubated with the HRP-conjugated goat anti-mouse or anti-rabbit IgG antibody (Cell Signaling Technology, USA).
Immunofluorescence (IF)
The GC cells were fixed with 4% paraformaldehyde and permeabilized with 0.025% Triton X-100. After blocking, the fixed cells were incubated with the E-cadherin, N-cadherin, and vimentin primary antibodies. The secondary antibodies were Fluorescein isothiocyanate isomer I (FITC)-labeled Goat Anti-Mouse IgG (H + L) and Cy3-labeled Goat Anti-Rat IgG (H + L) (Beyotime, China). 4’,6-diamidino2-phenylindole (DAPI) was used to label the cell nucleus. Fluorescence confocal images were captured using a laser-scanning confocal microscope (Carl Zeiss, Germany).
Cell migration and transwell assay
The invasion and migration assays were performed using a Transwell chamber (Corning, USA). The chambers were coated with 50 µL of Matrigel (BD, USA)/ PRMI-1640 medium (1:4, v/v) for 30 min at 37℃. A total of 2 × 104 cells were seeded into the upper chambers with 100 µL serum-free medium, while the cell culture dish was filled with 700 µL RPMI-1640 medium containing 10% FBS. After 24 h of incubation, the chamber membranes were fixed with 4% paraformaldehyde and stained with crystal violet dye. The number of invaded or migrated cells was quantified in five random visual fields and photographed under microscopic.
Wound healing assay
Cells were cultured in 12-welll plates until they reached confluence. After creating a straight line in the cell monolayer using a sterile pipette tip. Detached cells were removed by PBS. Images were captured after 24 h and the percentage of scratch area was quantified by using ImageJ.
Cell viability assay
The treatment and control cells were seeded in 96-well plates at a density of 1 × 103 cells/well and incubated at 37℃. The cell viability was measured at 6, 24, 48, 72, and 96 h using Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol.
Edu assay
The cell proliferation ability was inquired using a 5-ethynyl-20-deoxyuridine (Edu) assay kit (Ribobio, Guangzhou, China). The cells were seeded into 24-well plates at a density of 1 × 105 each well. Subsequent operations were carried out according to the manufacturer’s protocol. The proliferating cells are stained with Edu dye, and nuclei with Hoechst. Then, the results were visualized using a fluorescence microscope, and cell counting was performed using ImageJ software.
Xenograft mouse model
Four-week-old BALB/c female nude mice were purchased from the Experimental Animal Center of Zhejiang Province. We chose MGC803 to construct ME3-knockdown cell line and HGC-27 to construct ME3 expression cell line. An equal number of treated GC cells were resuspended in 100 µL PBS and subcutaneously injected into the left flank of mice, randomly divided into several groups. Tumor size was measured every five days. After four weeks of feeding, mice were euthanized, and subcutaneous transplanted tumors were removed for histology analysis. Tumor volume was calculated using the formula: 1/2(length×width2).
Six-week-old BALB/c female mice were randomly grouped and injected with 1 × 106 (in 100 µL PBS) control shRNA or sh-ME3/ Luc HGC-27 cells via the tail vein. Bioluminescent images were captured four weeks after cancer cell injection with the IVIS Spectrum to detect whether lung metastasis had occurred. Mice were euthanized with organs (lung, liver, and bone) removed for hematoxylin-eosin staining (HE) staining and other histology analysis.
All animal experiments were approved by the Ethics Committee for Laboratory Animals of the First Affiliated Hospital of Zhejiang University (Approval number: 2021 − 1431, 2022 − 1060).
Measurement of intracellular ROS
Intracellular ROS was probed using 2, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime) and MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen) according to manufacturers’ instruction. The staining of cells treated with hypoxia was conducted in a hypoxia incubator. The stained cells were washed with ice-cold HBSS, then observed under Zeiss LSM900 laser confocal microscope (Carl Zeiss, Germany). The fluorescence intensity was analyzed using ImageJ software.
Cell and mitochondria oxygen consumption rate analysis
The oxygen consumption rate was analyzed using the Oxygraph-2 K platform (Oroboros Instruments Corp, Austria). Briefly, 1 × 106 cells resuspended in RPMI-1640 medium were injected into the assay chamber. Oligomycin (1 µM), rotenone (1 µM), FCCP (6 µM), and antimycin (1 µM) were sequentially injected into the assay chambers when the oxygen consumption rate reached a plateau. The soluble oxygen concentration was recorded for data analysis.
Measurement of malate
After cultured in normoxia or hypoxia environment, cells were rinsed three times with cold PBS, followed by adding 800 µL perchloric acid per well. The plates were scraped, and the suspension was transferred into a 1.5 mL Eppendorf tube. For the hypoxia treatment group, the above protocol was performed in a hypoxia incubator with 1% O2. The samples were placed on ice, and potassium carbonate was used to adjust the pH value of the suspension. Afterward, the mixture was thoroughly mixed and centrifuged (14,000 rpm, 10 min, and 4℃) to remove the cell debris. The supernatant was collected for subsequent analysis.
Briefly, 100 µL of the sample was added to 900 µL of the reaction buffer (100 mmol/L Glycine, 170 mmol/L hydrazine, 2 mmol/L NAD+, 2.1 U/mL malic dehydrogenases, pH = 9.2). The sample was thoroughly mixed and incubated at room temperature for 1 h, and the absorbance was recorded at 340 nm against the blank (100 µL sample without enzyme).
Assay for α-KG
According to the manufacturer’s instructions, the α-KG in cells was measured using Abcam α-ketoglutarate Assay Kit (ab83431).
Statistical analysis
All data were analyzed using GraphPad Prism software (GraphPad Prism 8.0) and IBM SPSS Statistics software, version 21. The data were presented as the mean ± standard deviation (SD) from at least three independent experiments. The significant difference in quantitative data was assessed using an independent Student’s t-test or One-way analysis of variance (ANOVA), while qualitative data were compared using the Pearson χ2 tests. P-values for each analysis were marked on figures, and significance was defined at *p < 0.05.
