
Abstract
Lentiviral vectors have markedly enhanced gene therapy efficiency in treating congenital diseases, but their long-term safety remains controversial. Most gene therapies for congenital eye diseases need to be carried out at early ages, yet the assessment of related risks to ocular development posed by lentiviral vectors is challenging. Utilizing single-cell transcriptomic profiling on human retinal organoids, this study explored the impact of lentiviral vectors on the retinal development and found that lentiviral vectors can cause retinal precursor cells to shift toward photoreceptor fate through the up-regulation of key fate-determining genes such as PRDM1. Further investigation demonstrated that the intron and intergenic region of PRDM1 was bound by PHLDA1, which was also up-regulated by lentiviral vectors exposure. Importantly, knockdown of PHLDA1 successfully suppressed the lentivirus-induced differentiation bias of photoreceptor cells. The findings also suggest that while lentiviral vectors may disrupt the fate determination of retinal precursor cells, posing risks in early-stage retinal gene therapy, these risks could potentially be reduced by inhibiting the PHLDA1-PRDM1 axis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene therapy presents promising treatment options for numerous severe diseases by introducing normal protein-coding genes to treat hereditary diseases. Notably, this therapeutic approach has demonstrated improved retinal function in patients with Leber’s congenital amaurosis (LCA) [1, 2] and has shown anti-leakage activity in neovascular age-related macular degeneration [3].Currently, widely used viral vectors in gene therapy include adenovirus, lentivirus, and adeno-associated virus. Among these, lentivirus, an improved retrovirus derived from HIV, stands out for its effective gene transfer, stable expression [4], and relatively large transgene carrying capacity [5]. Nevertheless, previous studies have raised concerns about lentiviral vector-mediated transduction, highlighting possible adverse reactions such as tumor formation [6], immune response, retinal degeneration [7], and the exacerbation of retinal pigment epithelial atrophy [8]. Importantly, gene therapies for genetic developmental disorders, such as enhanced S-cone syndrome caused by NRL mutations [9] and photoreceptor ciliary defects caused by CEP290 mutations [10], need to be carried out at early ages. However, the assessment of related risks to ocular development posed by lentiviral vectors is challenging. A comprehensive understanding of the impact of lentiviral infection on the human developing retina is important for a thorough assessment of gene therapy in the retinal genetic diseases.
Due to ethical, technical, and biological limitations, it is not feasible to assess the impact of lentiviral infection on the developing system in vivo. While animal models such as mice have been widely used for in vivo experiments, significant differences in molecular characteristics and structure of rodent and human retinas [11] pose challenges in fully evaluating the impact of lentiviral vectors on retinal development. The development of human retinal organoid culture technology has provided a potential solution, as human retinal organoids can resemble the development of the retina and possess mature photoreceptor cells [12]. Compared to mice, retinal organoids have a more similar developmental sequence, cell types, and layered structure, providing an ideal model to replicate the development process of human embryonic retina in vitro [13,14,15]. Moreover, the lack of immune cells in the development of human retinal organoids eliminates the confounding effects of immune responses induced by lentivirus infections. This permits a clearer assessment of the lentiviral vectors’ influence on retinal neuron development without the variability introduced by immune-mediated reactions [16].
In this study, we used human retinal organoids as a model to evaluate the effects of lentiviral vectors on retinal development. Our results demonstrated that lentiviral infection can disturb the differentiation of retinal progenitor cells (RPCs) into photoreceptor cells. Single-cell transcriptome analysis revealed that lentiviral vectors can stimulate the up-regulation of the key gene PHLDA1, inducing the activation of gene transcription networks associated with photoreceptor cell specialization. Furthermore, by binding to the intron and intergenic regions of PRDM1, PHLDA1 can upregulate PRDM1 expression, thereby regulating the fate of photoreceptor cell specialization. The findings also suggest that while lentiviral vectors may disrupt the fate determination of retinal precursor cells, posing risks in early-stage retinal gene therapy, these risks could potentially be reduced by inhibiting the PHLDA1-PRDM1 axis.
Materials and methods
Cell culture
Y79 cells were cultured in RPMI 1640 medium supplemented with 15% fetal bovine serum. Human embryonic stem cell line H9 (kindly provided by Professor Qinghuai Liu, Nanjing Medical University) was cultured in Essential 8 (E8) medium (Invitrogen) on Vitronectin (VTN-N)-coated plates. Mesenchymal progenitor cells (MPCs) were derived from the human embryonic stem cell line H9 (hESC H9) using the STEMdiff™ Mesenchymal Progenitor Kit (Stemcell, 05240) both of cell lines were incubated at 37 °C with 5% CO2.
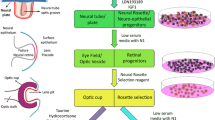
Three-dimensional (3D) retinal organoid differentiation from hESCs
The differentiation of human 3D retinal organoids was based on the differentiation protocol reported by the research of Sasai and David Gamm and further optimized [17, 18].
Retinal organoids were derived from the human embryonic stem cells (hESCs). Initially, hESCs were maintained on Vitronectin (VTN-N) matrix with E8 medium. When the confluence of hESCs reached 70%, Dispase (2 mg/mL) was used to dissociate to harvest embryoid bodies (EBs). And within the next four days, the culture medium was gradually transitioned from E8 medium to neural induction medium (NIM: DMEM/F12, [1:1], 1% N2 supplement, 1% MEM non-essential amino acids, and 2 mg/mL heparin sulfate). On the sixth day, 50 ng/mL BMP4 recombinant protein was introduced to the NIM medium. Subsequently, on the seventh day, NIM medium supplemented with 10% FBS was employed to facilitate the adherent growth of EBs. After 24 h, the medium was refreshed and subsequently half-replaced regularly. On the sixteenth day, EBs were lifted to obtain retinal organoids and retinal differentiation medium (RDM: DMEM/F12[3:1], 2% B27 supplement, 1% MEM non-essential amino acids, 1% penicillin-streptomycin) was employed for further culture. Starting from day 30, the RDM medium was enriched with 10% FBS, 100 μM taurine, 2 mM GlutaMAX, and 0.5 μM retinoic acid (abbreviated as RDM+3) for the long-term culture of retinal organoids.
