
Abstract
Silenced protein tyrosine phosphatase receptor type R (PTPRR) participates in mitogen-activated protein kinase (MAPK) signaling cascades during the genesis and development of tumors. Rat sarcoma virus (Ras) genes are frequently mutated in lung adenocarcinoma, thereby resulting in hyperactivation of downstream MAPK signaling. However, the molecular mechanism manipulating the regulation and function of PTPRR in RAS-mutant lung adenocarcinoma is not known. Patient records collected from the Cancer Genome Atlas and Gene Expression Omnibus showed that silenced PTPRR was positively correlated with the prognosis. Exogenous expression of PTPRR suppressed the proliferation and migration of lung cancer cells. PTPRR expression and Src homology 2 containing protein tyrosine phosphatase 2 (SHP2) inhibition acted synergistically to control ERK1/2 phosphorylation in RAS-driven lung cancer cells. Chromatin immunoprecipitation assay revealed that HDAC inhibition induced enriched histone acetylation in the promoter region of PTPRR and recovered PTPRR transcription. The combination of the HDAC inhibitor SAHA and SHP2 inhibitor SHP099 suppressed the progression of lung cancer markedly in vitro and in vivo. Therefore, we revealed the epigenetic silencing mechanism of PTPRR and demonstrated that combination therapy targeting HDAC and SHP2 might represent a novel strategy to treat RAS-mutant lung cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer carries the highest incidence and mortality of cancer-related diseases, and remains a major public-health issue in China [1]. Rat sarcoma virus (Ras) genes (Kirsten rat sarcoma viral oncogene homolog (KRas), neuroblastoma RAS viral oncogene homolog (NRas), Harvey rat sarcoma viral oncogene homolog (HRas)) are the most common genes driving cancer. KRAS is the most frequently mutated RAS isoform (22% of all tumors), and NRAS and HRAS account for 8% and 3%, respectively [2]. Mutations to 12, 13, and 61 codons convert RAS into an oncoprotein by impairing intrinsic and guanosine triphosphate (GTP)ase-activating protein-mediated GTPase activity [3]. The three isoforms of RAS protein show high similarity, but a distinct bias in mutation-site ‘signatures’ has been discovered. 80% of KRAS mutations occur at codon 12, whereas 60% of NRAS tumors harbor mutations at codon 61 [2]. RAS mutations are present in one-third of patients with lung adenocarcinoma [4]. Mutations of RAS activate the downstream mitogen-activated protein kinase (MAPK) pathway defined by a rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase kinase (MEK)-extracellular regulated protein kinases (ERK) signaling axis, which is a critical mediator of RAS-driven tumorigenesis and cancer development [5, 6].
Tyrosine phosphatases dephosphorylate proteins and participate in regulation of key signaling [7, 8]. Recent studies have shown dysfunction of tyrosine phosphatases to be involved in sustained activation of MAPK signaling in cancer. Loss of protein tyrosine phosphatase receptor type R (PTPRR) has been found in cancer of the colorectum, cervix, prostate gland, and ovary [8,9,10,11]. Silencing of PTPRR expression caused by aberrant methylation of DNA in the cervix has been reported to facilitate ERK1/2 hyperactivation [9]. Re-expression of PTPRR impairs ERK1/2 phosphorylation, restrains downstream signaling, and delays tumor progression [9, 11].
Despite the tumor-suppressor function of tyrosine phosphatases, tyrosine-protein phosphatase non-receptor type 11 (Ptpn11)-encoded Src homology 2 containing protein tyrosine phosphatase 2 (SHP2) dephosphorylates RAS protein, restores the canonical GTPase cycle, augments RAS binding affinity to effectors and, consequently, promotes MAPK signaling [12]. Recently, SHP2 has been postulated to be a potential drug target for RAS-driven cancers. Pharmacologic inhibition of SHP2 expression has been shown to suppress RAS-MAPK signaling and impair the growth of cancer cells in RAS-mutant cell lines, xenografts, and patient-derived organoids [13, 14]. However, 61 codon mutations of both KRAS and NRAS confer resistance to SHP2 inhibitors, which limited possible future applications of SHP2-targeted therapy on RAS-mutant lung cancers [15, 16].
Targeting the RAS-MAPK pathway has been suggested as a treatment for RAS-mutant lung cancer [17]. Given that the phosphatases PTPRR and SHP2 mediate the dephosphorylation and phosphorylation of ERK1/2, respectively, we wondered if interventions targeting phosphatases had therapeutic efficacy by preventing ERK1/2 overactivation in lung cancer. However, the function and regulation of PTPRR in lung adenocarcinoma, and the anti-tumor effect of targeting the phosphatases PTPRR and SHP2 simultaneously, are not known.
