
Abstract
N-methyl-d-aspartate receptors (NMDARs) are members of the glutamate receptor family and participate in excitatory postsynaptic transmission throughout the central nervous system. Genetic variants in GRIN genes encoding NMDAR subunits are associated with a spectrum of neurological disorders. The M3 transmembrane helices of the NMDAR couple directly to the agonist-binding domains and form a helical bundle crossing in the closed receptors that occludes the pore. The M3 functions as a transduction element whose conformational change couples ligand binding to opening of an ion conducting pore. In this study, we report the functional consequences of 48 de novo missense variants in GRIN1, GRIN2A, and GRIN2B that alter residues in the M3 transmembrane helix. These de novo variants were identified in children with neurological and neuropsychiatric disorders including epilepsy, developmental delay, intellectual disability, hypotonia and attention deficit hyperactivity disorder. All 48 variants in M3 for which comprehensive testing was completed produce a gain-of-function (28/48) compared to loss-of-function (9/48); 11 variants had an indeterminant phenotype. This supports the idea that a key structural feature of the M3 gate exists to stabilize the closed state so that agonist binding can drive channel opening. Given that most M3 variants enhance channel gating, we assessed the potency of FDA-approved NMDAR channel blockers on these variant receptors. These data provide new insight into the structure–function relationship of the NMDAR gate, and suggest that variants within the M3 transmembrane helix produce a gain-of-function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
N-methyl-d-aspartate receptors (NMDARs) are ligand-gated ion channels that mediate a slow Ca2+-permeable component of the excitatory synaptic current throughout the central nervous system [1]. NMDARs not only play an essential role in development, synaptic plasticity, and learning, but also are linked to a wide spectrum of neurological and psychiatric disorders such as epilepsy, Parkinson’s disease, autism, and schizophrenia [1,2,3,4,5]. NMDA receptors have also been considered potential targets to improve symptoms in a number of conditions, notably depression [6, 7]. The NMDARs are encoded by the GRIN gene family, which includes GRIN1, GRIN2A-D and GRIN3A-B. Functional NMDARs are heterotetramers comprising two glycine-binding GluN1 and two glutamate-binding GluN2 subunits. All NMDAR subunits share a similar structural arrangement, which contains an extracellular amino-terminal domain (NTD, also known as ATD), an agonist binding domain (ABD, also known as LBD, ligand binding domain), a pore-forming transmembrane domain (TMD) comprising three transmembrane helices (M1, M3, M4) and a re-entrant loop (M2), and an intracellular carboxyl-terminal domain (CTD) [1].
Recent advances in next-generation whole exome sequencing technology have led to the identification of a multitude of clinically relevant de novo and rare variants in GRIN genes in patients with neurological or psychiatric disorders, including epilepsy, intellectual disability (ID), developmental delay, and schizophrenia [1, 8, 9]. These rare variants are distributed across GRIN1, GRIN2A, GRIN2B, and GRIN2D and alter amino acid residues located in all four semi-autonomous domains (NTD, ABD, TMD, and CTD), with an enrichment in the ABD and the TMD. Regional analysis of missense intolerance of GluN1, GluN2A and GluN2B subunits in the healthy population showed that some sub-regions of ABD and TMD have more severe missense depletion, which suggests that genetic variation in these regions is more likely to be associated with diseases [10]. For example, the M3 transmembrane helix, including the highly conserved SYTANLAAF motif, plays an essential role in channel gating and is virtually intolerant to variation. Although pathogenic missense variants affecting this region are involved in a series of neurological and neurodevelopmental disorders, data describing the functional consequences of these variants is limited [11, 12]. To address this mismatch in terms of variants and function, we report the pharmacological and functional properties of 48 missense variants located in M3 transmembrane helix from 56 patients. We use these and other available data to assess the overall effect of these variants using a recently described comprehensive strategy to classify variants as gain-of-function (GoF) or loss-of-function (LoF) [13]. We find that variants in this region that allow surface expression are more likely to produce a gain-of-function, raising the idea that the sequence of the M3 gate was selected to stabilize the channel closed state, which is a pre-requisite for ligand-based activation.
Materials and methods
Consent and study approval
This study was approved by the Medical Ethics Committee and the Institutional Review Boards of University of Colorado School of Medicine and Children's Hospital Colorado (COMIRB 16–1520), Federico II University, Brown University, Duke University, Emory University, University of Melbourne, Seattle Children’s Research Institute, Cedars-Sinai Medical Center, University of Pennsylvania, and University of Leipzig Hospitals and Clinics. All data of this study were analyzed anonymously.
All in vitro studies were conducted according to the guidelines of Emory University.
Molecular biology
We studied recombinant cDNA encoding human GluN1-1a (referred to as GluN1; NCBI Reference Sequence NM_007327.3), GluN2A (NM_000833.4), and GluN2B (NM_000834.4). Site-directed mutagenesis was performed on complementary DNA (cDNA) encoding the human GRIN genes [14] using the QuikChange protocol with Pfu DNA polymerase (Stratagene La Jolla, CA, USA) to replicate the parental DNA strand with the desired mismatch incorporated into the primer. Methylated parental DNA was digested with Dpn I for 1 h at 37˚C and the nicked mutant DNA was transformed into TOP10 Competent Cells (Life Tech, Grand Islands, NY, USA). Bacteria were spun down and plasmid DNA isolated using the Qiagen Qiaprep Spin Miniprep kit (Hilden, Germany). Sequences were verified through the mutated region using dideoxy DNA sequencing (Eurofins MWG Operon, Huntsville, AL, USA). The plasmid vector hosting wild type (WT) human GluN1, human GluN2A, and human GluN2B was pCI-neo [15].
The cDNA was linearized using FastDigest (Thermo, Waltham, MA) restriction digestion at 37 °C for 1 h. Complementary RNA (cRNA) was synthesized in vitro from linearized wild type and mutant cDNA using the mMessage mMachine T7 kit (Ambion, Austin, TX, USA). Unfertilized Xenopus laevis stage VI oocytes were prepared from commercially available ovaries (Xenopus one Inc, Dexter, MI, USA). The ovary was digested with Collagenase Type 4 (Worthington-Biochem, Lakewood, NJ, USA) solution (850 μg/ml, 15 ml for a half ovary) in Ca2+-free Barth’s solution, which contained (in mM) 88 NaCl, 2.4 NaHCO3, 1 KCl, 0.82 MgSO4, 10 HEPES (pH 7.4 with NaOH) supplemented with 100 μg/mL gentamycin, 40 μg/mL streptomycin. The ovary was incubated in enzyme with gentle mixing at RT (room temperature, 23 ℃) for 2 h. The oocytes were rinsed 5 times with Ca2+-free Barth’s solution (35–40 ml of fresh solution each time) for 10 min each time, and further rinsed 4 more times with normal Barth’s solution (containing 0.33 Ca(NO3)2, 35–40 ml of fresh solution) on the mixer for 10 min each time. The sorted oocytes were kept at 16 ℃ in an incubator for future use. Xenopus laevis oocytes were injected with a 1:1 ratio of GluN1:GluN2A and GluN1:GluN2B cRNA that by weight was a total of 0.25–25 ng in 50 nL of RNAase-free water per oocyte [14]. Injected oocytes were maintained in normal Barth’s solution at 15–19 ℃.
Two-electrode voltage clamp current recordings
Two-electrode voltage clamp (TEVC) current recordings were performed one to three days post-injection at room temperature (23˚C) as previously described [14, 16]. The extracellular recording solution contained (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, and 0.01 EDTA (pH7.4 with NaOH). Solution exchange was computer controlled through an 8-valve positioner (Digital MVP Valve, Hamilton, CT, USA). Oocytes were placed in a dual track chamber that shared a single perfusion line, allowing simultaneous recording from two oocytes. All concentration–response solutions were made in the extracellular recording solution. Voltage control and data acquisition were achieved by a two-electrode voltage-clamp amplifier (OC725C, Warner Instruments, Hamden, CT, USA). The voltage electrode was filled with 0.3 M KCl and the current electrode with 3 M KCl. Oocytes were held under voltage clamp at a holding potential of –40 mV unless otherwise indicated. MTSEA (2-aminoethyl methanethiol sulfonate hydrobromide, Toronto Research Chemicals, Ontario, Canada) solution was prepared fresh and used within 30 min. All chemicals were obtained from Sigma–Aldrich unless otherwise stated.
Xenopus oocytes express a Ca2+-activated Cl− current that confounds voltage clamp recordings of NMDAR-mediated currents in the presence of extracellular Ca2+. We, therefore, substituted Ba2+ for Ca2+, because Ba2+ permeates NMDARs and is relatively ineffective at the Ca2+-activated Cl− current. Potential changes in the permeability to the divalent cation Ba2+ for WT and variant NMDARs were assessed by recording the current–voltage relationship in normal external recording solution (described above) and then again in external recording solution in which 90 mM NaCl was replaced with 60 mM BaCl2. The current–voltage relationship was determined from the difference in currents recorded during 15 mV step changes in the holding potential from 0 mV to − 90, − 75, − 60, − 45, − 30, − 15, 0, + 15, and + 30 mV before and during application of 100 µM glutamate plus 100 µM glycine for Na+ and for Ba2+ as the charge carrier in the same oocyte. Substitution of BaCl2 for NaCl was predicted to produce a − 6 mV shift in junction potential (pClamp, Molecular Devices), which was not corrected. The shift in the reversal potential between Ba2+ and Na+ solutions was determined for WT (∆VREV,WT) and for variant (∆VREV,VARIANT) NMDARs, and then the differences between variant and WT reversal potential shifts was determined as ∆∆VREV = (∆VREV,VARIANT – ∆VREV,WT). The mean ± SEM for ∆VREV for WT GluN1/GluN2A NMDARs was 12.1 ± 1.3 mV (n = 15), which corresponds to a 99% confidence interval of ± 3.4 mV. We, therefore, considered WT and variant reversal potential shifts in ∆VREV that were greater than ± 3.4 mV as evidence for potential changes in Ba2+ permeability, which we assume is reflective of Ca2+ permeability.