Lentiviral transfection
We employed two kinds of third-generation lentiviral vectors, a non-targeting human genome and another targeting hPHLDA1 knockdown, to infect developing retinal organoids. The maps of lentiviral vectors are provided in Supplementary Fig. 1. Human retinal organoids with similar sizes and morphological structures were selected and randomly divided into groups of 4–5. Each retinal organoid was transfected with 1 × 106 viral transduction units in RDM + 3 medium supplemented with 5 µg/ml polybrene. After 24 h, the virus-containing medium was removed, and the organoids were maintained under normal culture conditions for 1 week before evaluation. Additionally, Y79 cells from the RB cell line were seeded into a six-well plate at a density of 2 × 106 cells per well, and the lentivirus was diluted to 2 × 107 viral transduction units per milliliter in RPMI 1640 medium. Following a 24-hour transfection period, the virus-containing medium was removed, and the cells were cultured under normal conditions. The cells were harvested 3 days post-infection, and the knockdown of target genes was evaluated by RT-qPCR and Western blot analysis.
Immunofluorescence and imaging
Human retinal organoids were fixed in 4% paraformaldehyde for 30 min at 4 °C, washed with PBS, dehydrated in sucrose concentrations of gradients 6.25-12.5%-25% overnight at 4 °C, embedded in OCT, and sectioned (5 μm thickness) for storage at -80 °C. The entire immunofluorescence staining is performed at room temperature. Sections were incubated with blocking buffer(0.5% Triton X-100/PBS, 1% BSA)for 1 h, incubated with primary and secondary antibodies for 2 h for each, counterstained with DAPI(1:1000)washed, and finally mounted.
Antibody details are in Appendix Table-2. Confocal images were quantified using FIJI-ImageJ, comparing immunostaining intensity to DAPI values.
RT-qPCR
Total RNA was extracted using TRIzol™ Reagent (Invitrogen, 15,596,026), and 1 µg was reverse-transcribed to cDNA using HiScript III RT SuperMix for qPCR (Vazyme, R323-01). Use NCBI-Primer-BLAST to design RT-qPCR primers, and use the two-step method of AceQ Universal SYBR qPCR Master Mix (Vazyme, Q511-03) for RT-qPCR detection to obtain the Ct value. Relative gene expression was calculated using 2−△△Ct method, normalized against β-actin.
Western blot
Cells were lysed using RIPA lysis buffer (50 mM Tris (pH 7.4), 0.1% SDS) and boiled for 10 min at 105 ℃. Whole cell lysates were quantified via BCA kit (Thermo Fisher Scientific, 23,252) before mixing with 5×SDS loading buffer. The prepared samples were separated by SDS-PAGE, and the electrophoretically separated bands were transferred from the gel to PVDF membrane (Millipore, IPVH00010). Membranes were then blocked with 5% skim milk (Biofroxx, 1172GR500) at room temperature for 1 h and incubated with primary antibody at 4 °C overnight. After TBST washing, the membranes were incubated with horseradish peroxidase (HRP)-labeled secondary antibody at room temperature for 1 h. ECL luminescence solution (Millipore, WBKLS0100) and Tanon5200 fully automated chemiluminescence image analysis system was used to obtain bands and ImageJ was used for quantitative analysis.
TUNEL staining
The presence of DNA strand breaks was detected using fluorescent terminal dUTP nick end labeling (TUNEL; Serologals Corporation, Norcross, GA, USA) in the retinal organoids’ sections according to the manufacturer’s instructions. The number of apoptotic cells was counted by FIJI-ImageJ.
CUT&Tag
The Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme Biotech, TD903) was used for CUT&Tag analysis. The raw reads of CUT&Tag were trimmed to 40 bp, and low-quality reads were removed using Trimmomatic v0.32. Paired reads were aligned to the human genome (version hg38) using Bowtie2 v2.3.4.2 with the parameters: “-X 2000 -no-discordant –no-contain”. Reads with a mapping quality (MAPQ) below 10 and PCR duplicated reads were filtered out using Samtools and Picard. SCEAR was used to call CUT&Tag peaks with the parameters: “--broad --broad-cutoff 0.1 -B –SPMR”. The fold enrichment for the peaks compared to a random Poisson distribution (with lambda) had to be greater than 10. The normalized signals of CUT&Tag were represented as the fold change of the treatment over the lambda control (whole genome) using macs2 bdgcmp and converted to BigWig format using bedGraphToBigWig. Peaks were annotated using chipseeker.
RNA-seq analysis
The paired-end clean reads were aligned to the reference genome using STAR. The read counts for each gene were quantified using featureCounts. DESeq2 was employed for differential expression analysis, and the resulting p-values were adjusted using the Benjamini and Hochberg’s approach to control the false discovery rate. Genes with |log2 (Fold Change) | > 1 & adjusted P value < 0.05 were assigned as differentially expressed. GO and KEGG enrichment analysis of differentially expressed gene sets were implemented by the topG.
Retinal organoid cell dissociation for scRNA-seq
We selected 4–5 retinal organoids with similar structural layering for experiments at the rod development time point, treated with Accutase for 30 min at 37 °C to dissociate into single-cell suspensions, and then filtered through a 40 μm cell strainer, and finally resuspend the cells in PBS containing 0.04% bovine serum albumin. ScRNA-seq libraries were prepared using the Single Cell Gene Expression 3’V2 Kit (10×Genomics, Pleasanton, CA, USA) following the manufacturer’s protocol. Briefly, single cells were distributed onto latex gel beads (GEM) within a Chromium instrument, followed by cell lysis, reverse transcription, cDNA amplification, and library construction. All libraries were sequenced on the Illumina HiSeq 2500 platform.
ScRNA-seq analysis
Preprocessing of scRNA-seq data
We demultiplexed and aligned the raw scRNA-seq data to the human reference genome (GRCh38) using Cell Ranger (version 3.1) with default parameters. The expression level of each transcript was determined using UMI and transcripts were assigned to cells based on barcode. The genes filtered by the software were then used for subsequent analyses.
Processing and cell-type annotation of human retinal organoids scRNA-seq data
Briefly, different cell markers were used to annotate cell types. Principal component analysis (PCA) was performed on the integrated data to reduce dimensionality, and a k-nearest neighbor graph (k = 30) was constructed based on Euclidean distance in the salient PC space. Different cell clusters were identified using a Louvain-Jaccard graph-based method and labeled based on marker genes and cell type annotation from a previous study [16].
Identification of differentially expressed genes in scRNA-seq data sets
Model-based single-cell transcriptomics (MAST) [19]analysis was used to identify DEGs for each cell type. The identified DEGs were then tested against the asymptotic chi-square null distribution. Genes with FDR correct P < 0.05 were considered to be differentially expressed.