Here, we report that ectopic expression of silenced PTPRR suppressed the proliferation and migration of lung cancer cells. ERK1/2 dephosphorylation by PTPRR and inhibition of SHP2 cooperatively restrained MAPK signaling in RAS-mutant lung adenocarcinoma. Notably, the histone deacetylase (HDAC) inhibitor SAHA recovered PTPRR expression by increasing acetylation of histone H3K9 in the promoter region of PTPRR. Dual inhibition of HDAC and SHP2 had a synergistic effect in RAS-mutant lung cancer. These results suggest that targeting dysregulated phosphatases to suppress RAS-MAPK signaling could be an effective strategy to treat RAS-mutant lung adenocarcinoma.
Materials and methods
Patient records
Patient records were collected from the Cancer Genome Atlas (TCGA) to analyze PTPRR expression by TNMplot (www.tnmplot.com). Images of histological sections from normal lung tissue and lung adenocarcinoma were obtained from the Human Protein Atlas [18]. GSE30219 from Gene Expression Omnibus repository (GEO) was used to analyze correlation between PTPRR expression and prognosis of people suffering from lung adenocarcinoma by Kmplot (www.kmplot.com) [19, 20].
Cell culture
Human lung adenocarcinoma cell lines (NCI-H1299, NCI-H2087, A549, and NCI-H358) and immortalized human lung epithelial cell line BEAS-2B were obtained from China Infrastructure of Cell Line Resources (Beijing, China). NCI-H1299, NCI-H2087, A549, and NCI-H358 cells were cultured in RPMI-1640 (Corning Life Sciences, Tewksbury, MA, USA) supplemented with 10% inactivated fetal bovine serum (FBS; Gemini, West Sacramento, CA, USA). BEAS-2B cells were cultured by Bronchial Epithelial Cell Growth Medium BulletKit™ (Lonza, Basel, Switzerland). Mycoplasma testing (InvivoGen, San Diego, CA, USA) was undertaken before experimentation.
Chemicals
The compounds SAHA (Vorinostat™), trichostatin A (TSA), and SHP099 were purchased from TargetMol (Boston, MA, USA). For in vitro assays, compounds were dissolved in dimethyl sulfoxide and added to cell culture media to achieve final concentrations.
PTPRR overexpression in lung cancer cells
PTPRR complementary-DNA (NM_002849, 658aa) was cloned to the pCDH lentiviral expression vector and transfected to 293 T cells with pMD2G and pSPAX2 packaging plasmids. Transfections were undertaken using Lipofectamine™ 3000 following manufacturer instructions (Thermo Fisher Scientific, Waltham, USA). After 24 h, 48 h, and 72 h, the supernatant was harvested, filtered, and concentrated by adding lentivirus solution (Yeason, Shanghai, China). Lung cancer cells were seeded to 12-well plates and infected in the presence of polybrene (8 μg/mL; Cyagen, Suzhou, China). After 24 h, cells were placed under fresh medium with puromycin selection (1.5 μg/mL) to generate cells with PTPRR expression.
Knockdown of gene expression via small interfering (si)RNA or short hairpin (sh)RNA
Silencing of SHP2 expression in A549 cells and NCI-H1299 cells was mediated by siRNA with the indicated sequences. Oligonucleotides targeting human HDAC1, HDAC2, HDAC3, and PTPRR were designed to construct the pKO.1 lentiviral vector. The lentivirus was produced in 293 T cells by co-transfecting pMD2G and psPAX2 plasmids. Filtered supernatant was concentrated and used to infect target cells. The sequences of siRNA and shRNA are listed in Supplementary Table 1.
RNA extraction, cDNA synthesis, and real-time reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from each sample using a RNeasy kit (Qiagen, Hilden, Germany). RNA was reverse-transcribed to cDNA using TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen, Beijing, China) according to manufacturer protocols. Real-time RT-qPCR was undertaken using Hieff® qPCR SYBR Green Master Mix (Yeason Biotechnology, Shanghai, China) in a real-time thermal cycler (qTOWER3; Analytik, Jena, Germany). The primers we employed are listed in Supplementary Table 2.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was carried out with a SimpleChIP® Enzymatic Chromatin IP kit (9003; Cell Signaling Technology) according to manufacturer protocols. The antibody used in the process was anti-acetyl-histone H3 (Lys9) (PTM-156; PTM Bio). Input DNA (2%) and eluted DNA were subjected to real-time RT-qPCR. Three pairs of primers that targeted the promoter region of PTPRR were designed, and are shown in Supplementary Table 2.