Results
ME3 is downregulated in GC and correlated with poor prognosis
We examined ME3 expression in cancer and para-cancerous tissues to validate its role in GC. We collected tissues from patients after gastrectomy and retrieved their medical histories. We discovered that ME3 mRNA (Fig. 1a) and protein (Fig. 1b–c) levels were remarkably reduced in cancer tissues using RT-qPCR and WB, suggesting that ME3 was restrained at the transcriptional level during GC progression (Cohort A for RT-qPCR and Cohort B for WB). As mentioned, ME2 is an important isoenzyme of ME3, so we simultaneously detected ME2 protein expression in 23 pairs of patients’ tissue samples by WB assay and compared its correlation with the expression of ME3 in GC tissues. The result showed that there was no significant correlation between the expression of ME2 and ME3 in GC (Supplementary Fig. 1a–b). We also extracted ME3 expression data from the Gene Expression Profiling Interactive Analysis (GEPIA) online tool to re-confirm our findings. The result demonstrated that ME3 mRNA had a lower expression level in GC (Fig. 1d). Table 1 lists the clinical characteristics of 258 GC patients and their relationships with ME3 expression. Low ME3 expression was significantly associated with younger age (P = 0.02), poor differentiation (P = 0.049), and increased lymph node metastasis (P = 0.001).

Down-regulated ME3 expression is associated with poor outcomes of GC. (a) The expression of ME3 mRNA was determined based on Cohort A (N = 258); (b-c) The expression of ME3 protein was analyzed by western blotting in 23 pairs of GC tissues (T: tumor; N: normal control); (d) The expression of ME3 was verified by GEPIA (dataset from TCGA and the GTEx projects) (STAD: Stomach and Esophageal carcinoma); (e-f)IHC scores of 161 pairs of GC tissues in TMA (Cohort C) based on ME3 staining; (g) Representative IHC images of ME3 staining in gastric cancer or cancerous tissues (scale bar,100 μm; magnification, 100× and 200×); (h) Proportion of ME3 low- or high-expressing in gastric cancer tissues or para-cancerous tissues (based on IHC scores); (i-j) Kaplan-Meier analysis of overall survival (OS) and progression-free survival (PFS) of 161 GC patients (data from Cohort C); (k) Differences in ME3 expression between patients with simultaneous or diachronic distant metastases (Metastasis) and those without metastasis (No metastasis)
Then, we randomly selected 161 patients with GC (Cohort C), collected cancer and para-cancerous tissues, and created a GC tissue microarray (TMA). The ME3 expression was detected using an Immunohistochemistry (IHC) assay. The IHC result revealed that ME3 was down-regulated in cancer tissues (Fig. 1e–f). According to the IHC score, the patients were divided into the ME3-high and the ME3-low group. Figure 1g depicts the typical high and low ME3 protein expression from GC tissue and its adjacent counterpart. Based on the median IHC score, the percentage of samples with low ME3 expression was 21.1% in para-cancerous tissues and as high as 91.6% in cancerous tissues (Fig. 1h). Table 2 lists the clinical characteristics of 161 GC patients and their relationship with ME3 expression level. Low ME3 expression was significantly associated with advanced T staging (P < 0.0001), N staging (P = 0.018), TNM staging (P < 0.001), and tumor size (P = 0.048). The Kaplan-Meier analysis revealed that patients with low ME3 expression levels had poor overall survival (OS, P = 0.026) and disease-free survival (DFS, P = 0.0453, Fig. 1i–j). We divided patients into metastasis and non-metastasis groups based on the presence of simultaneous or heterogeneous distant metastasis to verify further the difference in the ME3 expression protein in patients with or without metastasis. The results displayed that ME3 expression was lower in the metastasis group than in the non-metastasis group (P < 0.05, Fig. 1k). This result suggests that reduced ME3 expression may be associated with distant metastasis in GC.
Downregulation of ME3 promotes tumor growth, migration, and invasion in vitro
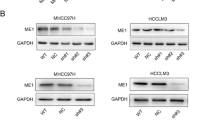
Then, we compared the ME3 mRNA and protein level (Fig. 2a) in several GC cell lines using GES-1 (a type of human gastric mucosal cell) as normal control. We used siRNA transfection or lentiviral infection to establish stable cell lines with ME3 knockdown to investigate the role of ME3 in GC cells. Figure 2b depicts the knockdown efficiency of lentiviral infection. We also detected ME2 expression when ME3 was knockdown at protein level and there was not significantly different of ME2 expression between ME3 knockdown group and control group (Supplementary Fig. 1c).

Down-regulated ME3 promotes tumor growth, invasion and migration abilities of GC in vitro. (a) ME3 expression (mRNA and protein) in different types of GC cell lines; (b) Efficiency of ME3 knockdown in AGS and MGC803 cell lines via lentiviral infection; (c-e) ME3 knockdown promotes GC cell growth. The proliferation capacity was detected by CCK-8, colony formation assay and EdU assays (scale bar,100 μm). (f-g) ME3 knockdown promotes invasion and migration abilities of AGS and MGC803 (scale bar,20 μm)
Supplementary Fig. 2a presents siRNA interference efficiency. Subsequent functional studies selected shME3-1 and shME3-3. The results of CCK-8, Edu dyeing, and clone formation assay revealed that ME3 knockdown significantly promoted cell proliferation and growth abilities (Fig. 2c–e). Meanwhile, the migration and invasion abilities were bolstered (Fig. 2f, g). Supplementary Fig. 2b-f depicts the corresponding phenotype assay results of ME3 siRNA knockdown in GC cells. We backfilled ME3 in GC cell lines with low ME3 expression via plasmid transfection and lentiviral infection to verify whether ME3 functioned as a cancer suppressor protein. ME3 stable-expressed cell lines were also constructed with lentiviral infection. Figure 3a presents the overexpression efficiency. And the ME2 expression was detected and it was shown that there wasn’t different between ME3 overexpression group and control group (Supplementary Fig. 1d). However, ectopic ME3 expression suppressed the proliferation, invasion, and migration ability (Fig. 3b–f). We also employed plasmid transfection to overexpress ME3 in GC cells, the corresponding phenotype experiments were conducted, and the results were consistent with those after lentiviral infection (Supplementary Fig. 3). In previous experiments, we discovered that either transfection with siRNA or stable knockdown of ME3 by lentivirus promoted GC cell proliferation in vitro. Thus, we utilized AGS and MGC803 cells with stable knockdown of ME3 expression after drug screening and ME3 overexpression plasmid to restore its expression. The CCK-8 assay result demonstrated that ectopic ME3 expression inverted the effect of promoting tumor proliferation caused by ME3 downregulation (Supplementary Fig. 3f). We then established a set of ME2 knockdown cell lines and examined the ME3 expression. It was found that the ME2 expression was not significantly affected the ME3 expression in GC cells (Supplementary Fig. 1e). So in later study, we focused on investigating the effect of ME3 expression on GC cells.