Cell culture and transfection
HEK293 cells (hereafter HEK cells; CRL 1573, ATCC, Manassas, VA, USA) for di-heteromeric receptor experiments were maintained in standard DMEM/Gluta-Max media (Fisher) with 10% dialyzed fetal bovine serum (R&D Systems) plus 10 U/ml and 10 µg/ml streptomycin (Fisher) at 37 ℃ and 5% CO2. The calcium phosphate method was used to transiently transfect the cells with plasmid cDNA encoding green fluorescent protein (GFP) and NMDAR GluN subunits in pcIneo (5:1:1 for GFP:GluN1:GluN2A or variant GluN2A, 1:1:1 for GFP:GluN1:GluN2B or variant GluN2B), as previously described [17]. Each well was transfected with 12.5 mM CaCl2, 2.5 mM BES solution (N,N-Bis(2-hydroxyethyl-2-aminethanesulfonic acid, N,N-Bis(2-hydroxyethyl)taurine; 14 mM NaCl; 75 µM Na2HPO4, pH 6.95) and 1 µg/ml of a DNA mixture for 4 h at 37 ℃, then washed and incubated overnight in standard media. NMDAR antagonists APV (d,l-2-amino-5-phosphonovalerate, 200 µM) and 7-CKA (7-chlorokynurenic acid, 200 µM) were added to reduce cell death caused by excessive activation of NMDARs.
Whole cell voltage clamp recording
Whole cell voltage clamp recordings were performed on transiently transfected HEK cells 12–72 h post transfection. The patch electrodes (resistance 3–5 MΩ) for whole cell voltage clamp current recordings were pulled from thin-walled glass micropipettes (TW150F-4, World Precision Instruments, Sarasota, FL, USA) by a dual-stage glass micropipette puller (PC-10, Narishige, Tokyo, Japan) and filled with internal solution (in mM) 110 D-gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 NaATP and 0.3 NaGTP, pH 7.35. Transfected HEK cells were perfused with external recording solution that contained (in mM) 150 NaCl, 10 HEPES, 22 D-mannitol, 3 KCl, 1 CaCl2, and 0.01 EDTA (pH 7.4, 230C). The current response was recorded with an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA) at a holding potential of − 60 mV at room temperature (23 ℃). A two-barreled theta-glass micropipette was used for rapid solution exchange controlled by a piezoelectric translator (Burleigh Instruments, Newton, NJ, USA).
Beta-lactamase assay
HEK cells were plated in 96-well plates (50,000 cells per well) and transiently transfected using Fugene6 with cDNA encoding β-lac-GluN1 variants and wild type GluN2, or wild type GluN1 and β-lac-GluN2 variants (Promega, Madison, WI) [10]. Cells treated with Fugene6 alone were used to define background absorbance. NMDAR antagonists (200 μM APV and 200 μM 7-CKA) were added at the time of transfection. Six wells were transfected for each condition; surface and total protein levels were measured in three wells each. After 24 h, cells were rinsed with Hank’s Balanced Salt Solution (HBSS, in mM, 140 NaCl, 5 KCl, 0.3 Na2HPO4, 0.4 KH2PO4, 6 glucose, 4 NaHCO3) supplemented with 10 mM HEPES, and then in HBSS/HEPES solution supplemented with 100 μM nitrocefin (100 μL; Millipore, Burlington, MA, USA) solution to each well for measuring the level of extracellular enzymatic activity, which reflected NMDAR surface expression. To determine the level of total enzymatic activity, the cells were lysed by a 30 min incubation in 50 μL H2O prior to the addition of 50 μL of 200 μM nitrocefin in HBSS/HEPES. The absorbance at 486 nm was read using a microplate reader every min for 30 min at 30 ℃. The rate of increase in absorbance was generated from the slope of a linear fit to the data.
Evaluation of FDA-approved NMDAR inhibitors
FDA-approved drugs that act as NMDAR open channel blockers (memantine, ketamine, dextromethorphan and its metabolite dextrorphan) were evaluated using TEVC recordings from Xenopus oocytes co-expressing GluN1 variants with the WT GluN2, and WT GluN1 with GluN2 variants. The composite concentration–response curves were recorded at a holding potential of − 40 mV and fitted to determine the IC50 values (see below).
Data and statistical analysis
Statistical analyses were performed in GraphPad Prism 8.0.1 (La Jolla, CA, USA) and OriginPro 9.0 (Northampton, MA, USA). Statistical significance was assessed using one-way ANOVA with Post hoc Dunnett’s Multiple Comparison Test, with p < 0.05 considered significant. Power was determined using GPower (3.1.9.2). Data are presented as mean ± SEM. Error bars represent SEM unless otherwise stated. The concentration–response relationship for agonists were fitted by Eq. 1, and the concentration–response relationship for inhibition by FDA-approved channel blockers was fitted by Eq. 2,
where N is the Hill slope, EC50 is the concentration of the agonist that produces a half-maximal effect, IC50 is the concentration of the inhibitor that produces a half-maximal effect, minimum is the degree of residual response at a saturating concentration of the inhibitor, constrained during fitting to be > 0, and Response is expressed as a percent of the fitted maximum. The maximal channel open probability (POPEN) when activated by maximally effective concentrations of agonist was estimated from the fold Potentiation observed in MTSEA using Eq. 3 [18]:
where γMTSEA and γCONTROL were the single channel chord conductance values estimated from GluN1/GluN2A receptors and fold Potentiation was defined as the ratio of current in the presence of MTSEA to current in the absence of MTSEA, and γMTSEA / γCONTROL was 0.67 [18]. Rise time for each response was determined as the time measured between 10 and 90% of the peak current. The current response time course was fitted using ChanneLab (Synaptosoft, Decatur, GA, USA) by Eq. 4,
The weighted deactivation tau (τw) was calculated by Eq. 5,
Synaptic charge transfer was estimated as the product of peak whole cell current response amplitude and the weighted deactivation tau (τw) for the termination of responses following rapid removal of glutamate. We calculated the relative fold-change in synaptic charge transfer with Eq. 6 [13]:
where τw is the mean weighted deactivation time constant, P is the receptor maximum open probability, Surf is surface protein levels, Mg is percentage inhibition by 1 mM Mg2+ at a holding potential between – 40 and – 60 mV, and RGLY and RGLU, Synaptic are a ratio of the relative response for variant to WT NMDARs to an extracellular concentration of glycine assumed to be 3 × 10−6 M and a synaptic concentration of 1 × 10−3 M glutamate, respectively.
We calculated the relative fold-change in non-synaptic charge transfer in a similar manner, with time dependence removed and 1 × 10−7 M glutamate for RGLU, Non-synaptic by Eq. 7 [13]:
We classified variants as gain-of-function (GoF) or loss-of-function (LoF) according to criteria described in Myers et al. [13]. For some variants in which we executed all six assays, the current amplitude of NMDAR responses in transfected HEK cells recorded under voltage clamp was too small to accurately determine the deactivation tau, and thus these variants were classified as Indeterminant* given we had data in all other assays but cannot determine the overall outcome. The small amplitude could be the result of altered receptor function leading to cell death or detachment of cells with a large number of NMDARs on the surface. In addition, some variants that were initially deemed as a conflict according to Myers et al. [13] were considered Indeterminant when the synaptic and non-synaptic charge transfers were both suprathreshold but in conflict.
Three-dimensional Missense tolerance determination
The three-dimensional missense tolerance ratios (3DMTR, [19]) of the diheteromeric NMDARs were calculated using homology models based on the non-active GluN1/GluN2B structure (pdb: 6WHS, [20]), using the 3DMTR application (https://github.com/riley-perszyk/3DMTR). In this analysis, the non-neuro gnomAD (v.2.1.1) dataset was used. The 3DMTR assesses the intolerance of each residue as a running average using the central residue along with the intolerance of the 30 closest residues in 3-dimensional space, determined from the structure of the receptor [19].
Results
Variants in M3 transmembrane helix and neurological and neurodevelopmental disorders
We identified 48 variants in the M3 transmembrane helix across GRIN1, GRIN2A, and GRIN2B in 56 patients with neurological and/or neurodevelopmental disorders (Table 1, Supplemental Table S1). 22 of these variants were reported in the peer-reviewed literature or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and others were newly discovered. According to the clinical data from 47 patients (see Supplemental Table S1), the most common phenotypes of these patients with M3 transmembrane variants are epilepsy (70%, 33/47), intellectual disability (70%, 33/47), developmental delay (51%, 24/47), movement disorders (28%, 13/47), language disorders (15%, 7/47), and autism spectrum disorder (ASD; 6.4%, 3/47). Clinical information for the remaining 9 patients was not available.
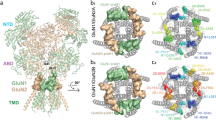
The residues in the M3 transmembrane helix are highly conserved in different subunits within the GRIN/GluN family (Fig. 1A) and contain the SYTANLAAF motif that is largely invariant throughout the glutamate receptor family. The intolerance of M3 to genetic variation was assessed with the 3D Missense Tolerance Ratio [19] across GRIN1, GRIN2A, GRIN2B, GRIN2C and GRIN2D genes, which revealed minimal regional variation for M3 for all subunits except GluN2C (Fig. 1A). This result is consistent with strong purifying selection [21] acting upon much of the M3 transmembrane helix and suggests that this region is highly intolerant to genetic variation. The variants studied here all fall into the intolerant region, and span the full M3 transmembrane domain, as shown in Fig. 1B, C.