Tumor xenograft model
Male Balb/c nude mice (8–10 weeks) were purchased from Charles River Laboratories (Beijing, China). NCI-H1299 pCDH- and PTPRR-overexpressed cells (5 × 107/mL) were prepared in PBS, mixed with Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA) and injected (s.c.) into the left flank and right flank of mice. To study the efficacy of combination therapy using SAHA and SHP099, NCI-H1299 or A549 tumor tissues were cut to pieces (1 mm × 1 mm) and inoculated to the right flank of male Balb/c nude mice. Once the tumor volume reached 100 mm3, mice were grouped randomly and received vehicle, SAHA (75 mg/kg, once-daily), SHP099 (75 mg/kg, every other day), or a combination of SAHA (75 mg/kg, once-daily) plus SHP099 (75 mg/kg, every other day). Tumor volume and bodyweight were monitored twice a week. When the length of tumor surpassed 15 mm, mice were sacrificed and tumor xenografts were harvested. The protocol for animal experiments was approved by the Ethics Committee for Animal Experiments of the Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College.
Statistical analysis
IC50 values were analyzed by nonlinear regression and calculated by Graphpad Prism 8. Student’s unpaired t test or one-way ANOVA were used to evaluate differences between groups. For all experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 were considered statistically significant.
Results
Re-expression of silenced PTPRR suppresses malignant phenotypes of lung cancer
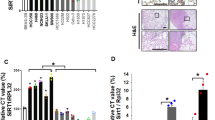
Lower PTPRR expression in lung adenocarcinoma tissues than that in adjacent normal lung tissues were observed in patient samples from the Cancer Genome Atlas and Human Protein Atlas (Fig. 1a). Representative results from immunochemical staining of tissues from the Human Protein Atlas also showed diminished expression of PTPRR in sections of lung tissue with adenocarcinoma (Fig. 1b). Similarly, PTPRR expressions in lung cancer cell line including NCI-H1299, NCI-H2087, and A549 were significantly downregulated relative to the lung normal cell line BEAS-2B (Fig. 1c). Clinical data from GEO dataset was used to investigate the relationship between PTPRR expression and tumor progression. As it shown in Fig. 1d, PTPRR expression was positively co-related with overall survival for patients suffering from lung adenocarcinoma.

PTPRR was underexpressed in lung cancer. a Diminished expression of PTPRR was found in lung adenocarcinoma tissues (n = 57) versus adjacent normal lung tissues (n = 57) by TMNplot. b Tissue sections documented on the Human Protein Atlas indicated low expression of PTPRR in malignant lung carcinoma. Scale bar, 200 μm c PTPRR expression in the immortalized human normal lung cell line BEAS-2B and three lung cancer-derived cell lines were examined by immunoblotting. d Kaplan–Meier survival analysis of GSE30219 data set from GEO showed positive correlation of PTPRR (Affymetrix ID: 206084_at) expression with overall survival. e Growth curve of NCI-H1299 cells and NCI-H2087 cells infected with pCDH or pCDH-PTPRR lentivirus. f and g Motility and migration capabilities of PTPRR-expressing cells were assessed by wound-healing (f) and Transwell™ assays (g). Scale bar, 100 μm. Data are shown as mean ± SD. n = 3, two-tailed unpaired Student’s t-test was used for statistical analysis. *p < 0.05 and ***p < 0.001
Exogenous expression of PTPRR suppressed the growth of human lung cancer (NCI-H1299, NCI-H2087) cells (Fig. 1e). Moreover, PTPRR overexpression inhibited the migration ability of lung cancer cells significantly as determined by the wound-healing assay (Fig. 1f) and Transwell assay (Fig. 1g). These findings suggested that expression of PTPRR phosphatase was silenced in lung adenocarcinoma, and had an important role in the proliferation and migration of cancer cells.