Up-regulated ME3 suppresses tumor growth, invasion and migration abilities of GC in vitro. (a) Efficiency of ME3 overexpression in HGC-27 and MGC803 cell lines via lentiviral infection; (b-d) Ectopic expression of ME3 depresses GC cell growth. The proliferation capacity was detected by CCK-8, colony formation assay and EdU assays (scale bar, 100 μm). (e-f) Ectopic expression of ME3 depresses invasion and migration abilities of HGC-27 and MGC803 (scale bar,20 μm)
Downregulation of ME3 promotes tumor growth and metastasis in vivo
Tumor xenograft models were constructed by subcutaneously injecting MGC803 cells with stable knockdown (sh-ME3-1, sh-ME3-3) or HGC-27 cells with ME3 overexpression (LV-OV) into NOD-SCID mice to confirm the role of ME3 in vivo. We discovered that the ME3 knockdown promoted tumorigenesis with prominently greater tumor volumes than the negative control group (Fig. 4a–c). However, the body weight of mice did not differ significantly among groups (Fig. 4d). Meanwhile, ectopic ME3 expression restrained tumorigenesis compared to the negative control group (Fig. 4e–g). Moreover, the growth of body weight in mice revealed no significant difference between the LV-OV and the LV-NC groups (Fig. 4h).

Down-regulated ME3 promotes tumor growth and metastasis of GC in vivo. (a) Subcutaneous transplanted tumor nodules of were collected to present the growth difference within the influence of ME3 knockdown; (b-c) Tumor growth curve of stable ME3 silenced MGC803 cell (or negative control) in the xenograft mouse model was based on the measurement of tumor size; (d) Body weight of mice with injection of stable ME3 silenced MGC803 cell was recorded; (e) Subcutaneous transplanted tumor nodules were collected to present the growth difference within the influence of ME3 overexpression; (f-g) Tumor growth curve of stable ME3 overexpressed HGC-27 cell (or negative control) in the xenograft mouse model was based on the measurement of tumor size; (h) Body weight of mice with injection of stable ME3 overexpressed HGC-27 cell was recorded; (i) Body weight of mice with tail vein injection of stable ME3 silenced and luciferase expressed MGC803 cell was recorded; (j) Bioluminescence intensity of tumors formed by ME3 knockdown MGC803 cell and negative control; (k) Classic image of lung sample with metastatic lung lesions; (l) HE staining results of biopsies of mouse lung and bone tissues (scale bar,100 μm)
MGC803 cells with stably expressing luciferase and ME3 knockdown (Luci-shME3-1 and Luci-shME3-3) and control group (Luci-sh-NC) were injected into BALB/c nude mice via tail vein. The mice’s weight was recorded regularly (Fig. 4i). We observed high fluorescence intensity in lungs, spines, and legs in the ME3 knockdown groups using in vivo animal bioluminescence imaging technology (Fig. 4j). Figure 4k displays the typical images of the lung from dissection. Paraffin section and HE staining were performed on the tissues obtained from mice autopsy. Figure 4l illustrates the HE staining results of the metastatic and non-metastatic lung tissues and the metastatic foci in bone. Based on the above results, ME3 knockdown GC cells were more prone to distant metastasis in vivo.
Alteration of ME3 influences ROS levels in GC cells
Studies have demonstrated that metabolic reprogramming occurs in undifferentiated stromal tumor cells with high glycolysis rates, elevated ATP production, and abnormal redox balance [36,37,38]. ME3 is a protein with metabolic enzymatic activity that catalyzes the oxidative decarboxylation of malate and reduces the cofactor NADP+ to NADPH [39, 40]. We first measured the NADPH/NADP+ ratio of GC cells under normoxia to explore the potential regulatory pathway of ME3 on GC. The ratio of NADPH/NADP+ was decreased in ME3 knockdown GC cells, while it was increased in GC cells with ME3 overexpression (Fig. 5a). However, a certain hypoxia environment in solid tumors frequently appears with the rapid proliferation of tumor cells [41, 42]. We cultured GC cells under hypoxia (1% O2) to mimic the hypoxia microenvironment and measured the NADPH/NADP+ ratio. We observed that the NADPH/NADP+ ratio was significantly lower in ME3 knockdown cells than in the control group. In contrast, the NADPH/NADP+ ratio was significantly higher in cells with ME3 overexpression (Fig. 5b). Meanwhile, we tested the enzyme activity of MEs in vitro and discovered that ME3 knockdown reduced the total activity of MEs, whereas ME3 overexpression promoted the total activity under normoxia (Fig. 5c). The results of in vitro experiment suggest that altered ME3 expression might affect the biochemical reactions in GC cells by changing the overall enzymatic activity of malic enzymes.

Down-regulated ME3 decreases NADPH/NADP+ ratio and increases ROS in GC cells under hypoxia. (a-b) Effect of knockdown or overexpression of ME3 on NADPH/NADP+ in GC cells in normoxia (20% O2) or hypoxia (1% O2) environment. The NADPH/NADP+ ratio was measured by using the NADPH/NADP+ test kit; (c) In vitro assay of malic enzyme (MEs) activity in GC cell lysates under normoxia conditions (20% O2); (d-e) Intracellular ROS in GC cell with ME3 knockdown or overexpression under normoxia or hypoxia conditions. The intracellular ROS was detected by DCFH-DA probe; (f) Downregulation of ME3 increased both intracellular total ROS (DCFH, green) and mitochondrial ROS (MitoSox, red), (scale bar, 100 μm)
NADPH, an important cofactor of glutathione reductase, is involved in maintaining glutathione (GSH) reduction and plays an essential role in regulating GSH concentration and oxidative stress balance in cells [43]. We cultured AGS and MGC803 with ME3 stably knockdown in normoxia (20% O2) and hypoxia (1% O2), respectively, to explore whether abnormal ME3 expression could influence intracellular redox homeostasis. The DCFH-DA probe was used to detect the total intracellular ROS. There is no significant difference between ME3 downregulation and upregulation in GC cells under normoxia (Fig. 5d). When cells were transferred to a hypoxia environment, it was discovered that the ROS level was significantly increased in GC cells with ME3 knockdown but significantly lower in the ME3 overexpression group (Fig. 5e). Intracellular ROS is primarily distributed in the cytoplasm and mitochondria; we utilized the MitoSOX Red mitochondrial superoxide indicator and the DCFH-DA reactive oxygen species detection kit to visualize mitochondrial and total intracellular ROS in GC cells under hypoxia, respectively. The image displayed that cytoplasm and mitochondrial ROS were significantly increased in GC cells with ME3 knockdown and significantly lower in the ME3 overexpression group (Fig. 5f). These results suggested that the ME3 knockdown could reduce NADPH/NADP+ ratio and increase ROS in GC cells under hypoxia.