Gene ontology analysis of DEGs
Gene ontology (GO) analysis was performed using clusterProfiler (version 4.0) [20]and visualized using the ggplot2 R package (https://github.com/tidyverse/ggplot2). GO terms or pathways were considered enriched at adjusted P < 0.05.
Statistical analysis
All experiments were performed at least three times. Statistical analysis of data between different groups used GraphPad Prism 8 software for two-tailed unpaired t-test. Data were presented as mean ± SEM. An unpaired two-tailed Student’s t-test was used to determine significance, denoted by ns, not significant; *p < 0.05; ***p < 0.001 and ****p < 0.0001.
Results
Lentiviral vectors disrupt the normal differentiation of retinal progenitor cells into photoreceptor cells
To further assess the impact of viral infection on retinal development, we infected retinal organoids with an empty lentiviral vector (Supplementary Fig. 1A). Given that different retinal cell types have distinct developmental windows, we performed lentiviral infections (MOI = 10) at both early (week 7) and late (week 13) developmental stages. One week post-infection, we observed that lentiviral infection did not cause any significant morphological or structural changes in the retinal organoids (Fig. 1A-D). Further analysis using immunofluorescence staining revealed that lentiviral infection led to an increase in neurogenic RPCs (ASCL1+ cells) (Supplementary Fig. 2G-H), cone photoreceptors (OTX2+, CRX+, PRDM1+ cells at week 8), and rod photoreceptors (OTX2+, CRX+, PRDM1+ cells at week 14) (Fig. 1E-H). However, other cell types remained unaffected, including retinal ganglion cells (RGCs; ISLET1+ cells), horizontal cells (HCs; ONECUT2+ cells), and amacrine cells (ACs; TFAP2A+ cells) (Supplementary Fig. 2A-F). These results suggest that lentiviral vectors can cause to aberrant differentiation of photoreceptor cells in retinal organoids.

Lentiviral vectors disrupt the normal differentiation of retinal progenitor cells into photoreceptor cells. A-D Bright-field images of retinal organoids at both early and late stages in mock and lentivirus-infected groups. Scale bars, 500 μm. E-H In immunofluorescence images, the early and late stages of retinal organoids exhibited a significant increase in the numbers of CRX+, PRDM1+, and OTX2+ photoreceptor cells within the lentivirus-infected group compared to the mock group. Scale bars, 20 μm
PHLDA1 mediates the effects of lentiviral infection on the specification of retinal progenitor cells into photoreceptor cells
To unravel the molecular mechanisms responsible for the abnormal development of photoreceptor cells induced by lentiviral vectors, we performed single-cell RNA sequencing on both unaffected (Mock) and lentiviral vectors infected (Lentivirus) retinal organoids, identifying transcriptional profiles of 14,086 cells totally. Through cell clustering and cell type annotation, we identified 9 retinal cell types, including proliferative RPCs, cones, rods, bipolar cells, photoreceptor /bipolar precursors, horizontal /amacrine precursors, amacrine cells, retinal ganglion cells, horizontal cells, etc. (Fig. 2A-B). Differential gene expression (DGE) analysis further revealed significant changes in the gene expression patterns of all retinal cell types following lentiviral infection of retinal organoids (Fig. 2C, Supplementary Fig. 3). To explore mechanisms underlying the abnormal differentiation of photoreceptor cells induced by lentiviral vectors, we first analyzed the expression patterns of photoreceptor fate-determining genes following lentiviral infection, and found an up-regulation of photoreceptor fate-determining genes (OTX2, CRX, PRDM1, RAX, RXRG and PHLDA1, etc.) [21,22,23] (Fig. 2D, Supplementary Fig. 4). We further observed a correlation between PHLDA1 and other critical genes such as CRX, OTX2, and PRDM1 via gene correlation analysis in all cells (Fig. 2E). By performing lentiviral infection on retinoblastoma cell line Y79, we found that the MOI of lentiviral vectors was positively correlated with the protein expression level of PHLDA1 (Fig. 2G-H). Subsequently, we confirmed that lentiviral infection significantly increased the number of PHLDA1+ cells in retinal organoids through immunofluorescence staining (Fig. 2I-J). We next collected transcriptome data from cells infected with various viruses including SARS-CoV-2, RSV, ZIKA, and VSV-G. The results showed consistent up-regulation of PHLDA1 across different cell types upon different viral infections, suggesting PHLDA1 is a key intracellular regulator upon viral infections (Fig. 2F). In summary, our results indicate that PHLDA1 may play an important role in driving abnormal differentiation of photoreceptor cells during lentiviral infection.

Lentiviral infection induces significant up-regulation of PHLDA1 expression. A-B UMAP analysis of scRNA-seq comparing the lentivirus-infected group to the mock group in retinal organoids (W13), including 9 distinct cell types: RPC, (retinal progenitor cell); Cone/Rod, (photoreceptor cell); BC, (bipolar cell); PR/BC pre, (photoreceptor cell/bipolar cell precursor); HC/AC pre, (horizontal cell/amacrine cell precursor); AC, (amacrine cell); RGC, (retinal ganglion cell); HC, (horizontal cell). C Differential gene expression patterns were observed in the lentivirus-infected cells of human retinal organoid. D Dot plot showing the up-regulation of photoreceptor fate-determining genes in retinal organoids caused by lentiviral infection. E Gene correlation analysis shows the key photoreceptor fate-determining genes such as CRX, PRDM1, and OTX2 exhibited a high positive correlation with PHLDA1 F Transcriptome analysis showing significantly up-regulation of PHLDA1 caused by the transfections of SARS-CoV-2, RSV, ZIKA, and VSV-G viruses compared with the corresponding mock group. G-H Western blot analysis of PHLDA1 in Y79 cells at varying multiplicity of infection (MOI = 10, MOI = 20). I-J The number of PHLDA1+ cells was significantly up-regulated in the lentivirus group compared to the mock group in both early (week 8) and late (week 14) developmental stages? human retinal organoids. Scale bars, 20 μm
Our previous research found that knockdown of PHLDA1 led to an increase in proliferative RPCs numbers [21], suggesting that PHLDA1 may promote RPC differentiation into photoreceptor cells. In this study, we conducted cell proliferation and apoptosis assays in retinal organoids infected with lentiviral vectors and found that lentiviral infection led to a decrease in RPCs (MKI67+) and an increase apoptotic cells (TUNEL+, from 0.45% to 1.07% for W8, and from 0.72% to 1.17% for W14, respectively) (Fig. 3A-D, Supplementary Fig. 5A-D), but the number of apoptotic cells is significantly lower than the decreased proliferating cells. Therefore, these results indicate that the increase in the number of photoreceptor cells may be due to abnormal RPC differentiation caused by lentiviral infection. Lentiviral vectors were employed to mediate PHLDA1 knockdown in Y79 cells. Further, RT-qPCR and Western blot analyses revealed that this approach significantly decreased PHLDA1 expression levels initially upregulated by the lentiviral vectors (Fig. 3E-F). Additionally, the lentiviral vector-mediated knockdown of PHLDA1 markedly inhibited the rise in photoreceptor cell numbers (RXRG+, NR2E3+, CRX+, OTX2+) typically triggered by lentiviral infection in retinal organoids (Fig. 3G-J, Supplementary Fig. 6–7). These results indicate that the interference with the fate of photoreceptor cells affected by lentiviral vectors is mainly through the up-regulation of PHLDA1, thereby triggering the fate deviation of RPCs towards photoreceptor cells.