PTPRR suppresses ERK1/2 phosphorylation and sensitizes RAS-mutant lung cancer cells to SHP2 inhibition
It has been reported that PTPRR removes phosphate groups from ERK1/2 and inhibits the growth of cervical cancer cells [9]. Similarly, exogenous expression of PTPRR in HEK293T cells downregulated ERK1/2 phosphorylation significantly (Fig. 2a). Gain-of-function mutation of the RAS proto-oncogene activates RAS-MAPK pathway abnormally, and is involved in the proliferation and signal transduction of lung cancer cells. Following ectopic PTPRR expression, NRAS Q61 mutant NCI-H1299 cells and NCI-H2087 cells showed a marked reduction of phospho-ERK1/2 while negligible changes were found in KRAS G12-mutant A549 cells and NCI-H358 cells (Fig. 2a), The activator protein-1 (AP-1) transcription factors are known to be targets of ERK signaling [21]. As one pivotal component of AP-1 complex, FOS-related antigen 1 (FRA-1) was reported to linearly responded to ERK1/2 activation and was inhibited by PTPRR expression in cervical cancer [9, 22] Likewise, downstream MAPK-dependent FRA-1 transcription showed the same trend with ERK1/2 phosphorylation in PTPRR-overexpressed lung cancer cells (Fig. 2b).

ERK1/2 activation in lung cancer cells was regulated by PTPRR and SHP2. a PTPRR inhibited ERK1/2 phosphorylation in NCI-H1299 cells and NCI-H2087 cells, but not in A549 cells or NCI-H358 cells. b Fra1 (downstream target of the MAPK pathway) expression was reduced by PTPRR expression in NCI-H1299 cells and NCI-H2087 cells. c ERK1/2 phosphorylation in lung cancer cells after SHP099 (50 μM) treatment for 24 h. d PTPRR downregulated ERK1/2 activation in SHP2-deficient cells significantly. e Western immunoblots in PTPRR-expressing NCI-H1299 cells and A549 cells treated with SHP099 (50 μM). f PTPRR-expressing NCI-H1299 cells and A549 cells were subjected to viability assessment with increasing concentrations of SHP099 for 72 h. Data are shown as mean ± SD. n = 3, two-tailed unpaired Student’s t-test was used for statistical analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. n.s., no significance
The Ptpn11-encoded tyrosine phosphatase SHP2 is an activator of MAPK signaling in cancer due to RAS mutations. The allosteric SHP2 inhibitor SHP099 reduced the cycling of RAS GTPase and impaired its binding to effector proteins. In contrast to the effects caused by PTPRR expression, addition of SHP099 suppressed ERK1/2 phosphorylation in KRAS G12-mutant cells but not in NRAS Q61-mutant cells (Fig. 2c). Therefore, we wondered whether lung cancer cells harboring a different RAS status or mutant “hotspot” codons could be treated with PTPRR re-expression in combination with SHP2 inhibition. For NRAS Q61-mutant NCI-H1299 cells, although silencing of SHP2 expression by siRNA transfection did not induce a noticeable change of phospho-ERK1/2, a stronger inactivation of ERK1/2 was detected in cells overexpressing PTPRR (Fig. 2d). In KRAS G12-mutant A549 cells, knockdown of SHP2 expression showed a mild effect, whereas PTPRR expression led to a significant reduction of ERK1/2 phosphorylation (Fig. 2d). Similar effects were observed by treating PTPRR-expressing NCI-H1299 cells and A549 cells with SHP099 for 24 h (Fig. 2e). Then, a wide range of SHP099 concentrations was used to determine the change in the half-maximal inhibitory concentration (IC50) following PTPRR expression. As expected, PTPRR-sensitized NCI-H1299 cells and A549 cells underwent SHP2 inhibition (NCI-H1299 cells: IC50 85.9 μM vs. 33.3 μM; A549 cells: IC50 17.4 μM vs. 3.54 μM) (Fig. 2f). Therefore, PTPRR in combination with a SHP2 inhibitor was an effective strategy to target lung cancer because it cooperatively increased the removal and reduced the addition of phosphate groups to ERK1/2.
Inhibition of HDAC restores PTPRR expression in lung cancer cells
It has been reported that DNA methyltransferase 3 beta (DNMT3B)-mediated high methylation of DNA is responsible for PTPRR silencing in colorectal cancer and cervical cancer [9]. A shRNA targeting DNMT3B was packaged to a lentivector and NCI-H1299 cells were infected. Surprisingly, PTPRR expression was not affected by knockdown of DNMT3B expression (Fig. 3a). Meanwhile, an inhibitor of DNA methyltransferase, decitabine, also did not induce an increase in mRNA expression of PTPRR (Fig. 3b). This result prompted us to speculate that silencing of PTPRR expression in lung cancer might be the result of another pattern of epigenetic modification.