ME3 promotes GC progression by regulating the stability of HIF-1α
Hypoxia and intracellular oxidative stress state are important features of solid tumors. The alteration of ME3 expression level affects NADPH/NADP+ and ROS in GC cells, especially under a hypoxic environment. HIF-1α is upregulated when cells are exposed to an anaerobic environment. It acts as a transcription factor, enters the nucleus, and activates various downstream signaling pathways [44]. It plays an essential role in promoting tumor progression and metastasis. However, the relationship between ME3 and HIF-1α expression and its regulatory mechanism is still unclear. We first analyzed the expression pattern using GEPIA online tool to further explore the role of ME3 and HIF-1α in GC progression. Figure 6a displays that HIF-1α is negatively correlated with ME3 (q-value = 6e-30, R = -0.43). The GC microarray Cohort C, consisting of 161 pairs of GC tissues and paired adjacent tissues randomly selected in our center, was adopted, and IHC staining and scoring were performed. The results demonstrated that HIF-1α was upregulated in GC tissues (P < 0.001, Fig. 6b–c). Figure 6d depicts typical IHC results for high- and low HIF-1 expression in GC tissues. We discovered a negative correlation between HIF-1α expression and ME3 in GC tissues by comparing the immunohistochemical staining scores of the same patient samples (Fig. 6e). We discovered that HIF-1α expression in the ME3 knockdown group was higher than in the control group, while it was lower in the ME3 overexpressed group tumor tissues than in the control group by detecting HIF-1α expression in subcutaneous tumor transplantation tissues from mice model. Figure 6f illustrates the classic IHC images.

HIF-1α is upregulated in GC cell and tissue, and negatively correlated with ME3. (a) HIF-1α was negatively correlated with ME3 expression. The expression of ME3 and HIF-1α were determined based on GEPIA online tool; (b-c) The expression of HIF-1α was based on IHC scores of Cohort C; (d) Representative IHC images of HIF-1α staining in gastric cancer or cancerous tissues (scale bar,100 μm; magnification, 100× and 200×); (e) HIF-1α protein was negatively correlated with ME3 based on the IHC score of Cohort C; (f) Representative IHC images of HIF-1α staining in subcutaneous tumor from mice model (scale bar, 100 μm)
We detected ME3 and HIF-1α mRNA and protein levels in knockdown and overexpressed GC cells using qRT-PCR and WB further to investigate the relationship between ME3 and HIF-1α expression (cells were cultured under normoxia or hypoxia for same time and samples were obtained at the same time). We observed that ME3 and HIF-1α mRNA expression was negatively correlated in the normoxia environment (Fig. 7a-b), while the HIF-1α expression level in each GC cell group was elevated under hypoxic conditions, especially in ME3 knockdown groups (Fig. 7c–d). We discovered that HIF-1α and ME3 manifested opposite expression trends at the protein level under normoxia through WB experiments (Fig. 7e). Although HIF-1α in ME3 overexpressed group was elevated at the mRNA level under hypoxia, HIF-1α was decreased at the protein level in the ME3 overexpression group (Fig. 7f). The protein level is influenced by transcription, translation speed, and protein degradation rate. We speculated that alteration of ME3 protein level might influence HIF-1α protein level by impacting its protein stability and degradation rate.

HIF-1α is upregulated under hypoxia and negatively correlated with ME3 expression. (a-b) HIF-1α mRNA was negatively correlated with ME3 mRNA under normoxia condition (20% O2); (c-d) HIF-1α mRNA was upregulated in hypoxia environment (1% O2) regardless of ME3 expression; (e-f) HIF-1α protein was negatively correlated with ME3 protein expression regardless of ME3 expression
Ectopic ME3 expression promoted HIF-1α degradation
Several published studies describe the regulatory mechanisms of HIF-1α protein expression, stability, and degradation. The α-KG, a key substrate of HIF-PHDs, participates in the progression of proline hydroxylation of HIF-1α and degradation via ubiquitination and proteasome pathway. We followed experiments to verify whether altering ME3 expression influenced HIF-1α protein fluctuation by regulating its degradation. We observed content of intracellular α-KG was influenced when ME3 changed the weather in a normoxia or hypoxia environment. Figure 8a–b display that the content of α-KG decreases when ME3 is knocked down, while α-KG increases when ME3 is overexpressed. We assumed ME3 overexpression might affect the stability of HIF-1α by regulating intracellular α-KG content.

Ectopic expression of ME3 promoted HIF-1α degradation via regulating α-KG production. (a) α-KG content in GC cell cultured in normoxia environment (20% O2); (b) α-KG content in GC cell cultured in hypoxia environment (1% O2); (c-d) Alteration of HIF-1α and ME3 protein after the addition of CHX in GC cells with ME3 ectopic expression after 24 h of incubation under hypoxia (1% O2). (e) Alteration of HIF-1α and ME3 protein with or without DMKG treatment after 24 h of incubation under hypoxia (1% O2)
We cultured HGC-27 and MGC803 in a hypoxia incubator (1% O2) for 24 h and then added CHX at different time intervals to depressed protein biosynthesis to verify whether overexpressing ME3 could promote HIF-1α degradation. The total protein was extracted, and a WB assay was adopted to detect HIF-1α and ME3 protein. We observed that total HIF-1α protein content was lower in cells with ME3 overexpressing than its control group after CHX treatment (Fig. 8c–d).