Locations of disease-associated missense variants in the M3 transmembrane helix. A A linear schematic showing domain architecture of the GRIN/GluN and protein residue sequence alignment for highly conserved M3 transmembrane domain (TMD) across GluN1 and GluN2 subunits. NTD indicates the N-terminal domain (also known as ATD, amino terminal domain); S1 and S2 denote the first and second polypeptide sequences comprising the agonist binding domain (ABD). The aligned amino-acid sequence of the M3 helix of each NMDAR subunits is listed with a raster plot depicting the missense and synonymous variants found in gnomAD along with the 3DMTR score for these stretches, shown as a colorimetric raster plot. The red dashed box indicates the M3 transmembrane helix. B Homology model of the GluN1/GluN2A receptor built from previously reported GluN1/GluN2B cryo-EM data [20]. C The residues harboring missense variants are highlighted with different colors in a side view; the side of the subunit facing the pore is indicated
Variants in the M3 transmembrane helix alter agonist potency
Two-electrode voltage clamp current recordings in Xenopus laevis oocytes expressing GluN1 variants with wild type (WT) GluN2A or GluN2B, and WT GluN1 with GluN2A or GluN2B variants were performed to evaluate the functional effects of variants in the M3 transmembrane helix. The half-maximally effective concentration (EC50) of agonists was determined by analyzing the glutamate and glycine concentration-effect relationships. Almost all M3 variant-containing NMDARs (37/48) showed a significantly increased glutamate and glycine potency (i.e., decreased EC50 values) including GluN1-A652T which increased glutamate potency when expressed only with GluN2B. Only 3/48 variants decreased glutamate potency by ~ twofold or less, 6/48 variants had no detectable effect on EC50 (Table 2), and 2/48 variants produced responses that were too small to measure. Some M3 variant-containing NMDARs exhibited over a 100-fold increase in potency compared to WT NMDARs, and a number of variants produced more than a tenfold increase glutamate potency (reduced EC50 values), including GluN1-V644M, GluN1-Y647C, GluN1-Y647S, GluN1-N650I, GluN1-A653G, GluN1-A653T, GluN1-F654C, GluN1-L655Q, GluN2A-S644G, GluN2A-T646A, GluN2A-T646R, GluN2A-L649V, GluN2A-M653I, GluN2A-M653V, GluN2A-I654T, GluN2B-N649S, and GluN2B-N649T (Fig. 2A–J, Table 2). One potential interpretation of these data is that the M3 transmembrane helix is invariant to enable the NMDAR to be activated by micromolar concentrations of glutamate, as virtually any change in the amino acid side chain is not tolerated and usually lowers glutamate EC50 values. This is consistent with the idea that the invariant SYTANLAAF region of the M3 transmembrane helix forms the gate that occludes the pore in the closed conformation. Figure 1C shows the location of these variants on an expanded cut-away view of the NMDAR pore.

Variants in the M3 transmembrane helix influence pharmacological and biophysical properties of NMDARs. A–F Representative two electrode voltage clamp current recordings from Xenopus oocytes expressing GluN1/GluN2A and GluN1/GluN2B wild type or variant NMDAR subunits, as indicated. The glutamate concentration–response relationship was determined by co-applying increasing concentrations of glutamate with maximally effective concentration of glycine (30-100 µM). G–J Composite concentration–response curves for glutamate in the presence of 30-100 µM glycine fitted with the Hill equation (see Methods). K–M Representative voltage clamp current recordings show the Mg2+ concentration–response relationship for GluN1/GluN2A wild type and variants (as indicated) determined by co-applying increasing concentrations of Mg2+ with 100 µM glutamate and 100 µM glycine. N, O Composite concentration–response curves are shown for Mg2+ inhibition recorded at a holding potential of –60 mV for wild type and variant GluN1/GluN2A. P Summary of the effects of GluN1 and GluN2A variants on proton sensitivity, evaluated by the ratio of current response at pH 6.8 to pH 7.6 at a holding potential of − 40 mV
Similar to what was observed for glutamate potency, virtually all variants (40/48) in the M3 transmembrane helix increased the glycine potency (Table 2), including GluN1-A652T which increased glycine potency when expressed only with GluN2B. Only 2/48 variants decreased glycine potency (increased EC50 values) by < twofold, 4/48 variants did not detectably alter glycine EC50, and 2/48 variants had responses too small to measure. NMDARs containing variants located in M3 transmembrane helix exhibited up to a 75-fold increase in glycine potency; the following variants increased glycine potency by greater than tenfold: GluN1-Y647C, GluN1-Y647S, GluN1-N650I, GluN1-A653G, GluN1-A653T, GluN1-L655Q, GluN2A-A643D, GluN2A-S644G, GluN2A-T646A, GluN2A-T646R, GluN2A-L649V, GluN2A-M653I, GluN2A-M653V, GluN2A-I654T, and GluN2B-Y646C (Table 2). These results suggest that NMDARs that contain M3 variant subunits can be activated with lower concentrations of agonists, and are consistent with the idea that the structural requirements for gating are precise, and selected to reduce ability of low concentrations of glutamate present in the extracellular space (< 80 nM; [22,23,24]) to open the pore, as most departures from the naturally occurring amino acid within the transmembrane M3 helix enhance the ability of low concentrations of glutamate and glycine to open the channel. While it is possible that the absence of patient-derived variants that decrease agonist potency may reflect detrimental consequences that are incompatible with life, multiple examples of patient-derived variants that reduce glutamate potency are known (e.g., Ref [10]).
Variants in the M3 transmembrane helix alter sensitivity to endogenous inhibitors
One of the most important features of NMDA receptors is the negative regulation by endogenous extracellular inhibitors such as extracellular protons and Mg2+ [1]. NMDARs can be inhibited at low extracellular pH with an IC50 value near physiological pH, suggesting they are normally under tonic proton inhibition [25,26,27]. A central feature of NMDAR function is its voltage-dependent block by extracellular Mg2+ (e.g., [28]). We first evaluated the effect of extracellular Mg2+ by recording the current responses evoked by 100 μM glutamate and 100 μM glycine with different concentrations of extracellular Mg2+ at a holding potential of -60 mV. The data showed that 27 variant NMDARs had a decreased sensitivity to Mg2+ (i.e., increased IC50 values), compared to only 5 that showed increased sensitivity (decreased IC50 values) to extracellular Mg2+ (Table 2). The variants GluN1-A637V, GluN1-G638V, GluN1-M641I, GluN2A-S632F, GluN2A-L642R, GluN2A-T646R, GluN2A-F652V, GluN2B-A636V showed particularly strong increases in IC50 values (i.e., decreases in potency) greater than 4-fold for Mg2+ block at –60 mV, a holding potential selected for proximity to the membrane potential of principal cells (Fig. 2K–O, Table 2). All of these residues with the exception of GluN2A-F652V lie deep in the pore, close to the apex of the reentrant M2 loop, and are well positioned to perturb the M2 residues and alter electrostatic potential on the protein surface (Fig. 1C), which is an important structural determinant of Mg2+ block [29,30,31,32]. The variants GluN1-A637S, GluN1-G638A, GluN1-M641V, GluN1-V644M, GluN1-A645S, GluN1-N650K, GluN2A-A635T, GluN2A-V639I, GluN2A-L642M, GluN2A-A643D, GluN2A-S644G, GluN2A-T646A, GluN2A-L649V, GluN2A-A650S, GluN2A-M653V, GluN2A-I654T, GluN2B-Y646C, and GluN2B-A652G all showed modest increases in IC50 values (i.e., decreases in potency) for Mg2+ block at -60 mV (Fig. 2K–O, Table 2). The reduction in sensitivity to Mg2+ block will increase current flow at resting membrane potentials and further increase receptor function. Only four variants (GluN1-Y647C, GluN1-N650I, GluN2A-M653I, GluN2B-A639V) had decreased IC50 values (i.e., increases in potency) for Mg2+ block at – 60 mV.
We also evaluated proton sensitivity by comparing the current amplitude recorded from oocytes at pH 6.8 and pH 7.6 at a holding potential of – 40 mV. The majority of the variant NMDARs decreased proton sensitivity because these variants passed more current at pH 6.8 relative to pH 7.6 than WT NMDARs. GluN1 variants with reduced proton sensitivity included GluN1-G638A, GluN1-G638V, GluN1-M641L, GluN1-M641V, GluN1-I643V, GluN1-V644M, GluN1-A645S, GluN1-A653T co-expressed with GluN2A and GluN1-Y647C, GluN1-L655Q co-expressed with GluN2B. In addition, GluN2A-A635T, GluN2A-V639I, GluN2A-L642M, GluN2A-L642R, GluN2A-S644G, GluN2A-T646A, GluN2A-T646R, GluN2A-L649V, GluN2A-A650S, GluN2A-I654T, GluN2B-A636V, GluN2B-A639V, GluN2B-I641T, GluN2B-A652G, GluN2B-A652P and GluN2B-I655F decreased proton sensitivity. These data suggest a reduced sensitivity of gating to inhibition by a physiological concentration of protons at pH 7.4 (which corresponds to ~ 50 nM protons, Fig. 2P, Table 2). A small subset of variants showed increased pH-sensitivity, which was manifest as a lower current response at pH 6.8 relative to pH 7.6 than was observed for WT NMDARs. These variants included GluN1-I642L, GluN1-I642T, GluN1-Y647C, GluN1-Y647S, GluN1-A652T, GluN2A-N648S, GluN2A-F652V, GluN2A-M653I, and GluN2A-M653V (Fig. 2P, Table 2). This is consistent with the idea that most departures from the naturally occurring residues increases channel activation, in this case by reducing tonic proton inhibition present at physiological pH.