Upregulated H3K9ac level in the promoter region initiates PTPRR transcription. a DNMT3B expression in NCI-H1299 cells was knocked down using the lentiviral vector pKO.1 expressing shDNMT3B. The change in PTPRR mRNA expression was measured by RT-qPCR. b NCI-H1299 cells were treated with decitabine for 24 h and the transcription of PTPRR was measured. c NCI-H1299 cells were infected by lentivirus containing the empty pLKO.1 vector or pLKO.1-shHDAC1, HDAC2, and HDAC3. d Lung cancer cells were treated with SAHA (0, 2.5, 5, 10, or 20 μM) or TSA (2 μM) for 12 h and the acetylation level of histone H3K9 was determined. e After treating NCI-H1299 cells and NCI-H2087 cells with TSA (2 μM) for 12 h, PTPRR expression was detected by real-time RT-qPCR. f Expression of PTPRR mRNA was detected in SAHA (0, 10, or 20 μM)-treated lung cancer cells. g ChIP-qPCR of H3K9ac enrichment around the promoter region of PTPRR in SAHA-treated NCI-H1299 cells. h Higher acetylation level of histone H3K9 was found in BEAS-2B cells than NCI-H1299, NCI-H2087, and A549 cells. Data are shown as mean ± SD. n = 3, two-tailed unpaired Student’s t-test was used for statistical analysis. *p < 0.05, **p < 0.01, and ***p < 0.001
Histone acetylation has a pivotal role in chromatin remodeling and regulation of gene expression. HDAC1, HDAC2, and HDAC3 are class-I HDACs that regulated cancer development by deacetylating histones and regulating cellular homeostasis [23]. Higher level of HDAC1, HDAC2, and HDAC3 were found in lung adenocarcinoma than adjacent normal lung tissues (Fig. S1). Suppression of HDAC1, HDAC2, and HDAC3 via shRNA released PTPRR expression in NCI-H1299 lung cancer cells, (Fig. 3c), suggesting that PTPRR expression was silenced in lung adenocarcinoma due to changes in histone coding. Inhibition of HDAC activity by the clinically available drug Vorinostat (SAHA) and a robust anti-HDAC agent (TSA) increased acetylation of histone H3 at Lys 9 residues significantly (Fig. 3d). Consequently, treatment with TSA and SAHA upregulated PTPRR expression markedly (Fig. 3e, f). Accordingly, chromatin immunoprecipitation by antibody against H3K9 acetylation and quantitative real-time PCR using primers targeting PTPRR promoter was performed. The results showed that H3K9 acetylation at the promoter region of PTPRR were dramatically elevated in the presence of SAHA compared to DMSO control (Fig. 3g). Moreover, histone H3K9 acetylation level of PTPRR promoter region in human lung BEAS-2B cells were significantly higher than that in lung cancer cells, which also explains the diminished PTPRR expression in lung adenocarcinoma (Fig. 3h). Taken together, these data suggested the role of histone acetylation in maintaining PTPRR gene expression by transcriptional regulation.
HDACi treatment restrains ERK1/2 signaling and sensitizes RAS-mutant lung cancer cells to SHP2 inhibition
As PTPRR dephosphorylates ERK1/2, we next studied the effect of HDAC inhibition on ERK1/2 signaling. Knockdown of HDAC1, HDAC2, and HDAC3 expression suppressed ERK1/2 activation in NCI-H1299 cells significantly (Fig. 4a). In accordance with the effect of PTPRR overexpression on RAS-mutant lung cancer cells, the HDAC inhibitor SAHA reduced ERK1/2 signaling in NRAS Q61-mutant cells dose-dependently, but showed little effect on KRAS G12-mutant cancer cells (Fig. 4b). In NCI-H1299 cells, inhibition of ERK1/2 signaling accompanied with PTPRR expression were induced by SAHA treatment and lasted from 6 to 36 h (Fig. 4c). The transcription of Fra-1, which is downstream of MAPK signaling, was impaired severely by SAHA treatment in NRAS-mutant lung cancer cells (Fig. 4d).