PHLDA1-mediated the effects of lentiviral infection on the specification of retinal progenitor cells into photoreceptor cells. A-D Immunofluorescence revealed that in both early (week 8) and late (week 14) developmental human retinal organoids, lentiviral infection resulted in a reduction of KI67+ retinal progenitor cells compared with the mock control group. Scale bars, 20 μm. E-F RT-qPCR and Western blot analysis of PHLDA1 in Y79 cells in the lentivirus group and shPHLDA1(KD) group. G-H Immunofluorescence showed that the knockdown of PHLDA1 markedly suppressed the lentivirus-induced increase in RXRG+ cells numbers in early-stage retinal organoids. Scale bars, 20 μm. I-J Immunofluorescence showed that knockdown of PHLDA1 significantly obstructed the rise in NR2E3+ cells counts caused by lentiviral infection in the late stages of retinal organoids. Scale bars, 20 μm
PHLDA1 regulates the expression of key transcription factors that determine photoreceptor cell fate
Structural analysis of PHLDA1 revealed the presence of QQ and PQ domains, which are commonly found in transcription factors, implicating a potential involvement of PHLDA1 in regulating gene expression [24, 25]. Based on these findings, we hypothesized that PHLDA1 may be involved in regulating the expression of genes crucial for determining the fate of photoreceptor cells during their differentiation process. To examine this hypothesis, we first performed immunofluorescence to determine the subcellular localization of PHLDA1. Our results indicated that PHLDA1 was present in both the cytoplasm and the nucleus (Supplementary Fig. 8A–C) [26], suggesting a possible role in gene regulation.
To further elucidate the role of PHLDA1 in gene expression, we established three stable Y79 cell lines following lentiviral infection and performed CUT&Tag experiments coupled with RNA-seq. Our analyses revealed that PHLDA1 was bound to DNA elements and influenced the expression of target genes. By integrating CUT&Tag with gene regulatory network analysis, we identified the potential role of PHLDA1 in mediating photoreceptor differentiation. This analysis showed that PHLDA1 was bound to DNA regulatory elements of key genes involved in determining the fate of photoreceptor cells, including RAX, PRDM1, NR2E3, and NRL (Fig. 4A-C). Correspondingly, RNA-seq data showed the significant up-regulation of genes crucial for determining the fate of photoreceptor cells, such as RAX, PRDM1, ATOH7, RXRG, and PDE6H following lentiviral infection. (Fig. 4D). We further investigated the binding sites of PHLDA1 in these genes, discovering they bind to the intronic and intergenic regions of PRDM1, as well as the promoter region of RAX (Fig. 4E-F).

PHLDA1 possesses transcription factor activity. A Bar graph depicted the genomic distribution of PHLDA1 binding sites. B Venn diagram compared the genes identified by CUT&Tag and RNA sequencing as differentially expressed between the lentivirus-infected and control (mock) groups. C Gene network diagram showed key genes for photoreceptor cell fate determination with PHLDA1 binding sites. D The volcano plot showed that compared with the mock group, several crucial genes associated with photoreceptor cell fate determination were notably up-regulated following the lentiviral infection of Y79 cells. E Trackplot showed that PHLDA1 could bind to the PRDM1 intergenic region, as well as to the RAX promoter region. F The bar-plot showed the difference in read counts between IgG and PHLDA1. G Single-cell transcriptome analysis revealed that the expression of PRDM1 and RAX in photoreceptor cells was significantly up-regulated in the lentivirus-infected group compared to the mock group. H-I Immunofluorescence staining images displayed the co-expression of PHLDA1 with PRDM1 in human retinal organoids at different development stages. Scale bars, 20 μm
Finally, we validated these findings by comparing single-cell RNA-seq data from lentivirus-infected retinal organoids with those from mock control. Our results indicated that lentiviral infection induced elevated expression of PHLDA1 in photoreceptor cells, accompanied by a significant up-regulation of PRDM1 and RAX expression. Moreover, PRDM1 showed up-regulation in different cells transfected with different viruses, consistent with PHLDA1 (Fig. 4G, Supplementary Fig. 9). Subsequently, through immunofluorescence staining, we found that PHLDA1 is predominantly co-expressed with PRDM1 and RAX at different developmental stages in retinal organoids (Fig. 4H-I, Supplementary Fig. 10A-B). And we also found that lentivirus vector infection significantly increased the co-location of PHLDA1 and RAX, and knockdown PHLDA1 reduced that, suggesting PHLDA1 can regulate the expression of RAX in retinal organoids (Supplementary Fig. 10C-D). Based on these findings, we hypothesize that PHLDA1 may possess transcriptional activity and disrupt the specialization of photoreceptor cells during lentiviral infection by activating the expression of RAX and PRDM1.