Variants in the M3 transmembrane helix alter divalent ion permeability
An important feature of NMDAR function is its permeability to the divalent cation Ca2+, which couples synaptic release of glutamate and depolarization-mediated reduction in Mg2+ block to engagement of second messenger systems, post-translational modifications of important proteins, and gene transcription [1]. Variant-induced changes in Ca2+ permeability could influence neuronal development, plasticity, and circuit function, and thus could contribute to the clinical phenotype (e.g., [32]). We, therefore, utilized a screen for potential changes to divalent permeability of NMDARs harboring M3 variants expressed in Xenopus oocytes. We evaluated the current–voltage relationship and the reversal potential in solutions that contained either 90 mM Na+ or 60 mM Ba2+ as a charge carrier for 20 representative variants (see Methods, [33]) to determine whether there might be systematic changes in Ca2+ permeability with variants in M3. We also measured the ratio of current recorded at -75 mV to + 30 mV for solutions containing either Na+ or Ba2+. A leftward (negative) Ba2+-induced shift in the variant NMDAR reversal potential relative to the reversal potential shift observed for WT NMDARs and a reduced current in Ba2+ with respect to Na+ at -75 mV for variant compared to WT are both consistent with reduced Ba2+ permeability. Both a rightward (positive) shift in the variant reversal potential in Ba2+ compared to WT NMDARs and an increase in current observed for variant compared to WT NMDAR in Ba2+ vs. Na+ at -75 mV are consistent with an increased Ba2+ permeability. No detectable shift in the reversal potential or the relative current at -75 mV in Ba2+ compared to Na+ would suggest minimal changes in Ba2+ permeability. We identified three variants that showed a negative reversal potential shift (GluN2A-T646A, GluN2A-T646R, GluN2A-N648S) compared to WT receptors, consistent with reduced Ba2+ permeability (see Methods, Supplemental Fig. S1, Supplemental Table S2). We note that the variant GluN2A-T646R, which places a positive charge in the permeation pathway, produces almost a complete loss of Ba2+-mediated inward current. We also identified five variants that showed a positive Ba2+-induced reversal potential shift (GluN1-A645S, GluN1-A653G, GluN1-A653T, GluN2A-V639I, GluN2A-I654T) compared to WT receptors, consistent with increased Ba2+ permeability (see Methods, Supplemental Fig. S1, Supplemental Table S2). These reversal potential changes were accompanied by the predicted changes in relative current for Ba2+ to Na+ at − 75 mV. All other variants showed less than a 3.4 mV difference in the Ba2+-induced reversal potential shift compared to WT (∆∆VREV), which we considered below our threshold for detection (see Methods). These data indicate that a subset of variants at specific M3 positions (GluN1-645,653 and GluN2A-639,646,648,654) can change Ba2+ permeability, which we suggest is predictive of changes in Ca2+ permeability. These results suggest more detailed and involved experiments to quantitatively assess the magnitude of these potential changes for the relative permeability of Ca2+ to Na+ [34] for a subset of M3 variants could be informative.
Variants in the M3 transmembrane helix alter receptor surface trafficking
Residues in the M3 domain are known to be important regulators of ER retention and surface delivery of NMDA receptors [35, 36]. To investigate if the M3 variants studied here can influence total NMDAR expression or surface expression, we measured the cell surface protein level and total protein level using a reporter assay in which beta-lactamase was fused to the extracellular ATD of WT GluN1 (β-lac-GluN1), WT GluN2A (β-lac-GluN2A), WT GluN2B (β-lac-GluN2B) or the ATD of M3 variant subunit cDNAs. The beta-lactamase cleavage of a cell-impermeable chromogenic substrate in the extracellular solution makes it possible to determine total and surface receptor expression by photometric measurement. ([10, 37]; see Methods). The data showed that NMDARs with a M3 variant subunit can reduce surface expression compared to WT, including GluN1-G638V, GluN1-M641I, GluN1-I642L, GluN1-Y647C, GluN1-Y647S, GluN1-N650K, GluN1-A653G, GluN1-L655Q, GluN2A-L642M, GluN2A-L642R, GluN2A-A643D, GluN2A-S644G, GluN2A-T646A, GluN2A-N648S, GluN2A-L649V, GluN2A-A650S, GluN2A-M653I, GluN2A-M653V, GluN2A-I654T, GluN2B-A636P, GluN2B-A636V, GluN2B-A639V, GluN2B-I641T, GluN2B-Y646C, and GluN2B-I655F. Only one variant exhibited over 50% increased surface expression compared to WT controls in this assay, GluN2B-A652G (154 ± 30% of WT, p < 0.05) (Table 2). In addition, several variants may alter total NMDAR expression in transfected HEK cells (Supplemental Table S3). These data illustrate the complex nature of variant effects, whereby some variants that alter functional properties in a manner that increases NMDAR currents will at the same time hinder trafficking to the surface, which may appear to reduce current. These data also support previous suggestion that the M3 transmembrane helix controls surface expression [35, 36], and emphasize the need for a comprehensive approach to evaluate all aspects of variant function and trafficking.
Variants in the M3 transmembrane helix alter response time course
To explore the effects of M3 variants on the deactivation response time course following rapid removal of glutamate from NMDARs, whole-cell patch clamp recordings were conducted on HEK cells transfected with variant GluN1 co-expressed with WT GluN2A, or WT GluN1 co-expressed with variant GluN2. Most of the human M3 variants derived from patients described here altered the deactivation time course compared to WT GluN1/GluN2A (44–51 ms) or WT GluN1/GluN2B (mean 524–663 ms; Fig. 3A–G, Table 3, Supplemental Table S4, S5). This is consistent with the increased agonist potency that often reflects a slower overall rate of agonist unbinding [38, 39]. The dissociation of agonist will often be a rate limiting step during the deactivation time course, although entry into a desensitized state can produce an additional slow component of the deactivation [38]. We observed three variants with strong desensitization in response to prolonged (e.g., 1.5 sec) application of agonists (GluN1-M641L, GluN1-M641V, GluN2B-I641T; see Supplemental Fig. S2, Supplemental Table S4). The magnitude of the desensitization of GluN1/GluN2B-I641T varied from cell to cell (Supplemental Fig. S2); the basis for the variability is unclear and may reflect sensitivity of this variant to post-translational modification or other intracellular processes that vary between individual cells.

Variants in the M3 transmembrane helix change NMDAR biophysical properties. A-D Representative whole cell current responses recorded under voltage clamp illustrate the deactivation time course for GluN1/GluN2A, GluN1/GluN2B, GluN1-A653G/GluN2A, GluN1-L655Q/GluN2B, GluN1/GluN2A-L649V, GluN1/GluN2B-I655F NMDARs in response to brief 2–6 ms application and rapid removal of 1 mM glutamate with 100 µM glycine present in all solutions. Variant responses were normalized to the peak amplitude of wild type NMDAR responses. Each variant showed a prolonged deactivation time course compared to WT NMDARs. E–G Summary of deactivation time course weighted tau for NMDAR variants. H, I Representative two electrode voltage clamp current recordings from Xenopus oocytes expressing GluN1/GluN2A wild type or variant NMDAR subunits (as indicated) showing current responses evoked by the application of 100 µM glutamate and 100 µM glycine followed by co-application of maximally effective glutamate and glycine plus 0.2 mM MTSEA (see Methods). N1-A7C indicates GluN1-A652C and 2A-A7C indicates GluN2A-A650C, which are covalently modified by MTSEA to lock the channel open. J–L Summary of calculated channel open probability (POPEN) evaluated by the degree of MTSEA potentiation at a holding potential of − 40 mV for GluN1 (J), GluN2A (K) and GluN2B variants (L) (see Methods). Data are shown as mean ± SEM
We evaluated the relationship between deactivation and potency, restricting our analysis to EC50 values greater than 30 nM, since values lower than this can be impacted by contaminant agonists, which can be present at concentrations of 10’s of nanomolar. Figure 4A shows the position within the M3 transmembrane helix of variants that alter EC50. Figure 4B shows the position within the M3 transmembrane helix of variants that alter tauweighted describing the deactivation of the time course. Figure 4D compares the relationship between the experimentally determined EC50 values and tauweighted. There was a significant correlation between these two parameters when expressed as the Log of the fold changes. The slope was − 0.84 and the intercept was − 0.08, suggesting an expected inverse relationship between EC50 and the time course of deactivation. The relationship between EC50 and tauweighted appears similar for GluN2A and GluN2B variants over this range of EC50 values. Actions on desensitization could influence the weighted time constant for deactivation by altering the rates or probability that a given receptor will enter a long-lived desensitized state from which it must return before unbinding, although we expect that relationship to be more complex. For GluN1 variants, increases in maximal open probability may reduce glutamate EC50 (Fig. 4C,F, see below) even though GluN1 binds glycine not glutamate. It is known that there is allosteric coupling between glutamate and glycine affinity [40]. Enhanced glutamate potency in GluN1 variants or GluN2 variants could also reflect enhanced gating, which would lead to similar shifts produced by variants in glutamate and glycine potency and was observed in this M3 variant dataset (Fig. 4E).

Effects of variants on tau deactivation, glutamate EC50, glycine EC50, and open probability (POPEN). A Measured EC50 values for GluN1 and GluN2A variants that were significantly different than WT controls are mapped onto a GluN1/GluN2A homology model of two subunits of the transmembrane pore (GluN1-gray, GluN2 yellow) based on the non-active GluN1/GluN2B structure from Chou et al. [20]. Variants that increase open probability are green, variants that decrease open probability are red, those without an effect are blue, and positions that have multiple variants at the same residue with opposing significantly different effects are orange. Residues that had multiple tested variants which either produced no significant alterations or significant effects in one direction were colored according to observed significant effects. B Measured deactivation tau weighted values for GluN1 and GluN2A variants that were significantly different than WT controls are colored as in (A). C Measured open probability values for GluN1 and GluN2A variants that were significantly different than WT controls are colored as in (A). The site at which a Cys residue is substituted and modified by MTSEA on GluN1 (grey) and on GluN2A (yellow) are shown in magenta with an asterisk. D The logarithm (Log, base ten) of the ratio of variant to WT tauweighted is plotted against the Log of the ratio of variant to WT glutamate EC50. The slope was − 0.84, the intercept was − 0.08, and the value for R2 was 0.77. E The Log of the ratio of variant to WT glycine EC50 is plotted against the Log of the ratio of variant to WT glutamate EC50. The slope was 1.24, the intercept was -0.05, and the value for R2 was 0.92. F The measured glutamate EC50 value is plotted against Po for variants (GluN1 and GluN2A) that increase open probability compared to WT GluN1/GluN2A NMDARs (see Supplemental Fig. S3 for the full dataset). Three regressions are shown; linear fit (red, r2 = 0.50), equation S1 (blue, r2 = 0.46, see Supplemental Fig. S3), and equation S2 (black, r2 = 0.54, see Supplemental Fig. S3). Error bars depict SEM for Tau and E values and 99% confidence intervals for EC50 values
Variants in the M3 transmembrane helix alter channel maximal open probability
To evaluate the effects of M3 variants on the maximal open probability for an agonist-bound NMDAR, we measured the MTSEA (2-aminoethyl methanethiosulfonate hydrobromide)-induced potentiation on NMDARs that contained a cysteine mutation introduced into the SYTANLAAF region of GluN1 (GluN1-A652C), GluN2A (GluN2A-A650C) or GluN2B (GluN2B-A651C; [18, 41]). We recorded current responses by TEVC at a holding potential of − 40 mV to 100 μM glutamate and 100 μM glycine, followed by co-application of glutamate and glycine with 200 μM MTSEA, which covalently modified the introduced Cys residue to lock the receptor into the open conformation. Maximal open probability can be calculated as the reciprocal of the degree of potentiation, corrected for a change in single channel conductance. These data suggest that multiple M3-variant-containing NMDARs have significantly higher calculated open probability compared to WT (Fig. 3H–L, Table 3). Notably, GluN2A-T646R showed no MTSEA potentiation (current responses with MTSEA were smaller than the control response), indicating that this variant may interfere with the ability of MTSEA to covalently label and lock the NMDA receptors into the open state. Although most variants increased maximal open probability, there were a few variants (GluN1 Tyr647, Asn650, Ala652 and GluN2A Asn648, Phe652) that decreased open probability, that were close to the residues that were converted to a cysteine (GluN1-A652C, GluN2A-A650C) that is covalently modified by MTSEA to lock the channel open. It is not clear if the reduced open probability in this assay reflects a confounding effect for these variants on the assay readout due to their proximity to the modified residue, or if these residues do indeed lower open probability. Figure 4C illustrates how the results from this assay map onto the pore-lining residues.