Accordingly, we incubated GC cells with ME3 knockdown in a hypoxic chamber for 24 h (1% O2) to verify the effect of α-KG on HIF-1α stability. DMKG (a cell membrane permeable ester that can be hydrolyzed by intracellular esterase to produce α-KG) was added at a final concentration of 1 mM for 24 h. Blank control without DMKG was set up for each group (Fig. 8e). We discovered that HIF-1α expression was higher in ME3 knockdown cells without exogenous DMKG than in their control group. In cells treated with DMKG, HIF-1α expression was slightly reduced or not significantly different, and this change was related to the active duration of DMKG. Therefore, we speculated that the alteration of ME3 level may influence α-KG content and affect HIF-1α stability and total HIF-1α protein, thus affecting GC progression.
ME3 expression affected GC mitochondria’s oxidative phosphorylation rate
The above experiments verified that α-KG concentration, NADPH/NADP+ ratio, and intracellular ROS altered when ME3 expression changed. We also measured the content of malate that served as the substrate of ME3 after normoxia or hypoxia treatment. The results indicated that ME3 knockdown led to malate accumulation in GC cells, whereas ME3 overexpression led to the reduction of malate in normoxia and hypoxia environments (Figures a–b). We revealed GC cell proliferation ability promoted when ME3 was knockdown. Cell growth and proliferation need adequate energy, and mitochondria are an important hub of material and energy metabolism [11, 45]. We measured the mitochondrial oxidative phosphorylation rate of GC cells. The oxygen consumption rate and ATP production were significantly accelerated when ME3 was knocked down. ME3 upregulation led to a reduction in oxygen consumption rate and ATP generation (Fig. 9c).

Downregulation of ME3 promotes mitochondria oxidative phosphorylation rate and EMT process. (a-b) Malate concentration in GC cells under normoxia (20% O2) or hypoxia (1% O2); (c) Alteration of ME3 expression affected mitochondrial oxidative phosphorylation in normoxia condition; (d) KEGG analysis result of ME3 co-expressed gene set of STAD (based on Oncomine database); (e-h) Down-regulated ME3 promoted the progress of EMT (scale bar, 10 μm)
ME3 downregulation promotes GC progression by regulating EMT progression
We obtained the co-expressed genes of ME3 from the Oncomine database for bioinformatic analyses to better explore the molecular mechanisms of ME3-driven GC growth and progression. The results indicated that ME3 was involved in cell adhesion and tissue migration (Supplementary Fig. 4). Supplementary Fig. 4 illustrates the Gene Ontology (GO) enrichment analysis and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis result. According to studies, the epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET) processes in tumor cells are closely related to distant metastasis. When the EMT-related protein expression is altered, tight junction-related proteins also change, which is involved in the progress of tumor cells shedding from in-site into circulatory systems and distant engraftment [46,47,48,49]. We tested the alteration of EMT-related biomarkers when ME3 was knockdown or overexpressed using WB. When ME3 was downregulated, the expression of N-Cadherin, Vimentin, and E-Cadherin elevated, whereas E-Cadherin was downregulated (Fig. 9d). ME3 overexpression upregulated E-Cadherin while downregulating N-Cadherin and Vimentin (Fig. 9e). Meanwhile, ME3-overexpression plasmid transfection restored ME3 expression in AGS and MGC803 cells (with ME3 stably knocked down). After ME3 overexpression, the EMT-related biomarkers N-Cadherin and Vimentin were reduced, while E-Cadherin was increased (Fig. 9f). Additionally, the alteration of N-Cadherin in GC cells with ME3 knockdown or overexpression was detected using cell immunofluorescence, and the results were consistent with those of WB (Fig. 9g).
ME3 may affect GC progression via mTOR and STAT3 signaling
The above results suggested that ME3 protein down-regulation could stabilize HIF-1α and promote GC progression and metastasis. According to previous research, complex networks of signaling pathways are involved in tumorigenesis and progression. mTOR signaling activation and HIF-1α stabilization are important for T cell receptor (TCR) signaling downstream, such as T cell metabolic reprogramming and prolonged lifespan of peripheric T cells. Interleukin-6 (IL-6) is a proinflammatory cytokine that promotes HIF-1α transcription via the JAK-STAT3 signaling pathway in the hepatocellular carcinoma model [50]. Our study measured mTOR and STAT3 signaling-related proteins using WB. The result revealed mTOR pathway was activated and the STAT3 pathway was restrained when ME3 was knockdown, and vice versa (Fig. 10a).

ME3 might regulate GC progression via mTOR and STAT3 signaling pathway. (A-B) mTOR and STAT3 signaling altered when ME3 expression changed; (C) A schematic model illustrating our findings on ME3 regulation in GC progression
Discussion
Abnormal metabolism is an essential feature of malignant tumors [5]. Intracellular redox homeostasis is destroyed due to rapid tumor cell proliferation and limited material supplements. Intracellular ROS could act as a “second messenger” that coordinates activating multiple pathways, carcinogenic phenotype occurrence, promoting tumor proliferation and angiogenesis, and affecting the microenvironment of tumor cells, leading to component alteration of tumor niche and abnormal inflammatory response. These changes provide favorable conditions for tumor progression [51]. Hypoxia is a common microenvironmental factor in rapidly proliferative solid tumors [52]. Our preliminary proteomic sequence analysis results demonstrated that ME3 expression was reduced in patients with high metastatic tendencies. Notably, additional pathological analysis of our clinical samples revealed that ME3 protein expression in patients with GC was closely related to tumor infiltration depth, lymph node metastasis, and poor prognosis. We discovered that ME3 expression was significantly lower in the group with distant metastases than in the group without distant metastases by comparing the tissue ME3 protein levels of patients with simultaneous and (or) diachronic distant metastases to those without distant metastases. This result implies that ME3 expression deficiency may be one of the key steps in promoting distant metastasis development in GC. It was demonstrated that ME3 low expression promoted the malignant phenotype of GC cells. Combining previous research results and the enzymatic function of ME, we speculated that the aberrant ME3 expression in a hypoxic environment might promote the progression and metastasis of GC by regulating the activity of oxidative stress and the HIF-1α signaling pathway. We propose a molecular regulation model for ME3 low expression promoting GC progression and metastasis (Fig. 10b): (1) down-regulation of ME3 expression in the mitochondria increased malate flux in the TCA cycle by reducing malate shunt, which led to the acceleration of the TCA cycle and metabolism, thus providing sufficient energy for the rapid proliferation of GC cells. (2) ME3 oxidatively decarboxylates malate to form pyruvate in the mitochondria. This process is accompanied by the reduction of NADP+ to NADPH, and the knockdown of ME3 expression in hypoxic environment makes the NADPH/NADPH in gastric cancer cells Under hypoxic environment, knockdown of ME3 expression decreased the NADPH/NADP+ ratio in GC cells and increased intracellular ROS, which activated the HIF-1α signaling pathway. Under normoxia, there was no significant change in intracellular ROS, although malate and NADPH/NADP+ changed accordingly. (3) In hypoxic environment, α-KG content was decreased in ME3-knockdown GC cells and inhibited HIF-1α hydroxylation at the corresponding sites by PHDs, thus reducing degradation of HIF-1α by proteasome pathway. The expression of downstream target proteins is activated by the dimerization of HIF-1β and HIF-1α into the nucleus, promoting the growth and metastasis of gastric cancer cells. However, the pathway of how ME3 expression is reduced in GC tissues needs to be further explored.