Reducing PHLDA1 expression neutralized the lentiviral vector-induced disturbance in the fate specification of retinal progenitor cells
Previous studies have demonstrated that PRDM1 (encoding for BLIMP1 transcription factor) plays a pivotal role in inhibiting the differentiation of photoreceptor/bipolar precursors into bipolar cells while promoting their differentiation into photoreceptors [27]. Building upon this foundation, we hypothesized that the PHLDA1-PRDM1 axis was critically involved in regulating the aberrant differentiation of photoreceptors. To validate this hypothesis, we initially investigated the influence of PHLDA1 on PRDM1 expression in the Y79 cell line. Through lentiviral vector-mediated shRNA, we successfully knocked down PHLDA1, which subsequently led to a significant reduction in PRDM1 expression (Fig. 5A-B). Subsequently, we further knocked out PHLDA1 in Y79 cells, with the results demonstrating that PHLDA1 knockout significantly inhibited the up-regulated expression of PRDM1 induced by lentiviral infection (Fig. 5C-F, Supplementary Fig. 11). These findings suggest that PHLDA1 is likely to modulate the expression of PRDM1 in photoreceptor precursors. Additionally, we performed lentivirus-mediated knockdown of PHLDA1 in retinal organoids during the developmental stages of cones and rods, respectively, and found a significant reduction in the number of PRDM1+ cells (Fig. 5G-J). Notably, we also observed an overproduction of photoreceptor cells, as evidenced by an increased count of PRDM1+ cells at both developmental stages, following lentivirus infection. Interestingly, the number of PRDM1+ cells post-PHLDA1 knockdown was comparable to that in the mock group (Fig. 5G-J), implying that PHLDA1 may play a dominant role in modulating photoreceptor production during lentiviral infection. In summary, our data indicate that PHLDA1 is a key regulator in the specification into photoreceptors within RPCs, and its knockdown seems to mitigate the effects of lentiviral vectors on RPCs fate determination.

Lentiviral infection activates PHLDA1-PRDM1 and promotes photoreceptor cell specialization. A-B RT-qPCR and Western blot analyses demonstrated that silencing PHLDA1 in Y79 cells markedly suppressed the lentivirus-induced up-regulation of PRDM1 expression. C-D Details on the specific regions knocked out and the mutation sites of PHLDA1 in this study. E-F RT-qPCR and Western blot demonstrated that knockout of PHLDA1 significantly reduced the lentivirus-mediated up-regulation of PRDM1 expression in Y79 cells. G-J Immunofluorescence imaging showing the PRDM1+ photoreceptor cells in W9 and W14 retinal organoids across different groups: control (Mock), lentivirus-infected(Lentivirus), and PHLDA1 knockdown (shPHLDA1#1, shPHLDA1#6). Scale bars, 20 μm
Discussion
In this study, we found that lentiviral vectors’ stimulation led to abnormal development of photoreceptor cells in human retinal organoids. Further analysis of single-cell transcriptome data revealed that the expression of the photoreceptor cell differentiation regulatory gene PHLDA1 was up-regulated after virus infection. Furthermore, through CUT&Tag experiments, we validated the transcription activation properties of PHLDA1 and discovered its ability to regulate the expression of key transcription factors involved in photoreceptor cell fate determination, such as RAX and PRDM1. This regulatory function of PHLDA1 promotes the differentiation of RPCs into photoreceptor cells. We identified the phenotypic and molecular mechanisms of abnormal development of photoreceptor cells in the human retina caused by lentiviral vectors.
By analyzing transcriptome data from various cell types infected with different viruses, we found that PHLDA1 was commonly upregulated (Fig. 2F), suggesting that PHLDA1 up-regulation may be a common response of various cell types to viral infection. Several potential molecular mechanisms may contribute to virus-induced PHLDA1 up-regulation, such as ER stress, autophagy and inflammation. Firstly, studies have shown that PHLDA1 expression is significantly upregulated following treatment with various ER-stress-inducing agents, including dithiothreitol (DTT), thapsigargin, tunicamycin, and farnesol, whereas agents that attenuate ER stress, such as salubrinal (a selective eIF2⍺ inhibitor) and intracellular calcium chelator BAPTA, mitigate the up-regulation of PHLDA1 [28,29,30]. Secondly, autophagy is considered as a defense strategy in organisms and plays an antiviral function. Upon viral infection, host cells rapidly initiate autophagy to degrade viral particles or virus components. Studies have shown that Rapamycin, a well-known autophagy activator, upregulates PHLDA1 expression in T-47 mammary cells [31]. Thirdly, in immune cells, such as microglia [32] and bone marrow-derived macrophages [33], lipopolysaccharide (LPS) treatment can increase the expression level of PHLDA1, which was mediated by the TLR2/4 signaling pathway. PHLDA1 could activate NF-κB signaling to promote the expression of pro-inflammatory cytokines, including TNF-α, IL-1β, iNOS, and COX-2. Some studies have revealed how lentiviral infection causes the above pathways. For example, the envelope protein VSV-G is used to package viruses, but research has found that it can activate signaling pathways through the TLR4 and CD14 receptors, triggering the production of type I interferons [34]. Recent studies have identified that P53 is the key regulatory factor that regulates the PHLDA1 expression by recruiting CBP and P300 to its promoter region [35], which may potentially serve as a hub of virus-induced PHLDA1 up-regulation. P53 can be activated by viral infection through various pathways. For example, cellular stress (such as oxidative stress and endoplasmic reticulum stress) can also promote P53 phosphorylation [36]. Inflammatory factors induced by viral infection (such as interferons and TNF-α) can further promote P53 activation and expression [37, 38]. Moreover, viral DNA or RNA can also induce P53 activation. For instance, HIV RNA can be recognized by the intracellular RIG-I and MDA5 receptors to promote type I interferon production, which promotes P53 activation [39, 40]. In conclusion, viral infection can induce PHLDA1 up-regulation through multiple pathways, including ER stress, interferons and pro-inflammatory, and these pathways finally activate P53, which leads to increased PHLDA1 expression.
In this study, we found that PHLDA1 possessed potential transcriptional regulatory activity as demonstrated by CUT&Tag experiments revealing its ability to bind to the RAX promoter region and the PRDM1 intronic region. Knockdown or knockout of PHLDA1 significantly reduced the expression levels of both RAX and PRDM1. Previous research has established the crucial role of RAX in photoreceptor cell development. RAX directly binds and activates the EELPOT enhancer on the OTX2 gene, thereby activating OTX2 transcription. Conditional knockout of RAX in photoreceptor precursors results in decreased OTX2 expression [41]. Furthermore, the absence of RAX activity in the early retinal progenitors, due to deletion by the Pax6α-Cre driver, leads to the loss of cone and late-born retinal neurons, such as rods and bipolar cells. Therefore, PHLDA1 may promote the differentiation of RPCs into photoreceptor/bipolar precursors by upregulated RAX expression. Our results also show that PHLDA1 can bind to and regulate the expression level of PRDM1, a gene known to play an important role in regulating the differentiation of OTX2+ cells into photoreceptor cells while inhibiting their differentiation into bipolar cells through the suppression of VSX2 expression [27]. Moreover, ectopic expression of PRDM1 in immature bipolar cells leads to their differentiation into photoreceptor cells [42]. Therefore, these findings suggest that PHLDA1 may maintain the specific differentiation of nascent OTX2+ cells into photoreceptor cells rather than bipolar cells by promoting PRDM1 expression. In summary, these pieces of evidence suggest that PHLDA1 potentially facilitates the differentiation of photoreceptor cells via a biphasic mechanism. Initially, it upregulates RAX in RPCs, promoting their differentiation into OTX2+ photoreceptor/bipolar precursors; subsequently, PHLDA1 enables the differentiation of OTX2+ cells into mature photoreceptors by upregulating PRDM1. Further research is required to elucidate the molecular mechanisms through which PHLDA1 mediates the photoreceptor/bipolar fate bifurcation.