There is a well understood relationship between EC50 and open probability for all receptors for which agonist binding and channel opening are separated as independent steps [42]. We investigated this relationship by plotting the EC50 for variants versus the maximal open probability (Supplemental Fig. S3A). The relationship between open probability and EC50 is less clear for maximal open probability values lower than 0.1 [42], and strong reductions to values below 0.1 often reflect processes like desensitization that are only indirectly related to channel opening and closing rates. We observed a trend towards lower glutamate EC50 values at higher open probabilities in the data, as expected (Supplemental Fig. S3A). We analyzed GluN1 and GluN2A variants (expressed as GluN1/GluN2A) that showed higher open probabilities than WT and had similar variant-induced fold shifts in glutamate and glycine EC50, reasoning that variants that primarily increase open probability will equally impact glutamate and glycine potency. We excluded variants with a ratio of the fold differences for glutamate and glycine that was greater than the mean ± STD (2.7 fold). The data for variants satisfying this criteria were fitted by a linear regression (r2 = 0.50). We also considered simple gating models that allowed for the relationship between open probability and EC50 to be quantified (see Supplemental Fig. S3). These models produced a similar fit to the data (r2 = 0.46–0.54; Fig. 4E, Supplemental Fig. S3), and suggest that there is an underlying relationship between the effects between M3 gain-of-function variants that primarily increase open probability and measured changes in EC50.
Estimating the impact of M3 transmembrane helix variants on NMDAR function
Genetic variants identified in patients with neurologic disorders usually lead to complex functional changes at the protein, which for NMDARs result in differences in current responses and other properties of the variant receptor, as well as compensatory changes in circuits and systems. To estimate the overall effect of rare variants on NMDAR function and predict their effect on neuronal function (in the absence of compensation), we developed a strategy that considered six measured parameters in addition to an estimation of the relative net change in synaptic and non-synaptic NMDAR charge transfer for variant compared to WT receptors [10, 32]. We completed this analysis for all 48 variants presented here. The majority of the M3 transmembrane helix variants (28/48) shows an enhanced function (Table 3, Fig. 5). The functional enhancement is mainly generated by increased agonist potency, prolonged deactivation time course, and increased maximal open probability, although several variants also showed reduced voltage-dependent Mg2+ block. This finding suggests that the arrangement of some amino acids in this region seems critically selected to allow the channel to remain closed so that opening can be driven by agonist binding. Many amino acid substitutions in M3 appear to perturb the receptor in a manner that reduces the activation energy required for gating and leads to apparent gain-of-function that is manifest as larger charge transfer both for responses to synaptic glutamate profile (27/48 showed greater than 2.5-fold increase [13]) and/or steady-state low glutamate concentration (32/48 showed greater than 2.5-fold increase [13]), as one would expect to find extrasynaptically [22,23,24]. We found that 9/48 variant appeared to reduce NMDAR function, 3 of which were influenced by strong reduction (e.g. > 90%) in trafficking to the plasma membrane.

Assessment of M3 variant-mediated changes in parameters supporting GoF and LoF status. Glutamate and glycine potency ratios are given as WT/variant EC50 because EC50 is reciprocally related to potency. Mg2+ IC50, open probability (POPEN), weighted tau (τw), and surface expression fold effects are given as variant/WT. Two sets of the pooled τw data for same day controls were used to obtain WT parameter values for comparison to variants (see Supplemental Table S5). A high (H) or moderate (M) confidence (see Myers et al. [13]) for the change is indicated; red is LoF and blue is GoF. The number of high (H) and moderate (M) changes are given Count when there is no conflict in the direction of change for two parameters. Conflicting and subthreshold variants were re-classified as Possible GoF or Possible LoF if the fold change in the synaptic (left number) or non-synaptic (right number) of relative charge transfer ratio (Variant/WT) was > 2.5-fold or < 0.4-fold. aCalculation of relative for charge transfer (Variant/WT) was from Myers et al., [13]. n.d. indicates response was too small to measure (tstm). *Indicates that experiments to assess tau were run and currents recorded, but they were too small to allow reliable determination of tau and thus we cannot determine whether the variant alters overall function
Rescue pharmacology of variants in M3 transmembrane helix
The functional data described here suggest that a significant proportion of M3 variants produces a gain-of-function, which means that these variant NMDARs will be more active on the cell surface. That is, the variant NMDARs are more likely to drive larger inward currents and produce greater Ca2+ entry into the cell each time they are activated compared to WT. To determine whether FDA-approved NMDAR channel blockers might be useful to potentially mitigate some of the effects of overactive NMDARs, we used TEVC recordings from Xenopus oocytes expressing WT and variant NMDARs to determine the IC50 values for these compounds (Figs. 6, 7; Table 4). In general, most M3 variant NMDARs showed a decreased sensitivity (increased IC50 values) to memantine, ketamine, dextromethorphan, and the metabolite dextrorphan. This was especially evident for GluN2A-L642M, GluN2A-L642R, GluN2A-A643D, GluN2A-T646A and GluN2B-A639V (Figs. 6, 7; Table 4). However, a subset of variants residing near the upper portion of the internal cavity increased blocker potency, raising the possibility that they could be useful for selectively reducing current through variant but not WT receptors (e.g., [43, 44]). This phenomenon is consistent with the effect of variants in this region, as illustrated in structural studies of therapeutically-relevant channel blockers such as memantine and ketamine [45].

Variants in the M3 transmembrane helix influence sensitivity to FDA-approved NMDAR channel blocker memantine. A-F Representative two electrode voltage clamp current recordings from Xenopus oocytes expressing GluN1/GluN2A and GluN1/GluN2B WT or variant NMDAR subunits, as indicated. The FDA-approved NMDAR channel blocker memantine concentration–response relationship was determined by co-applying increasing concentrations of memantine (Mem) with maximally effective concentrations of glutamate and glycine (100 µM) at a holding potential of − 40 mV. G-I Composite concentration–response curves of memantine were assessed by TEVC recordings of Xenopus oocytes for GluN1 (G), GluN2A (H) and GluN2B (I) variants in the presence of maximally effective concentrations of agonists (100 µM glutamate and 100 µM glycine) at a holding potential of − 40 mV. Data are shown as mean ± SEM

Variants in the M3 transmembrane helix influence sensitivity to FDA-approved NMDAR channel blockers ketamine, dextromethorphan and its metabolite dextrorphan. Composite concentration–response curves of FDA-approved NMDAR channel blockers ketamine (A–C), dextromethorphan (D–F) and its CYP2D6 metabolite dextrorphan (G–I) were assessed by TEVC recordings of Xenopus oocytes in the presence of maximally effective concentrations of agonists (100 µM glutamate and 100 µM glycine) at holding potential of – 40 mV. Data are shown as mean ± SEM
Discussion
In this study, we describe 48 missense variants in the M3 transmembrane helix encoded by various GRIN genes from 56 patients. According to the available clinical data, the most common phenotypes of these patients with M3 transmembrane variants are epilepsy, intellectual disability, and developmental delay. Evaluation of the genetic variation across the functional domains of GluN1 and GluN2 revealed that most residues within the M3 transmembrane helix (except those in GluN2C) are subject to purifying selection [21] (Fig. 1), suggesting that functional variation therein is likely to be detrimental and pathogenic. This analysis strongly suggests that future variants found in the M3 transmembrane helix are likely to be dysfunctional and potentially contribute to the patients’ clinical phenotype.