ME3 is a mitochondria-localized malic enzyme. ME3 has received little attention in regulating tumor development compared to the other malic enzymes. ME1 is localized in the cytoplasm and catalyzes the same biochemical reaction as ME3. ME2 differs from ME1 and ME3 because it uses NAD+ as a cofactor to produce NADH, which regulates cellular redox homeostasis. In non-small cell lung cancer (NSCLC), ME2 knockdown led to tumor cell cycle arrest, elevated ROS level, promotion of cell death, and increased sensitivity to cisplatin [53]. ME3 is elevated in pancreatic cancer and is associated with poor prognosis [54]. In our study, we discovered for the first time that ME3 expression was reduced in GC tissues, and cells’ growth and metastatic ability were significantly enhanced after ME3 knockdown. The accelerated tumor proliferation was inverted using plasmid to overexpress ME3 in ME3-stable-knockdown GC cells. Recent studies discovered that drugs based on extraneous transcribed mRNA might have potential therapeutic efficacy in treating malignant and infectious diseases [55]. Thus, we assumed that our study might provide new clues for the design of targeted therapy for GC. Malic enzymes catalyze the biochemical reaction of malate to pyruvate, shunting malate, an intermediate product of the TCA cycle, into other biochemical reactions in mitochondria. The present study revealed that malate content was increased in GC cells with ME3 knockdown in normoxic and hypoxic environments. We discovered that GC cells in the knockdown group had accelerated oxidative phosphorylation rates and increased ATP production. Since intracellular substance and energy metabolism is a complex process, and malate is an essential intermediate product of the TCA cycle. Considering the above experiment results, we assumed ME3 knockdown inhibited the reaction of malate to pyruvate, leading to the malate shunt reduction and its flux accumulation in the TCA cycle. It benefited the TCA circulation and increased the oxygen consumption rate, thus providing energy for cell proliferation.
The mitochondrial oxidative respiratory chain mainly produces ROS under normal physiological conditions, and an appropriate amount of ROS is important for maintaining cell proliferation, differentiation, and other physiological processes. An excessive amount of ROS may activate apoptosis, autophagy, and other related signaling pathways, eventually leading to cell death. However, cancer cells have a higher intracellular ROS concentration than normal tissue cells due to increased metabolic demand and accelerated rate of oxidative phosphorylation. A high level of ROS can contribute to new intracellular redox homeostasis by activating certain signaling pathways that upregulate the production of antioxidant components and promote tumor progression [56]. However, excessive ROS production in mitochondria can inhibit mitochondrial autophagy and cause cell death because many chemotherapeutic agents depend on ROS for tumor-killing activity [57, 58]. ME3 contributes to the progress that is companied by the reduction of NADP+ to NADPH. ME3 expression has been involved in regulating NADPH levels in mitochondria and intracellular redox homeostasis [59, 60]. We measured the total ROS content in GC cells cultured under normoxia. We discovered that it did not change significantly, probably because the changes in NADP+ and NADPH caused by ME3 expression under normoxia were insufficient to affect the overall redox homeostasis in cells. This also confirmed that cells can self-regulate redox homeostasis within a certain range. This may be explained by the fact that changes in NADPH content caused by ME3 expression alteration in the normoxia environment were insufficient to affect the overall redox homeostasis; the cells can still compensate for such changes via their intrinsic regulatory mechanisms. In this study, we discovered for the first time that ME3 is important for regulating ROS under hypoxia. As mentioned previously, a moderate increase in ROS is beneficial for stimulating cell growth and proliferation. Low ME3 expression can increase intracellular ROS, promoting GC cell proliferation and metastatic ability by activating other signaling pathways, such as HIF-1α.
Numerous studies have demonstrated that HIF-1α is an important regulatory molecule in hypoxia and oxidative stress stimulation. HIF-1α, as the primary oxygen concentration response molecule, is sensitive to changes in oxygen concentration. HIF-1α and HIF-1β form a dimer and enter the nucleus as a transcription factor that drives the downstream gene expression by selectively binding to hypoxia response elements (HREs) under hypoxia [61]. HIF-1α protein is negatively regulated by ubiquitination and degraded by the proteasome pathway under normoxia conditions [21, 62]. We discovered that HIF-1α expression was upregulated in ME3 knockdown and overexpression groups of cells at the transcriptional level under a hypoxic environment, but the intracellular HIF-1α content was more abundant in GC cells with the ME3 knockdown group at the protein level. Hypoxia and ROS upregulation are important signals to activate the HIF-1 signaling pathway [63]. We hypothesized that ME3 knockdown in hypoxic increased HIF-1α expression by promoting the HIF-1α transcription, maintaining the HIF-1α stability, and slowing down its degradation. Cells primarily produce α-KG via the TCA cycle. It regulates numerous pathophysiological processes, such as skeletal muscle protein synthesis, lipid metabolism, and inflammatory responses, either as a substrate for other biochemical reactions or by directly regulating enzymatic activity [64,65,66]. It has been reported that the collagen prolyl-4-hydroxylase α1 subunit (P4HA1) can regulate α-KG and succinate in breast cancer cells to maintain the stability of HIF-1α protein, thus enhancing the stemness of BC cells [67]. The present study observed that the intracellular α-KG content changed when the ME3 expression was altered, especially in GC cells with ME3 knockdown in a hypoxic environment. We hypothesized that ME3 affected intracellular HIF-1α content by regulating the level of α-KG content and HIF-1α degradation rate.