HDAC inhibition restrains ERK1/2 signaling. a Lentivirus packaging empty pLKO.1 vector or shRNAs targeting HDAC1, HDAC2, and HDAC3 were used to infect NCI-H1299 cells. b Lung cancer cells were treated with SAHA (0, 5, 10, or 20 μM) for 24 h. c NCI-1299 cells were treated with SAHA (10 μM) for different times to assess expression of PTPRR and ERK1/2 phosphorylation. d NCI-H1299 and NCI-H2087 cells were treated with SAHA for 12 h and subjected to real-time RT-qPCR to determine fra-1 mRNA expression. ***p < 0.001. n.s., no significance
To demonstrate the central role of PTPRR in SAHA-induced inhibition of MAPK signaling, we interfered with expression of PTPRR by shRNA in NCI-H1299 cells with or without SAHA treatment. Silencing of PTPRR expression rescued SAHA-induced ERK1/2 inhibition, thereby indicating that PTPRR was the main regulator of MAPK signaling under HDAC inhibition (Fig. 5a). To ascertain if the HDAC inhibitor SAHA could sensitize RAS-mutant lung cancer cells to SHP2 inhibition, lung cancer cell lines (NRAS Q61-mutant NCI-H1299, KRAS G12-mutant A549) were selected to determine the effect of SHP2-targeting siRNAs in combination with SAHA. As expected, silencing of SHP2 expression by siRNAs had a negligible effect on ERK1/2 phosphorylation in NCI-H1299 cells (Fig. 5b). However, in combination with SAHA, ERK1/2 activation in NCI-H1299 cells was impaired severely. Similarly, in A549 cells, although SAHA did not alter ERK1/2 signaling, addition of siRNAs targeting SHP2 showed a significant synergistic effect (Fig. 5b). Meanwhile, knockdown of SHP2 expression plus SAHA represented reduced proliferation of cancer cells (Fig. 5c).

SAHA treatment sensitizes RAS-mutant lung cancer cells to SHP2 inhibition. a Lentivirus containing shRNAs targeting PTPRR were used to infect NCI-H1299 cells with or without SAHA treatment (10 μM). b siRNAs were transfected to NCI-H1299 cells and A549 cells for 48 h, SAHA (10 μM) was added and incubated for 12 h. c Silencing of SHP2 expression in NCI-H1299 and A549 cells in combination with SAHA treatment (10 μM) restrain cell proliferation. Relative cell growth was determined by crystal violet staining and solubilized in 10% acetic acid. Data are shown as mean ± SD. n = 3, two-tailed unpaired Student’s t-test was used for statistical analysis. **p < 0.01 and ***p < 0.001
SAHA synergizes with SHP099 in RAS-mutant lung cancer cells
It has been found that SAHA treatment and SHP2 inhibition were complementary in regulating MAPK signaling. Hence, we assessed the sensitivity of RAS-mutant lung cancer cell lines to the SHP2 inhibitor SHP099 upon SAHA treatment. Interestingly, addition of SAHA at low doses sensitized lung cancer cells to SHP099 significantly, with IC50 values lower in combinations compared with those using SHP099 alone (Fig. 6a). To determine the synergistic effect of SAHA and SHP099, we utilized the Chou–Talalay method to provide the combination index (CI) equation that allows quantitative evaluations of drug combinations, where CI < 1 indicates a synergistic inhibition [24]. As the fraction affected-CI plot shown in Fig. 6b, SAHA and SHP099 exerted synergistic effects when fraction of cells affected by treatment was 0.5. Mechanistically, treatment of SAHA plus SHP099 led to markedly reduced ERK1/2 phosphorylation in lung cancer cells (NRAS Q61-mutant NCI-H1299, KRAS-G12 mutant A549) (Fig. 6c). Incorporation of 5-Ethynyl-2’-deoxyuridine (EdU) and staining enabled accurate and image-based analyses of cell proliferation. The number of cells labeled with EdU was reduced dramatically upon treatment with SAHA plus SHP099 (Fig. 6d). Similarly, although a reduction in clonogenic ability by SAHA was not consistent in NCI-H1299 cells harboring a NRAS mutation and A549 cells with a KRAS mutation, combinations with SHP099 resulted in elimination of cell-colony formation (Fig. 6e). These results provided evidence that combining SAHA with SHP099 had a synergistic effect, and led to stronger inactivation of MAPK signaling in RAS-mutant lung cancer cells.