As a downstream target gene of PHLDA1, it has been that PRDM1 is an important transcriptional repressor that plays multiple roles in viral infection. Additionally, viral infection can induce PRDM1 expression [43]. By binding to the IFN-β promoter, it inhibits its transcription, thereby exerting a negative feedback regulation after IFN-I induction [44, 45] and promoting viral replication. In plasmacytoid dendritic cells (pDCs), PRDM1 promotes type I interferon production and enhances the antiviral response [46]. Our findings suggest that PHLDA1 may have a role in promoting the interferon pathway in other immune systems, however, the specific functions of this pathway in other viral infection systems still require further research.
The use of lentiviral vectors in gene therapy has always been controversial [47, 48]. Our findings show that lentiviral vectors, as a tool for gene therapy, can affect the fate determination of RPCs and increase the number of photoreceptor cells, even though they do not cause significant pathological changes under traditional infections (MOI = 10). This early abnormal development of the retina resulting in changes in cell numbers may not necessarily lead to significant visual impairment in the early stages but could potentially have a delayed and cumulative effect, ultimately influencing retinal function in a mature retina. Our study has found that lentiviral infection can cause cell fate bias which warrants caution for the clinical use of lentiviral gene intervention. Clinicians need to carefully select delivery vectors and optimize clinical protocols to avoid risks and enhance social responsibility. In addition, we found that the use of lentiviral vectors can activate the PHLDA1-PRDM1 axis and affect retinal development, which provides a new reference indicator for evaluating viral vector risks or cell fate changes in other retinal development models. In our study, it was found that the use of lentiviral vectors induced up-regulation of PHLDA1 during the differentiation of photoreceptor cells. PHLDA1 has been identified as a gene that is associated with a variety of tumors [49, 50] and as one that promotes tumor growth [51, 52]. Through the process of gene therapy for disease rescue, lentiviral vectors infection induces the up-regulation of PHLDA1 expression, which may cause some potential safety risks, including the possibility of promoting tumorigenesis and leading to adverse prognosis. These issues need to be paid attention to and solved in the application of gene therapy. Thus, long-term patient follow-up is required after gene therapy to obtain accurate information and evaluate whether there is an association between lentiviral vectors and tumor development, recurrence, and prognosis.
Clearly, our study provides evidence that lentivirus affects the specification of RPCs into photoreceptor cells by activating the PHLDA1-PRDM1 axis. This suggests that lentiviral vectors might have adverse effects in clinical applications. As of 2023, the number of HIV-infected individuals has reached as high as 39 million, with an annual infection rate of 1.5 million [53]. Currently, there is insufficient attention given to the impact of HIV infection on the retina, despite the fact that HIV can be detected in the nervous system [54, 55]. However, we observed the impact of lentiviral vectors on retinal development in a model that was highly relevant to the retina. As the number of HIV infections rises, comprehending the effects of HIV infection on fetal neural system development becomes particularly important. Mother-to-child transmission is one of the major pathways of HIV transmission, and the virus may be transmitted to the fetus through vertical transmission when an infected mother gives birth. Therefore, it is crucial to pay attention to the impact of mother-to-child transmission of HIV infection on fetal neurodevelopment. This will help to formulate corresponding prevention and intervention measures to minimize potential adverse effects.
One limitation of this study is that, although we discovered that PHLDA1 possessed potential transcriptional activation functions and could bind to the promoter region of RAX and the intronic region of PRDM1, and knockdown/knockout of PHLDA1 significantly reducing the expression levels of these genes, additional research is required to elucidate whether PHLDA1 directly binds to DNA or regulates the transcription of downstream genes through interactions with other proteins. Furthermore, while our findings indicate that viral infection can induce aberrant photoreceptor development within a short time frame (1 week), the long-term consequences on retinal development necessitate further investigation.
Data availability
We obtained the raw sequencing data of different virus infector from the NCBI Sequence Read Archive under accession number GSE147507、GSE78711 and GSE168125. ScRNA-seq data from retinal organoids have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE136929. CUT&Tag and RNA sequence data from Y79 have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE251752.The data and material that support the findings of this study are available from the corresponding author on reasonable request.