Several studies have elucidated the molecular mechanisms associated with GC invasion and metastasis, including activation of the HIF-1α signaling pathway, tumor neovascularization, and EMT. It has been demonstrated that hypoxia-tolerant GC cells expressed higher levels of EMT-related protein markers Snail, Twist, ZEB, and Vimentin than their parental cells and exhibited stronger invasive ability and spindle-shaped morphology in vitro [68]. Moreover, hypoxia-tolerant GC cells were more likely to develop multiple peritoneal metastases and lymph node metastases in mice. The mTOR pathway activation could regulate the HIF-1α protein expression and is associated with tumor angiogenesis [69]. In this study, the enrichment analysis of ME3 co-expressed genes revealed that ME3 expression might be associated with epithelial adhesion. The mTOR and STAT3 may be involved in the ME3 promotion of GC progression and metastasis.
In summary, our study reported the ME3 expression in GC for the first time. We demonstrated that the ME3 knockdown promoted the proliferation and migration of GC cells in vitro and in vivo. We hypothesize that the potential mechanism of ME3 on the biological behavior of GC cells is as follows: ME3 knockdown accelerated the rate of oxidative phosphorylation and increased ATP production to meet the energy demand of cell proliferation. ME3 expression may promote the proliferation and migration of GC cells via mTOR, STAT3, and other signaling pathways. Particularly, ME3 downregulation in a hypoxic environment could increase intracellular ROS levels while decreasing α-KG levels, activating the HIF-1α signaling pathway, and maintaining the HIF-1α stability. The above mechanism allows HIF-1α signaling to remain continuously activated, stimulating various downstream signaling pathways and promoting GC tumor growth and metastasis. We hope that our research results will provide a theoretical and research basis for further understanding of the role of ME3 on GC development through the regulation of metabolites, intracellular oxidative stress levels and related signalling pathways and provide clues for further exploration of prognostic and therapeutic targets to improve the prognosis of patients with GC.
Our present study has some limitations. For example, we do not know whether ME3 is involed in GC carcinogensis and the upstream regulational mechanism was not sufficiently clear and need further study; other potential related metabolites mediated the progression of GC need to be further studied.
Conclusion
We provide experimental and clinical evidence that ME3 plays a role in GC progression. We demonstrate that ME3 promotes proliferation and metastasis via accelerating the TCA cycle and regulating α-KG to stabilize HIF-1α in GC cells, and ME3 may be a novel therapy and prognositic target in gastric cancer.
Data availability
The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.
Abbreviations
- AJCC:
-
American Joint Committee on Cancer
- GO:
-
Gene ontology
- BP:
-
Biology process
- CC:
-
Cellular component
- MF:
-
Molecular function
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- NADP+ :
-
β-nicotinamide adenine dinucleotide phosphate
- NADPH:
-
β-nicotinamide adenine dinucleotide phosphate, reduced form
- TCGA:
-
The Cancer Genome Atlas
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- STAD:
-
Stomach Adenocarcinoma
- EMT:
-
Epithelial-mesenchymal transition
- MET:
-
Mesenchymal-epithelial transition
- OS:
-
Overall survival
- PFS:
-
Progression-free analysis
- FDR:
-
False discovery rate
- α-KG:
-
α-ketoglutarate
- DMKG:
-
Dimethyl α-ketoglutarate
- OXPHOS:
-
Oxidative phosphorylation
- ROS:
-
Reactive oxygen species
- IHC:
-
Immunohistochemistry
- ATP:
-
Adenosine triphosphate
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
References
Bray F et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424
Thrift AP, El-Serag HB (2020) Burden of gastric Cancer. Clin Gastroenterol Hepatol 18(3):534–542
Smyth EC et al (2020) Gastric cancer. Lancet 396(10251):635–648
Sung H et al (2021) Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Missiroli S et al (2020) Cancer metabolism and mitochondria: finding novel mechanisms to fight tumours. EBioMedicine 59:102943
Cao Y et al (2020) Mitochondrial ROS accumulation inhibiting JAK2/STAT3 pathway is a critical modulator of CYT997-induced autophagy and apoptosis in gastric cancer. J Exp Clin Cancer Res 39(1):119
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947
Sabharwal SS, Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14(11):709–721
Gao P et al (2007) HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12(3):230–238
Weinberg F et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107(19):8788–8793
Sullivan LB et al (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51(2):236–248
Dong S et al (2022) ROS/PI3K/Akt and Wnt/beta-catenin signalings activate HIF-1alpha-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res 41(1):15
Ferreira F et al (2018) Early redox activities modulate Xenopus tail regeneration. Nat Commun 9(1):4296
Prabhakar NR, Semenza GL (2015) Oxygen sensing and Homeostasis. Physiol (Bethesda) 30(5):340–348
Matsuo Y et al (2014) Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J Hepatobiliary Pancreat Sci 21(2):105–112
Trastour C et al (2007) HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer 120(7):1451–1458
Deberardinis RJ et al (2008) Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 18(1):54–61
Carnero A, Lleonart M (2016) The hypoxic microenvironment: a determinant of cancer stem cell evolution. BioEssays 38(Suppl 1):S65–74
Sheta EA et al (2001) Cell density mediated pericellular hypoxia leads to induction of HIF-1alpha via nitric oxide and Ras/MAP kinase mediated signaling pathways. Oncogene 20(52):7624–7634
Wang XH et al (2021) Hypoxia-induced FOXO4/LDHA axis modulates gastric cancer cell glycolysis and progression. Clin Transl Med 11(1):e279
Kaelin WG Jr., Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30(4):393–402
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5(5):343–354
Shan T et al (2013) β2-AR-HIF-1α: a novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr Mol Med 13(6):1023–1034
Griffiths EA et al (2007) Hypoxia-inducible factor-1alpha expression in the gastric carcinogenesis sequence and its prognostic role in gastric and gastro-oesophageal adenocarcinomas. Br J Cancer 96(1):95–103
Stoeltzing O et al (2004) Role of hypoxia-inducible factor 1alpha in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst 96(12):946–956
Zhang R et al (2010) Subcellular distribution of S100A4 and its transcriptional regulation under hypoxic conditions in gastric cancer cell line BGC823. Cancer Sci 101(5):1141–1146