Combination therapy of SAHA and SHP099 strongly impairs the proliferation of lung cancer cells. a Increasing concentrations of SHP099 plus SAHA (1.25 μM) were added to a panel of RAS-mutant lung cancer cells. Cell viabilities were assessed by Cell-titer Glo at 72 h post-treatment. b Fraction affected (Fa)-combination index (CI) plots were obtained by determining the combination of a series concentrations of SAHA and SHP099. c Single and combination treatment of SAHA (10 μM) and SHP099 (25 μM) were added to four RAS-mutant lung cancer cell lines and western immunoblots obtained. d EdU staining of NCI-H1299 cells and A549 cells treated with SAHA (10 μM), SHP099 (50 μM), or their combination for 24 h. Scale bar, 100 μm. e NCI-H1299 cells and A549 cells were treated with SAHA (1.25 μM), SHP099 (50 μM for NCI-H1299, 10 μM for A549) or their combinations for 7 days and subjected to crystal-violet staining. Data are the mean ± SD. n = 3, two-tailed unpaired Student’s t-test was used for statistical analysis. *p < 0.05, **p < 0.01, and ***p < 0.001
Combination of SAHA and SHP099 reduces the tumor burden in vivo
NCI-H1299 cells stably expressing the pCDH empty vector or ectopic PTPRR were injected into the left flank and right flank of male Balb/c nude mice to study the effect of PTPRR expression on tumor progression. Similar to the results of the in vitro study, PTPRR expression in NCI-H1299 cells led to markedly reduced tumor volume and tumor weight, thereby indicating that re-expression of PTPRR was sufficient to restrain the growth of tumor xenografts (Fig. 7a–d). To determine the efficacy of SAHA and SHP099, male Balb/c nude mice bearing NCI-H1299 xenografts were administered (p.o.) the vehicle control, SAHA, SHP099, or SAHA plus SHP099. Treatment with SAHA or SHP099 showed modest efficacy upon NCI-H1299 xenografts, whereas combination treatment resulted in an improved response and significantly delayed tumor growth without causing severe body weight loss (Fig. 7e-g). In the A549 xenograft model, combination treatment of SAHA and SHP099 also significantly suppressed tumor progression (Fig. 7i-l). Consistent with those detected in vitro, immunoblots of representative tumor lysates indicated significant suppression of ERK1/2 phosphorylation upon combination treatment, which was reduced dramatically compared with that in the vehicle control or if a single drug was used (Fig. 7m, n). These results supported the notion of clinical application of a HDAC inhibitor in combination with SHP2 inhibition.

Dual inhibition of HDAC and SHP2 reduced the tumor burden in RAS-mutant NCI-H1299 and A549 lung cancer xenografts. a NCI-H1299 cells expressing empty pCDH or ectopic PTPRR were mixed with Matrigel™ and transplanted to the left flank and right flank of male Balb/c nude mice, respectively. b Tumor volumes of NCI-H1299 pCDH and PTPRR were tracked for 30 days (n = 4). c Tumor weight of xenografts were obtained at the endpoint (n = 4). d Representative image of NCI-H1299 pCDH and PTPRR tumor xenografts (n = 4). e Tumor volume of NCI-H1299 xenografts treated with SAHA (75 mg/kg, p.o.), SHP099 (75 mg/kg, p.o.), or their combination (n = 5). f Tumor weight of NCI-H1299 xenografts (n = 5). g Representative image of NCI-H1299 tumor xenografts (n = 5). h Body weight change of mice bearing NCI-H1299 xenografts (n = 5). i Tumor volume of A549 xenografts treated with SAHA (75 mg/kg, p.o.), SHP099 (75 mg/kg, p.o.), or their combination (n = 5). j Tumor weight of A549 xenografts (n = 5). k Representative image of A549 tumor xenografts (n = 5). l Body weight change of mice bearing A549 xenografts (n = 5). m and n Expression of phosphorylated ERK1/2 and total ERK1/2 in NCI-H1299 (m) and A549 (n) tumor lysates was measured by western blotting. Data are presented as the mean ± SEM. One-way ANOVA was used for statistical analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. n.s., no significance
Discussion
By analyzing datasets from public database, lower PTPRR expression was observed in patient samples with lung adenocarcinoma compared with normal adjacent lung tissues, and positively correlated with the prognosis. Together with studies reporting PTPRR expression to be silenced and identified as a prognostic marker in cancer of the cervix, ovary, and oral squamous cell carcinoma [9, 10, 25], our research reinforced the tumor-suppressor role of PTPRR in cancer development.
DNA methylation can silence PTPRR expression in cervical cancer [9]. However, knockdown of DNMT3B expression or treatment with an inhibitor of DNA methyltransferase, decitabine, did not alter PTPRR expression in lung adenocarcinoma. Interference of HDACs by treatment with shRNA and a HDAC inhibitor enhanced acetylation of histone H3 Lys 9, which were sufficient to boost PTPRR expression epigenetically. Furthermore, the ChIP assay validated that acetylation of histone H3K9 was enriched in the promoter region of PTPRR gene, which suggested a different regulatory mechanism of silencing of PTPRR expression in lung cancer.