An important point of consideration is the fact that properties assessed in this study are from NMDARs that contain two copies of the variant subunit, which will be a minority of receptors in patients. Most receptors in patients will contain a single copy of the variant subunit. This raises the question of whether the results obtained from the study of receptors with two copies of the variant subunit are predictive of receptors with a single copy of the variant subunit. A number of studies evaluating NMDARs that contain one or two copies of the variant subunit suggest that the changes in properties found in NMDARs with two copies of the variant subunit are similar to those that occur in receptors with a single copy, although the magnitude of the change is often less. For example, 15 GRIN variants including GluN1-G620R [32], GluN2A-P552R [46, 47], GluN2A-L611Q, GluN2A-N614S, GluN2A-N615K [32], GluN2A-S644G [48], GluN2A-D731N [49], GluN2A-L812M [16], GluN2A-M817V [14], GluN2B-E413G [10], and GluN2B-W607C, GluN2B-G611V, GluN2B-N615K, GluN2B-V618G, GluN2B-V620M [32] that have been studied show qualitatively similar changes in diheteomeric receptor properties occur when a single copy of a variant subunit is present compared to when two copies of a variant subunit are present. Consistent with this result, we find similar effects on glutamate and glycine potency for NMDARs with two copies of the GluN2A-L642M variant subunit and NMDARs harboring a single copy of this variant subunit (see Supplemental Fig. S4). In addition, GluN1/GluN2A/GluN2B triheteromeric receptors that contained one copy of either GluN2A-E551K, GluN2A-P552R, GluN2A-S644G, GluN2A-L649V, GluN2A-L812M, GluN2B-G543R, GluN2B-A639V, GluN2B-M818T, or GluN2B-A819T [50] show similar changes compared to diheteromeric receptors with two copies of the variant subunit. Some of these published variants (GluN2B-A639V, GluN2A-S644G, GluN2A-L649V) for which properties in receptors with two copies are predictive of effects on receptors with a single copy of the variant subunit [48, 50] were evaluated here in this study, and this previously published work confirms that their properties transfer to NMDARs with a single variant subunit. These data support the idea that the qualitative properties identified in NMDARs with two copies of the variant subunit are predictive of properties with a single copy of the subunit.
The impact of M3 variants on NMDAR function is comprehensive and includes changes in agonist potency, sensitivity to negative allosteric modulators, receptor surface trafficking, deactivation time course, and divalent ion permeability. Combining relative changes for most of these assays, we can predict the potential for gain- or loss-of-function for these variants [10, 32]. Most of the M3 variants evaluated in this study showed an increased potency to glutamate and glycine, suggesting that variants within the M3 domain often make the receptor more effective. For models in which binding is separated from gating steps, the coupling of EC50 to efficacy (i.e., open probability here) is implicit in the solution to the system of equations describing the model. For example, Colquhoun [42] shows that increases in efficacy alone without changes in association or dissociation rates decrease agonist EC50. This likely reflects a lower activation energy by the M3 variant for progression along steps that lead to pore opening, which is well-known to increase the potency of agonist by shifting the equilibrium toward open states. Although the consequent increased current expected from M3 variation could be homeostatically compensated by overall lower surface expression, we predict that variants within this region found in the future are most likely to be GoF, provided that they reach the neuronal surface. However, we also find some notable exceptions in these variants. For example, at the locus GluN2A-L642, different amino acid substitutions such as methionine and arginine produce a large difference in the sensitivity to Mg2+ block. GluN2A-L642M showed no significant difference compared to WT GluN2A, whereas GluN2A-L642R is insensitive to extracellular Mg2+, which is likely a consequence of the introduction of a positive charge near the Mg2+ binding site. GluN2A-T646A and GluN2A-T646R showed a similar difference, again consistent with the introduction of a charge within the pore that perturbs Mg2+ binding. These data suggest that variants within or close to the conserved SYTANLAAF motif whose side chains face the ion channel pore could have differential effects dependent on the characteristics of the side chain.
For patients of drug-resistant epilepsy with GRIN gene variants, FDA-approved NMDAR blockers might provide some reduction in excitation and possibly epileptiform activity, with potential utility as antiseizure medications [43, 48, 51, 52]. The evaluation of FDA-approved NMDAR channel blockers suggests that most of the GoF M3 variant NMDARs decreased the sensitivity to ketamine, memantine, dextrorphan and dextromethorphan, suggesting that these drugs are unlikely to be suitable for patients with certain GRIN variants, since the drugs do not reach levels in brain necessary to block variant NMDARs. By contrast, some variants near the extracellular facing surface of the internal pocket (Fig. 1B) increase sensitivity to channel blockers (GluN1-Y647C, GluN1-L655Q, GluN2A-N648S, and possibly GluN2B-A652G), and patients with these variants might find that these channel blockers mitigate some of the consequences of the variants. This is consistent with recent structural work that suggests that this region of the intra-pore cavity can interact with channel blockers [45]. However, the shallow current–voltage curves produced by monovalent organic channel blockers, while mitigating excess activation, are unlikely to possess the voltage-dependence of Mg2+ which is likely necessary to allow the receptors to act as coincidence detectors, and thus full restoration of synaptic plasticity might not be possible with these drugs [44].
In summary, this study shows for the first time that the most common functional consequences of variation in an intolerant portion of the NMDAR comprising the M3 transmembrane helix, which is known to control channel gating, are enhanced activity categorized as a gain-of-function. These data suggest that future variants in the M3 region of GRIN1 or any GRIN2 gene identified in patients will have similar effects and lead to similar clinical phenotypes. It further stresses the significance of the combination of clinical data, genomic sequencing, functional validation and in vitro rescue pharmacology to provide not only a better understanding of pathogenesis but precise strategy of treatment as well. In addition, it provides insight into the requirements for residues at some positions within this gating region, as random variants arising from single nucleotide polymorphisms often increase channel activation. This would be consistent with the need for the pore to remain closed except when coupled to energy produced by agonist binding.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABD:
-
Agonist binding domain
- ASD:
-
Autism spectrum disorder
- CTD:
-
Cytosolic carboxyl terminal domain
- DEE:
-
Developmental and epileptic encephalopathy
- EOEE:
-
Early-onset epileptic encephalopathy
- EPI:
-
Epilepsy/seizures
- ADHD:
-
Attention deficit hyperactivity disorder
- ID:
-
Intellectual disability
- IS:
-
infantile spasms
- GoF:
-
Gain-of-function
- LD:
-
Language disorder
- LoF:
-
Loss-of-function
- MD:
-
Movement disorder
- NMDAR:
-
N-methyl-d-aspartate receptor
- NTD:
-
N-terminal domain, also ATD
- TMD:
-
Transmembrane domains (M1, M3, M4, and a reentrant loop M2)
References
Hansen KB, Wollmuth LP, Bowie D, Furukawa H, Menniti FS, Sobolevsky AI, Swanson GT, Swanger SA, Greger IH, Nakagawa T, McBain CJ, Jayaraman V, Low CM, Dell’Acqua ML, Diamond JS, Camp CR, Perszyk RE, Yuan H, Traynelis SF (2021) Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol Rev 73(4):298–487. https://doi.org/10.1124/pharmrev.120.000131
Myers SJ, Yuan H, Kang JQ, Tan FCK, Traynelis SF, Low CM (2019) Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Res. https://doi.org/10.12688/f1000research.18949.1
Cenci MA, Skovgard K, Odin P (2022) Non-dopaminergic approaches to the treatment of motor complications in Parkinson’s disease. Neuropharmacology 210:109027. https://doi.org/10.1016/j.neuropharm.2022.109027
Geoffroy C, Paoletti P, Mony L (2022) Positive allosteric modulation of NMDA receptors: mechanisms, physiological impact and therapeutic potential. J Physiol 600(2):233–259. https://doi.org/10.1113/JP280875
Li S, Stern AM (2022) Bioactive human Alzheimer brain soluble Abeta: pathophysiology and therapeutic opportunities. Mol Psychiatry 27(8):3182–3191. https://doi.org/10.1038/s41380-022-01589-5
Kritzer MD, Mischel NA, Young JR, Lai CS, Masand PS, Szabo ST, Mathew SJ (2022) Ketamine for treatment of mood disorders and suicidality: a narrative review of recent progress. Ann Clin Psychiatry 34(1):33–43. https://doi.org/10.12788/acp.0048
Moore TJ, Alami A, Alexander GC, Mattison DR (2022) Safety and effectiveness of NMDA receptor antagonists for depression: a multidisciplinary review. Pharmacotherapy 42(7):567–579. https://doi.org/10.1002/phar.2707
Camp CR, Yuan H (2020) GRIN2D/GluN2D NMDA receptor: unique features and its contribution to pediatric developmental and epileptic encephalopathy. Eur J Paediatr Neurol 24:89–99. https://doi.org/10.1016/j.ejpn.2019.12.007
Benke TA, Park K, Krey I, Camp CR, Song R, Ramsey AJ, Yuan H, Traynelis SF, Lemke J (2021) Clinical and therapeutic significance of genetic variation in the GRIN gene family encoding NMDARs. Neuropharmacology 199:108805. https://doi.org/10.1016/j.neuropharm.2021.108805
Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, Strong KL, Hu C, Kusumoto H, Zhang J, Adams DR, Millichap JJ, Petrovski S, Traynelis SF, Yuan H (2016) Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet 99(6):1261–1280. https://doi.org/10.1016/j.ajhg.2016.10.002
Vyklicky V, Krausova B, Cerny J, Ladislav M, Smejkalova T, Kysilov B, Korinek M, Danacikova S, Horak M, Chodounska H, Kudova E, Vyklicky L (2018) Surface expression, function, and pharmacology of disease-associated mutations in the membrane domain of the human GluN2B subunit. Front Mol Neurosci 11:110. https://doi.org/10.3389/fnmol.2018.00110
Kolcheva M, Kortus S, Krausova BH, Barackova P, Misiachna A, Danacikova S, Kaniakova M, Hemelikova K, Hotovec M, Rehakova K, Horak M (2021) Specific pathogenic mutations in the M3 domain of the GluN1 subunit regulate the surface delivery and pharmacological sensitivity of NMDA receptors. Neuropharmacology 189:108528. https://doi.org/10.1016/j.neuropharm.2021.108528
Myers SJ, Yuan HJ, Perszyk RE, Zhang J, Kim S, Nocilla KA, Allen JP, Bain JM, Lemke JR, Lal D, Benke TA, Traynelis SF (2023) Classification of missense variants in the N-methyl-d-aspartate receptor GRIN gene family as gain- or loss-of-function. Hum Mol Genet 32(19):2857–2871. https://doi.org/10.1093/hmg/ddad104
Chen W, Tankovic A, Burger PB, Kusumoto H, Traynelis SF, Yuan H (2017) Functional evaluation of a de novo GRIN2A mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol Pharmacol 91(4):317–330. https://doi.org/10.1124/mol.116.106781
Hedegaard M, Hansen KB, Andersen KT, Brauner-Osborne H, Traynelis SF (2012) Molecular pharmacology of human NMDA receptors. Neurochem Int 61(4):601–609. https://doi.org/10.1016/j.neuint.2011.11.016
Yuan H, Hansen KB, Zhang J, Pierson TM, Markello TC, Fajardo KV, Holloman CM, Golas G, Adams DR, Boerkoel CF, Gahl WA, Traynelis SF (2014) Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat Commun 5:3251. https://doi.org/10.1038/ncomms4251
Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jorgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF (2013) Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Mol Pharmacol 84(1):114–127. https://doi.org/10.1124/mol.113.085803
Yuan H, Erreger K, Dravid SM, Traynelis SF (2005) Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J Biol Chem 280(33):29708–29716. https://doi.org/10.1074/jbc.M414215200
Perszyk RE, Kristensen AS, Lyuboslavsky P, Traynelis SF (2021) Three-dimensional missense tolerance ratio analysis. Genome Res 31(8):1447–1461. https://doi.org/10.1101/gr.275528.121
Chou TH, Tajima N, Romero-Hernandez A, Furukawa H (2020) Structural basis of functional transitions in mammalian NMDA receptors. Cell. 182(2):357-371 e313. https://doi.org/10.1016/j.cell.2020.05.052
Cvijovic I, Good BH, Desai MM (2018) The effect of strong purifying selection on genetic diversity. Genetics 209(4):1235–1278. https://doi.org/10.1534/genetics.118.301058
Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27(36):9736–9741. https://doi.org/10.1523/JNEUROSCI.3009-07.2007
Herman MA, Nahir B, Jahr CE (2011) Distribution of extracellular glutamate in the neuropil of hippocampus. PLoS ONE 6(11):e26501. https://doi.org/10.1371/journal.pone.0026501
Moldavski A, Behr J, Bading H, Bengtson CP (2020) A novel method using ambient glutamate for the electrophysiological quantification of extrasynaptic NMDA receptor function in acute brain slices. J Physiol 598(4):633–650. https://doi.org/10.1113/JP278362
Tang CM, Dichter M, Morad M (1990) Modulation of the N-methyl-D-aspartate channel by extracellular H+. Proc Natl Acad Sci U S A 87(16):6445–6449. https://doi.org/10.1073/pnas.87.16.6445
Traynelis SF, Cull-Candy SG (1990) Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345(6273):347–350. https://doi.org/10.1038/345347a0
Vyklicky L Jr, Vlachova V, Krusek J (1990) The effect of external pH changes on responses to excitatory amino acids in mouse hippocampal neurones. J Physiol 430:497–517. https://doi.org/10.1113/jphysiol.1990.sp018304
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307(5950):462–465. https://doi.org/10.1038/307462a0
Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Gunther W, Seeburg PH, Sakmann B (1992) Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 257(5075):1415–1419. https://doi.org/10.1126/science.1382314
Kawajiri S, Dingledine R (1993) Multiple structural determinants of voltage-dependent magnesium block in recombinant NMDA receptors. Neuropharmacology 32(11):1203–1211. https://doi.org/10.1016/0028-3908(93)90014-t
Wollmuth LP, Kuner T, Sakmann B (1998) Intracellular Mg2+ interacts with structural determinants of the narrow constriction contributed by the NR1-subunit in the NMDA receptor channel. J Physiol 506(1):33–52. https://doi.org/10.1111/j.1469-7793.1998.00033.x
Li J, Zhang J, Tang W, Mizu RK, Kusumoto H, XiangWei W, Xu Y, Chen W, Amin JB, Hu C, Kannan V, Keller SR, Wilcox WR, Lemke JR, Myers SJ, Swanger SA, Wollmuth LP, Petrovski S, Traynelis SF, Yuan H (2019) De novo GRIN variants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases. Hum Mutat 40(12):2393–2413. https://doi.org/10.1002/humu.23895
Dingledine R, Hume RI, Heinemann SF (1992) Structural determinants of barium permeation and rectification in non-NMDA glutamate receptor channels. J Neurosci 12(10):4080–4087. https://doi.org/10.1523/JNEUROSCI.12-10-04080.1992
Moody G, Musco A, Bennett J, Wollmuth LP (2023) An integrated approach to evaluate the functional effects of disease-associated NMDA receptor variants. Neuropharmacology 240:109703. https://doi.org/10.1016/j.neuropharm.2023.109703
Horak M, Chang K, Wenthold RJ (2008) Masking of the endoplasmic reticulum retention signals during assembly of the NMDA receptor. J Neurosci 28(13):3500–3509. https://doi.org/10.1523/JNEUROSCI.5239-07.2008
Kaniakova M, Krausova B, Vyklicky V, Korinek M, Lichnerova K, Vyklicky L, Horak M (2012) Key amino acid residues within the third membrane domains of NR1 and NR2 subunits contribute to the regulation of the surface delivery of N-methyl-D-aspartate receptors. J Biol Chem 287(31):26423–26434. https://doi.org/10.1074/jbc.M112.339085
Lam VM, Beerepoot P, Angers S, Salahpour A (2013) A novel assay for measurement of membrane-protein surface expression using a beta-lactamase. Traffic 14(7):778–784. https://doi.org/10.1111/tra.12073
Lester RA, Jahr CE (1992) NMDA channel behavior depends on agonist affinity. J Neurosci 12(2):635–643. https://doi.org/10.1523/JNEUROSCI.12-02-00635.1992
Vance KM, Simorowski N, Traynelis SF, Furukawa H (2011) Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun 2:294. https://doi.org/10.1038/ncomms1295
Mayer ML, Vyklicky L Jr, Clements J (1989) Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature 338(6214):425–427. https://doi.org/10.1038/338425a0
Jones KS, VanDongen HM, VanDongen AM (2002) The NMDA receptor M3 segment is a conserved transduction element coupling ligand binding to channel opening. J Neurosci 22(6):2044–2053
Colquhoun D (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125(5):924–947. https://doi.org/10.1038/sj.bjp.0702164
Xu Y, Song R, Chen W, Strong K, Shrey D, Gedela S, Traynelis SF, Zhang G, Yuan H (2021) Recurrent seizure-related GRIN1 variant: molecular mechanism and targeted therapy. Ann Clin Transl Neurol 8(7):1480–1494. https://doi.org/10.1002/acn3.51406
Song RZJ, Perszyk RE, Camp CR, Tang W, Kannan V, Li J, Chen J, Liang SH, Traynelis SF, Yuan H (2023) Differential responses of disease-related GRIN variants located in pore-forming M2 domain of N-methyl-D-aspartate receptor to FDA-approved inhibitors. J Neurochem. https://doi.org/10.1111/jnc.15942
Chou TH, Epstein M, Michalski K, Fine E, Biggin PC, Furukawa H (2022) Structural insights into binding of therapeutic channel blockers in NMDA receptors. Nat Struct Mol Biol 29(6):507. https://doi.org/10.1038/s41594-022-00772-0
Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF, Yuan H (2017) Molecular mechanism of disease-associated mutations in the Pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genet 13(1):e1006536. https://doi.org/10.1371/journal.pgen.1006536
Gibb AJ, Ogden KK, McDaniel MJ, Vance KM, Kell SA, Butch C, Burger P, Liotta DC, Traynelis SF (2018) A structurally derived model of subunit-dependent NMDA receptor function. J Physiol 596(17):4057–4089. https://doi.org/10.1113/JP276093
Amador A, Bostick CD, Olson H, Peters J, Camp CR, Krizay D, Chen W, Han W, Tang W, Kanber A, Kim S, Teoh J, Sah M, Petri S, Paek H, Kim A, Lutz CM, Yang M, Myers SJ, Bhattacharya S, Yuan H, Goldstein DB, Poduri A, Boland MJ, Traynelis SF, Frankel WN (2020) Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain 143(7):2039–2057. https://doi.org/10.1093/brain/awaa147
Gao K, Tankovic A, Zhang Y, Kusumoto H, Zhang J, Chen W, XiangWei W, Shaulsky GH, Hu C, Traynelis SF, Yuan H, Jiang Y (2017) A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE 12(2):e0170818. https://doi.org/10.1371/journal.pone.0170818
Han W, Yuan H, Allen JP, Kim S, Shaulsky GH, Perszyk RE, Traynelis SF, Myers SJ (2022) Opportunities for Precision Treatment of GRIN2A and GRIN2B Gain-of-Function Variants in Triheteromeric N-Methyl-D-Aspartate Receptors. J Pharmacol Exp Ther 381(1):54–66. https://doi.org/10.1124/jpet.121.001000
Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, Golas G, Simeonov DR, Holloman C, Tankovic A, Karamchandani MM, Schreiber JM, Mullikin JC, DftNCSP Ph, Tifft CJ, Toro C, Boerkoel CF, Traynelis SF, Sgahl WA (2014) GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol 1(3):190–198. https://doi.org/10.1002/acn3.39
Mir A, Qahtani M, Bashir S (2020) GRIN2A -related severe epileptic encephalopathy treated with memantine: an example of precision medicine. J Pediatr Genet 9(4):252–257. https://doi.org/10.1055/s-0039-3401028