Rohwer N et al (2009) HIF-1alpha determines the metastatic potential of gastric cancer cells. Br J Cancer 100(5):772–781
Chang GG, Tong L (2003) Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry 42(44):12721–12733
Murai S et al (2017) Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis 6(5):e329
Ranzani AT et al (2017) Identification of specific inhibitors of Trypanosoma Cruzi Malic enzyme isoforms by Target-based HTS. SLAS Discov 22(9):1150–1161
Jiang P et al (2013) Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493(7434):689–693
Lu YX et al (2018) ME1 regulates NADPH homeostasis to promote gastric Cancer growth and metastasis. Cancer Res 78(8):1972–1985
Dey P et al (2017) Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 542(7639):119–123
Sobhanifar S et al (2005) Reduced expression of hypoxia-inducible factor-1alpha in perinecrotic regions of solid tumors. Cancer Res 65(16):7259–7266
Tormos KV et al (2011) Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab 14(4):537–544
Upadhyay M et al (2013) The Warburg effect: insights from the past decade. Pharmacol Ther 137(3):318–330
Li L, Li W (2015) Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther 150:33–46
Saha N, Jeremiah SJ, Povey S (1978) Further data on mitochondrial malic enzyme in man. Hum Hered 28(6):421–425
Sauer LA, Dauchy RT, Nagel WO (1979) Identification of an NAD(P)+-dependent ‘malic’ enzyme in small-intestinal-mucosal mitochondria. Biochem J 184(1):185–188
Maynard MA, Ohh M (2007) The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci 64(16):2170–2180
Minchenko OH et al (2016) Effect of hypoxia on the expression of nuclear genes encoding mitochondrial proteins in U87 glioma cells. Ukr Biochem J 88(3):54–65
Xiao W et al (2018) NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal 28(3):251–272
Gabriely G et al (2017) Role of AHR and HIF-1alpha in Glioblastoma Metabolism. Trends Endocrinol Metab 28(6):428–436
Magnani ND et al (2020) Role of Mitochondria in the Redox Signaling Network and its outcomes in high impact inflammatory syndromes. Front Endocrinol (Lausanne) 11:568305
Wu Y et al (2018) Gold Nanorod Photothermal Therapy alters cell junctions and actin network in Inhibiting Cancer cell collective Migration. ACS Nano 12(9):9279–9290
Cui B et al (2013) Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res 73(12):3649–3660
Song P et al (2019) Estrogen receptor β inhibits breast cancer cells migration and invasion through CLDN6-mediated autophagy. J Exp Clin Cancer Res 38(1):354
Aban CE et al (2021) Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT Sci Rep, 11(1): p. 2048
Hamaguchi Y et al (2016) Longer warm ischemia can accelerate tumor growth through the induction of HIF-1α and the IL-6-JAK-STAT3 signaling pathway in a rat hepatocellular carcinoma model. J Hepatobiliary Pancreat Sci 23(12):771–779
Costa A, Scholer-Dahirel A, Mechta-Grigoriou F (2014) The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol 25:23–32
Kitajima Y, Miyazaki K (2013) The critical impact of HIF-1a on gastric Cancer Biology. Cancers (Basel) 5(1):15–26
Ren JG et al (2014) Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci Rep 4:5414
Zhang Q et al (2019) Effects of ME3 on the proliferation, invasion and metastasis of pancreatic cancer cells through epithelial-mesenchymal transition. Neoplasma 66(6):896–907
Heine A, Juranek S, Brossart P (2021) Clinical and immunological effects of mRNA vaccines in malignant diseases. Mol Cancer 20(1):52
Snezhkina AV et al (2019) ROS generation and antioxidant Defense systems in normal and malignant cells. Oxid Med Cell Longev 2019:p6175804
Galadari S et al (2017) Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med 104:144–164
Gupta N et al (2020) Free radicals as a double-edged Sword: the Cancer Preventive and therapeutic roles of Curcumin. Molecules, 25(22)
Fan J et al (2014) Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510(7504):298–302
Lewis CA et al (2014) Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell 55(2):253–263
Wang GL et al (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92(12):5510–5514
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3(10):721–732
Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Activation of the HIF pathway in cancer. Curr Opin Genet Dev 11(3):293–299
Hammarqvist F et al (1991) Alpha-ketoglutarate preserves protein synthesis and free glutamine in skeletal muscle after surgery. Surgery 109(1):28–36
Vaz FM, Wanders RJ (2002) Carnitine biosynthesis in mammals. Biochem J 361(Pt 3):417–429
Cummins EP et al (2006) Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A 103(48):18154–18159
Xiong G et al (2018) Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun 9(1):4456
Kato Y et al (2010) Establishment and characterization of a new hypoxia-resistant cancer cell line, OCUM-12/Hypo, derived from a scirrhous gastric carcinoma. Br J Cancer 102(5):898–907
Lang SA et al (2007) Mammalian target of rapamycin is activated in human gastric cancer and serves as a target for therapy in an experimental model. Int J Cancer 120(8):1803–1810
Acknowledgements
The authors thank Dr. Jionghui Gu and Dr. Minfeng Ying for technical assistance.
Funding
This work was supported by project of the regional diagnosis and treatment center of the Health Planning Committee (No. JBZX-201903).
Author information
Authors and Affiliations
Contributions
HYY participated in the conception and design, figure, data analysis and paper writing; YY contributes to literature search and design; CXL and ZSY contributes to analysis and methodology; CYR and WHY contributes to review; LXD contributes to data curation; HX contributes to supervision; TLS contributes to funding acquisition, supervision and project guidance. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Gastric cancer tissue and para-cancerous tissue samples were randomly collected from patients undergoing gastrectomy between 2015 and 2020 in the First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang Province, China (Approval Number: 2021 − 869). All the participants have signed “written informed consent” to take part in this study. All animal experiments were approved by the Ethics Committee for Laboratory Animals of the First Affiliated Hospital of Zhejiang University (Approval number: 2021 − 1431, 2022 − 1060).
Consent for publication
All authors gave their consent for publication.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Yang, Y., Chen, X. et al. Downregulation of malic enzyme 3 facilitates progression of gastric carcinoma via regulating intracellular oxidative stress and hypoxia-inducible factor-1α stabilization. Cell. Mol. Life Sci. 81, 375 (2024). https://doi.org/10.1007/s00018-024-05388-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05388-9