Abbreviations
- PHLDA1:
-
Pleckstrin homology-like domain, family A, member 1
- PRDM1:
-
PR domain zinc finger protein 1
- LCA:
-
Leber’s congenital amaurosis
- NRL:
-
Neural retina leucine zipper
- CEP290:
-
Centrosomal protein 290
- NR2E3:
-
Nuclear receptor subfamily 2 group E member 3
- OTX2:
-
Orthodenticle homeobox 2
- CRX:
-
Cone–rod homeobox protein
- RAX:
-
Retina and anterior neural fold homeobox gene
- RXRG:
-
Retinoid X receptor gamma
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- RSV:
-
Respiratory syncytial virus
- VSV-G:
-
Vesicular stomatitis virus G
- hESC:
-
Human embryonic stem cell
- RPCs:
-
Retinal progenitor cells
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- PQ:
-
Proline-glutamine
- QQ:
-
Polyglutamine
- HIV:
-
Human immunodeficiency virus
References
Maguire AM, Simonelli F, Pierce EA et al (2008) Safety and efficacy of gene transfer for leber’s congenital amaurosis. N Engl J Med 358:2240–2248. https://doi.org/10.1056/NEJMoa0802315
Bainbridge JWB, Smith AJ, Barker SS et al (2008) Effect of gene therapy on visual function in leber’s congenital amaurosis. N Engl J Med 358:2231–2239. https://doi.org/10.1056/NEJMoa0802268
Campochiaro PA, Lauer AK, Sohn EH et al (2017) Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (gem) study. Hum Gene Ther 28:99–111. https://doi.org/10.1089/hum.2016.117
Wang X, Ma C, Rodríguez Labrada R et al (2021) Recent advances in lentiviral vectors for gene therapy. Sci China Life Sci 64:1842–1857. https://doi.org/10.1007/s11427-021-1952-5
Balaggan KS, Ali RR (2012) Ocular gene delivery using lentiviral vectors. Gene Ther 19:145–153. https://doi.org/10.1038/gt.2011.153
Goyal S, Tisdale J, Schmidt M et al (2022) Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med 386:138–147. https://doi.org/10.1056/NEJMoa2109167
Ikeda Y, Goto Y, Yonemitsu Y et al (2003) Simian immunodeficiency virus-based lentivirus vector for retinal gene transfer: a preclinical safety study in adult rats. Gene Ther 10:1161–1169. https://doi.org/10.1038/sj.gt.3301973
Parker MA, Erker LR, Audo I et al (2022) Three-year safety results of sar422459 (eiav‐abca4) gene therapy in patients with abca4‐associated stargardt disease: an open‐label dose‐escalation phase i/iia clinical trial, cohorts 1‐5. Am J Ophthalmol 240:285–301. https://doi.org/10.1016/j.ajo.2022.02.013
Wright AF, Reddick AC, Schwartz SB et al (2004) Mutation analysis ofnr2e3 andnrl genes in enhanced s cone syndrome. Hum Mutat 24:439. https://doi.org/10.1002/humu.9285
den Hollander AI, Koenekoop RK, Yzer S et al (2006) Mutations in the cep290 (nphp6) gene are a frequent cause of leber congenital amaurosis. Am J Hum Genet 79:556–561. https://doi.org/10.1086/507318
Grünert U, Martin PR (2020) Cell types and cell circuits in human and non-human primate retina. Prog Retin Eye Res 78:100844. https://doi.org/10.1016/j.preteyeres.2020.100844
O’Hara-Wright M, Gonzalez-Cordero A (2020) Retinal organoids: a window into human retinal development. Development 147. https://doi.org/10.1242/dev.189746
Sasai Y (2013) Next-generation regenerative medicine: organogenesis from stem cells in 3d culture. Cell Stem Cell 12:520–530. https://doi.org/10.1016/j.stem.2013.04.009
Finkbeiner C, Ortuño-Lizarán I, Sridhar A et al (2022) Single-cell atac-seq of fetal human retina and stem-cell-derived retinal organoids shows changing chromatin landscapes during cell fate acquisition. Cell Rep 38:110294. https://doi.org/10.1016/j.celrep.2021.110294
Sridhar A, Hoshino A, Finkbeiner CR et al (2020) Single-cell transcriptomic comparison of human fetal retina, hpsc-derived retinal organoids, and long-term retinal cultures. Cell Rep 30:1644–1659. https://doi.org/10.1016/j.celrep.2020.01.007
Cowan CS, Renner M, De Gennaro M et al (2020) Cell types of the human retina and its organoids at single-cell resolution. Cell 182:1623–1640. https://doi.org/10.1016/j.cell.2020.08.013
Nakano T, Ando S, Takata N et al (2012) Self-formation of optic cups and storable stratified neural retina from human escs. Cell Stem Cell 10:771–785. https://doi.org/10.1016/j.stem.2012.05.009
Zhong X, Gutierrez C, Xue T et al (2014) Generation of three-dimensional retinal tissue with functional photoreceptors from human ipscs. Nat Commun 5. https://doi.org/10.1038/ncomms5047
Finak G, McDavid A, Yajima M et al (2015) Mast: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell rna sequencing data. Genome Biol 16. https://doi.org/10.1186/s13059-015-0844-5
Wu T, Hu E, Xu S et al (2021) Clusterprofiler 4.0: a universal enrichment tool for interpreting omics data. Innov (N Y) 2:100141. https://doi.org/10.1016/j.xinn.2021.100141
Xiao Y, Mao X, Hu X et al (2022) Single-cell transcriptomic profiling of human retinal organoids revealed a role of igf1-phlda1 axis in photoreceptor precursor specification. Invest Ophthalmol Vis Sci 63:9. https://doi.org/10.1167/iovs.63.12.9
Cepko CL (2015) The determination of rod and cone photoreceptor fate. Annu Rev Vis Sci 1:211–234. https://doi.org/10.1146/annurev-vision-090814-121657
Swaroop A, Kim D, Forrest D (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci 11:563–576. https://doi.org/10.1038/nrn2880
Butland SL, Devon RS, Huang Y et al (2007) Cag-encoded polyglutamine length polymorphism in the human genome. BMC Genomics 8:126. https://doi.org/10.1186/1471-2164-8-126
WILLIAMSON MP (1994) The structure and function of proline-rich regions in proteins. Biochem J 297:249–260. https://doi.org/10.1042/bj2970249
NAGAI MA (2016) Pleckstrin homology-like domain, family a, member 1 (phlda1) and cancer. Biomed Rep 4:275–281. https://doi.org/10.3892/br.2016.580
Brzezinski JA, Lamba DA, Reh TA (2010) Blimp1 controls photoreceptor versus bipolar cell fate choice during retinal development. Development 137:619–629. https://doi.org/10.1242/dev.043968
Hossain GS, van Thienen JV, Werstuck GH et al (2003) Tdag51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the development of atherosclerosis in hyperhomocysteinemia. J Biol Chem 278:30317–30327. https://doi.org/10.1074/jbc.M212897200
Carlisle RE, Heffernan A, Brimble E et al (2012) Tdag51 mediates epithelial-to-mesenchymal transition in human proximal tubular epithelium. Am J Physiol Ren Physiol 303:F467–F481. https://doi.org/10.1152/ajprenal.00481.2011