Ohba C, Shiina M, Tohyama J, Haginoya K, Lerman-Sagie T, Okamoto N, Blumkin L, Lev D, Mukaida S, Nozaki F, Uematsu M, Onuma A, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Kato M, Ogata K, Saitsu H, Matsumoto N (2015) GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 56(6):841–848. https://doi.org/10.1111/epi.12987
Pironti E, Granata F, Cucinotta F, Gagliano A, Efthymiou S, Houlden H, Salpietro V, Di Rosa G (2018) Electroclinical history of a five-year-old girl with GRIN1-related early-onset epileptic encephalopathy: a video-case study. Epileptic Disord 20(5):423–427. https://doi.org/10.1684/epd.2018.0992
Lemke JR, Geider K, Helbig KL, Heyne HO, Schutz H, Hentschel J, Courage C, Depienne C, Nava C, Heron D, Moller RS, Hjalgrim H, Lal D, Neubauer BA, Nurnberg P, Thiele H, Kurlemann G, Arnold GL, Bhambhani V, Bartholdi D, Pedurupillay CR, Misceo D, Frengen E, Stromme P, Dlugos DJ, Doherty ES, Bijlsma EK, Ruivenkamp CA, Hoffer MJ, Goldstein A, Rajan DS, Narayanan V, Ramsey K, Belnap N, Schrauwen I, Richholt R, Koeleman BP, Sa J, Mendonca C, de Kovel CG, Weckhuysen S, Hardies K, De Jonghe P, De Meirleir L, Milh M, Badens C, Lebrun M, Busa T, Francannet C, Piton A, Riesch E, Biskup S, Vogt H, Dorn T, Helbig I, Michaud JL, Laube B, Syrbe S (2016) Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86(23):2171–2178. https://doi.org/10.1212/WNL.0000000000002740
Fry AE, Fawcett KA, Zelnik N, Yuan H, Thompson BAN, Shemer-Meiri L, Cushion TD, Mugalaasi H, Sims D, Stoodley N, Chung SK, Rees MI, Patel CV, Brueton LA, Layet V, Giuliano F, Kerr MP, Banne E, Meiner V, Lerman-Sagie T, Helbig KL, Kofman LH, Knight KM, Chen W, Kannan V, Hu C, Kusumoto H, Zhang J, Swanger SA, Shaulsky GH, Mirzaa GM, Muir AM, Mefford HC, Dobyns WB, Mackenzie AB, Mullins JGL, Lemke JR, Bahi-Buisson N, Traynelis SF, Iago HF, Pilz DT (2018) De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141(3):698–712. https://doi.org/10.1093/brain/awx358
Epi KC, Phenome E, Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR (2013) De novo mutations in epileptic encephalopathies. Nature 501(7466):217–221. https://doi.org/10.1038/nature12439
Brock S, Laquerriere A, Marguet F, Myers SJ, Hongjie Y, Baralle D, Vanderhasselt T, Stouffs K, Keymolen K, Kim S, Allen J, Shaulsky G, Chelly J, Marcorelle P, Aziza J, Villard L, Sacaze E, de Wit MCY, Wilke M, Mancini GMS, Hehr U, Lim D, Mansour S, Traynelis SF, Beneteau C, Denis-Musquer M, Jansen AC, Fry AE, Bahi-Buisson N (2023) Overlapping cortical malformations in patients with pathogenic variants in GRIN1 and GRIN2B. J Med Genet 60(2):183–192. https://doi.org/10.1136/jmedgenet-2021-107971
Strehlow V, Heyne HO, Vlaskamp DRM, Marwick KFM, Rudolf G, de Bellescize J, Biskup S, Brilstra EH, Brouwer OF, Callenbach PMC, Hentschel J, Hirsch E, Kind PC, Mignot C, Platzer K, Rump P, Skehel PA, Wyllie DJA, Hardingham GE, van Ravenswaaij-Arts CMA, Lesca G, Lemke JR, group GAs, (2019) GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain 142(1):80–92. https://doi.org/10.1093/brain/awy304
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, Dhindsa RS, Hitomi Y, Schoch K, Spillmann RC, Heimer G, Marek-Yagel D, Tzadok M, Han Y, Worley G, Goldstein J, Jiang YH, Lancet D, Pras E, Shashi V, McHale D, Need AC, Goldstein DB (2015) Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 17(10):774–781. https://doi.org/10.1038/gim.2014.191
Fernandez-Marmiesse A, Kusumoto H, Rekarte S, Roca I, Zhang J, Myers SJ, Traynelis SF, Couce ML, Gutierrez-Solana L, Yuan H (2018) A novel missense mutation in GRIN2A causes a nonepileptic neurodevelopmental disorder. Mov Disord 33(6):992–999. https://doi.org/10.1002/mds.27315
von Stulpnagel C, Ensslen M, Moller RS, Pal DK, Masnada S, Veggiotti P, Piazza E, Dreesmann M, Hartlieb T, Herberhold T, Hughes E, Koch M, Kutzer C, Hoertnagel K, Nitanda J, Pohl M, Rostasy K, Haack TB, Stohr K, Kluger G, Borggraefe I (2017) Epilepsy in patients with GRIN2A alterations: Genetics, neurodevelopment, epileptic phenotype and response to anticonvulsive drugs. Eur J Paediatr Neurol 21(3):530–541. https://doi.org/10.1016/j.ejpn.2017.01.001
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, McKnight D, Bai R, Suchy S, Friedman B, Tahiliani J, Pineda-Alvarez D, Richard G, Brandt T, Haverfield E, Chung WK, Bale S (2016) Clinical application of whole-exome sequencing across clinical indications. Genet Med 18(7):696–704. https://doi.org/10.1038/gim.2015.148
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367(20):1921–1929. https://doi.org/10.1056/NEJMoa1206524
Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, Wright S, Tsintsadze V, Michel A, Doummar D, Lascelles K, Strug L, Waters P, de Bellescize J, Vrielynck P, de Saint MA, Ville D, Ryvlin P, Arzimanoglou A, Hirsch E, Vincent A, Pal D, Burnashev N, Sanlaville D, Szepetowski P (2013) GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 45(9):1061–1066. https://doi.org/10.1038/ng.2726
Platzer K, Yuan H, Schutz H, Winschel A, Chen W, Hu C, Kusumoto H, Heyne HO, Helbig KL, Tang S, Willing MC, Tinkle BT, Adams DJ, Depienne C, Keren B, Mignot C, Frengen E, Stromme P, Biskup S, Docker D, Strom TM, Mefford HC, Myers CT, Muir AM, LaCroix A, Sadleir L, Scheffer IE, Brilstra E, van Haelst MM, van der Smagt JJ, Bok LA, Moller RS, Jensen UB, Millichap JJ, Berg AT, Goldberg EM, De Bie I, Fox S, Major P, Jones JR, Zackai EH, Abou Jamra R, Rolfs A, Leventer RJ, Lawson JA, Roscioli T, Jansen FE, Ranza E, Korff CM, Lehesjoki AE, Courage C, Linnankivi T, Smith DR, Stanley C, Mintz M, McKnight D, Decker A, Tan WH, Tarnopolsky MA, Brady LI, Wolff M, Dondit L, Pedro HF, Parisotto SE, Jones KL, Patel AD, Franz DN, Vanzo R, Marco E, Ranells JD, Di Donato N, Dobyns WB, Laube B, Traynelis SF, Lemke JR (2017) GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet 54(7):460–470. https://doi.org/10.1136/jmedgenet-2016-104509
Freunscht I, Popp B, Blank R, Endele S, Moog U, Petri H, Prott EC, Reis A, Rubo J, Zabel B, Zenker M, Hebebrand J, Wieczorek D (2013) Behavioral phenotype in five individuals with de novo mutations within the GRIN2B gene. Behav Brain Funct 9:20. https://doi.org/10.1186/1744-9081-9-20
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu HM, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CL, Tang S (2015) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17(7):578–586. https://doi.org/10.1038/gim.2014.154
Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, Tang S, Helbig I (2016) Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 18(9):898–905. https://doi.org/10.1038/gim.2015.186
Lewis SA, Shetty S, Gamble S, Heim J, Zhao N, Stitt G, Pankratz M, Mangum T, Marku I, Rosenberg RB, Wilfong AA, Fahey MC, Kim S, Myers SJ, Appavu B, Kruer MC (2023) Intrathecal magnesium delivery for Mg++-insensitive NMDA receptor activity due to GRIN1 mutation. Orphanet J Rare Dis 18(1):225. https://doi.org/10.1186/s13023-023-02756-9
Acknowledgements
We thank Wei Han, Ian Hwang, Phuong Le, Daniel Teuscher, and Lingling Xie for their excellent support and for sharing unpublished data.
Funding
This work was supported by the NIH (NS111619 to SFT; HD082373, MH127404, AG075444, AG079956 and AG081401 to HY; AG072142 to SJM), the CureGRIN Foundation to SFT, Austin’s Purpose to SFT, and GRIN Therapeutics to SJM, an Uplifting Athletes Young Investigator grant to REP, and the Fashion Industries Guild Endowed Fellowship for the Undiagnosed Diseases Program, the Cedars-Sinai Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases, and the Cedars-Sinai institutional funding program to TMP. This work was also supported by a grant from SFARI (732132 to SFT, TAB, JL).
Author information
Authors and Affiliations
Contributions
HY, SJM, and SFT: designed the study, analyzed the data. KLP, GC, JG, JS, VS, SP, RTJ, HKE, XB, DRL, IH, TMP, CFB, JRL, and TAB: conducted the neuropsychological assessment, performed the genetic analyses, and/or reviewed the patients’ clinical histories. YX, RS, REP, WC, SK, JPA, KAN, JZ, WX, AT, EDM, RS, RKM, MMK, CH, HK, JP, SJM, and HY: conducted functional studies and analyzed the data. All authors wrote and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
SFT is a member of the SAB for Sage Therapeutics, Eumentis Therapeutics, the GRIN2B Foundation, the CureGRIN Foundation, and CombinedBrain. SFT is consultant for GRIN Therapeutics and Neurocrine, a cofounder of NeurOp, Inc. and Agrithera, and a member of the Board of Directors for NeurOp Inc. HY is the PI on a research grant from Sage Therapeutics to Emory and SJM is PI on a grant from GRIN Therapeutics to Emory. TAB—Consultancy for AveXis, Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda, Taysha, CureGRIN, GRIN Therapeutics, Alcyone, Neurogene, and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department.
Ethical approval
This study was approved by the Medical Ethics Committee and the Institutional Review Boards of University of Colorado School of Medicine and Children's Hospital Colorado (COMIRB 16–1520), Federico II University, Brown University, Duke University, Emory University, University of Melbourne, Seattle Children’s Research Institute, Cedars-Sinai Medical Center, University of Pennsylvania, and University of Leipzig Hospitals and Clinics. All in vitro studies were conducted according to the guidelines of Emory University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, Y., Song, R., Perszyk, R.E. et al. De novo GRIN variants in M3 helix associated with neurological disorders control channel gating of NMDA receptor. Cell. Mol. Life Sci. 81, 153 (2024). https://doi.org/10.1007/s00018-023-05069-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05069-z