Joo JH, Liao G, Collins JB et al (2007) Farnesol-induced apoptosis in human lung carcinoma cells is coupled to the endoplasmic reticulum stress response. Cancer Res (Chicago Ill) 67:7929–7936. https://doi.org/10.1158/0008-5472.CAN-07-0931
Durbas M, Pabisz P, Wawak K et al (2018) Gd2 ganglioside-binding antibody 14g2a and specific aurora a kinase inhibitor mk-5108 induce autophagy in imr-32 neuroblastoma cells. Apoptosis 23:492–511. https://doi.org/10.1007/s10495-018-1472-9
Han C, Yan P, He T, Brain et al (2020) Behav Immun 88:640–653. https://doi.org/10.1016/j.bbi.2020.04.064
Park ES, Jeon H, Lee N et al (2023) Tdag51 promotes transcription factor foxo1 activity during lps-induced inflammatory responses. Embo J 42:e111867. https://doi.org/10.15252/embj.2022111867
Georgel P, Jiang Z, Kunz S et al (2007) Vesicular stomatitis virus glycoprotein g activates a specific antiviral toll-like receptor 4-dependent pathway. Virology 362:304–313. https://doi.org/10.1016/j.virol.2006.12.032
Song X, Zhou L, Yang W et al (2023) Phlda1 is a p53 target gene involved in p53-mediated cell apoptosis. Mol Cell Biochem. https://doi.org/10.1007/s11010-023-04752-w
Lazo PA, Santos CR (2011) Interference with p53 functions in human viral infections, a target for novel antiviral strategies? Rev Med Virol 21:285–300. https://doi.org/10.1002/rmv.696
Takaoka A, Hayakawa S, Yanai H et al (2003) Integration of interferon-a/b signalling to p53 responses in tumour suppression and antiviral defence. Nat (London) 424:516–523. https://doi.org/10.1038/nature01850
Wu H, Lozano G (1994) Nf-kappa b activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem 269:20067–20074
Vermeire J, Roesch F, Sauter D et al (2016) Hiv triggers a cgas-dependent, vpu- and vpr-regulated type i interferon response in cd4 + t cells. Cell Rep 17:413–424. https://doi.org/10.1016/j.celrep.2016.09.023
Cai X, Chiu Y, Chen ZJ (2014) The cgas-cgamp-sting pathway of cytosolic dna sensing and signaling. Mol Cell 54:289–296. https://doi.org/10.1016/j.molcel.2014.03.040
Muranishi Y, Terada K, Inoue T et al (2011) An essential role for rax homeoprotein and notch–hes signaling inotx2 expression in embryonic retinal photoreceptor cell fate determination. J Neurosci 31:16792–16807. https://doi.org/10.1523/JNEUROSCI.3109-11.2011
Goodson NB, Park KU, Silver JS et al (2020) Prdm1 overexpression causes a photoreceptor fate-shift in nascent, but not mature, bipolar cells. Dev Biol 464:111–123. https://doi.org/10.1016/j.ydbio.2020.06.003
Kaczmarek Michaels K, Natarajan M, Euler Z et al (2015) Blimp-1, an intrinsic factor that represses hiv-1 proviral transcription in memory cd4 + t cells. The Journal of immunology (1950) 194:3267–3274. https://doi.org/10.4049/jimmunol.1402581
KELLER AD, MANIATIS T (1991) Identification and characterization of a novel repressor of β-interferon gene expression. Genes Dev 5:868–879. https://doi.org/10.1101/gad.5.5.868
Ren B, Chee KJ, Kim TH et al (1999) Prdi-bf1/blimp-1 repression is mediated by corepressors of the groucho family of proteins. Genes Dev 13:125–137. https://doi.org/10.1101/gad.13.1.125
Ko Y, Chan Y, Liu C et al (2018) Blimp-1-mediated pathway promotes type i ifn production in plasmacytoid dendritic cells by targeting to interleukin-1 receptor-associated kinase m. Front Immunol 9. https://doi.org/10.3389/fimmu.2018.01828
Milone MC, O Doherty U (2018) Clinical use of lentiviral vectors. Leukemia 32:1529–1541. https://doi.org/10.1038/s41375-018-0106-0
Dunbar CE, High KA, Joung JK et al (2018) Gene therapy comes of age. Science 359. https://doi.org/10.1126/science.aan4672
Yeh I, McCalmont TH, LeBoit PE (2012) Differential expression of phlda1 (tdag51) in basal cell carcinoma and trichoepithelioma. Br J Dermatology (1951) 167:1106–1110. https://doi.org/10.1111/j.1365-2133.2012.11165.x
Bonatto N, Carlini MJ, de Bessa Garcia SA et al (2018) Phlda1 (pleckstrin homology-like domain, family a, member 1) knockdown promotes migration and invasion of mcf10a breast epithelial cells. Cell Adh Migr 12:37–46. https://doi.org/10.1080/19336918.2017.1313382
Wang J, Yao N, Hu Y et al (2022) Phlda1 promotes glioblastoma cell growth via sustaining the activation state of Ras. Cell Mol Life Sci 79:520. https://doi.org/10.1007/s00018-022-04538-1
Sakthianandeswaren A, Christie M, D’Andreti C et al (2011) Phlda1 expression marks the putative epithelial stem cells and contributes to intestinal tumorigenesis. Cancer Res 71:3709–3719. https://doi.org/10.1158/0008-5472.CAN-10-2342
Barton F, Haynes KWPB, Bette Korber KWAJ, Beatrice H, Hahn FAAG (2023) Strategies for hiv-1 vaccines that induce broadly neutralizing antibodies
González-Scarano F, Martín-García J (2005) The neuropathogenesis of aids. Nat Rev Immunol 5:69–81. https://doi.org/10.1038/nri1527
Davis LE, Hjelle BL, Miller VE et al (1992) Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 42:1736–1739. https://doi.org/10.1212/wnl.42.9.1736
Acknowledgements
We thank Professor Zhuang Jing for providing the human RB cell line Y79 cells and the staff of Core Facilities at State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center for technical support.
Funding
This work was funded by National Key R&D Program of China, (2023YFC2506100); The National Natural Science Foundation of China (32171445); The Science and Technology Program of Guangzhou (202201020624); “Precision Medicine and Stem Cells” Project of Key Field R&D Program of Guangdong province (2023B1111020006).
Author information
Authors and Affiliations
Contributions
Y.H., X.H., Y.X. conceived and designed the study; X.H., J.C., W.D., X.C., S.Z., Z.C. performed experiments; Y.H., X.H., Y.X. analyzed the data and performed statistical analyses. Y.H., X.H., Y.X., J.C. and W.D. interpreted the data and wrote the manuscript in discussion with all authors. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have been involved in writing the manuscript and consented to publication.
Conflict of interest
The funders had no conflicts of interest in the design of the study; in the collection, analysis or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, X., Chen, J., Dai, W. et al. PHLDA1-PRDM1 mediates the effect of lentiviral vectors on fate-determination of human retinal progenitor cells. Cell. Mol. Life Sci. 81, 305 (2024). https://doi.org/10.1007/s00018-024-05279-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05279-z