Several studies have mentioned that HDAC inhibitors suppressed ERK phosphorylation, but the underlying molecular mechanism is not known [26, 27]. In our study, silenced expression of PTPRR was restored by histone acetylation in lung cancer, and interference with PTPRR expression reversed (at least in part) SAHA-induced ERK1/2 inhibition. Hence, our investigations suggested a key role of PTPRR in suppression of MAPK signaling by HDAC inhibition.
We speculated that MAPK signaling inhibition is only one of the mechanisms of HDAC inhibition, as SAHA treatment showed little effect on ERK1/2 phosphorylation while severely inhibited the proliferation of KRAS-mutant cells. It is worth noticing that SAHA was identified as a pan HDAC inhibitor and suppressed both class I and class II HDAC [28]. Aside from histone acetylation, non-histone acetylation and protein non-acetyl acylations also have been found regulated by SAHA or pan HDAC inhibition, which facilitated tumor suppression though diverse cellular processes including gene transcription, cell cycle, DNA damage repair, signaling pathways, and cytoskeleton organization [29, 30].
Allosteric inhibitors targeting KRAS G12C directly have been applied to clinical therapeutics [31], but treatment of patients with lung adenocarcinoma harboring different RAS mutations is a challenge. Studies have shown that SHP2 is required for RAS-driven cancer progression, and that targeting SHP2 could be therapy for RAS-mutant cancer [13, 32]. It has been reported that, compared with the G12 mutation, the Q61 mutation of RAS shows impaired GTP hydrolysis and a minimal impact by phosphorylation [15]. Meanwhile, the RAS Q61 mutation binds RAF preferentially and activates MAPK signaling rather than other downstream cascades [33]. Therefore, the Q61 mutation not only protects RAS from SHP2 regulation, it also drives persistent MAPK hyperactivation. In line with recent reports, our results supported the notion that the Q61 mutation of NRAS in lung adenocarcinoma was a predictor of resistance to SHP2 inhibitors. Interestingly, we revealed that PTPRR-mediated dephosphorylation of ERK1/2 could aid suppression of Q61 mutant RAS-driven persistent MAPK activation, highlighting a novel strategy to treat RAS-mutant lung cancers.
As one of the main regulators during tumor progression, suppression of the MAPK pathway by dual inhibition of SHP2 and MEK/ERK has been demonstrated to be effective in multiple types of cancer cell [16, 34,35,36]. Our findings revealed that dysregulated phosphatases PTPRR and SHP2 maintained aberrant activation of ERK1/2 cooperatively and investigated the synergistic efficacy of HDAC and SHP2 co-inhibition. Hence, targeting of disordered phosphatases to suppress RAS-MAPK signaling may be a promising strategy against RAS-mutant lung cancer (Fig. 8).

Graphical summary of suppressing RAS-MAPK pathway by regulating dysfunctional phosphatase PTPRR and SHP2. Histone acetylation facilitates PTPRR expression in lung adenocarcinoma. A combination of the HDAC inhibitor (SAHA) and SHP2 inhibitor (SHP099) suppressed RAS-mutant lung cancer progression markedly through regulation of ERK1/2 signaling. The figure was created with BioRender.com
Availability of data and materials
All data generated or analyzed during this study are included in this published article and supplementary information file. All additional information will be made available upon reasonable request from the authors.
Abbreviations
- PTPRR:
-
Protein tyrosine phosphatase receptor type R
- RAS:
-
Rat sarcoma viral oncogene homolog
- MAPK:
-
Mitogen-activated protein kinase
- HDAC:
-
Histone deacetylase
- SHP2:
-
Src homology 2 domain-containing protein tyrosine phosphatase
- ERK1/2:
-
Extracellular signal-regulated kinase ½
- MEK:
-
Mitogen-activated protein kinase kinase
- DNMT3B:
-
DNA methyltransferase 3 beta
References
Zheng R, Zhang S, Wang S et al (2022) Lung cancer incidence and mortality in China: updated statistics and an overview of temporal trends from 2000 to 2016. J Nat Cancer Cent 2:139–147
Prior IA, Lewis PD, Mattos C (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res 72:2457–2467
Simanshu DK, Nissley DV, McCormick F (2017) RAS proteins and their regulators in human disease. Cell 170:17–33
Network CGAR (2018) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543
Huang L, Guo Z, Wang F et al (2021) KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther 6:386
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310
Ostman A, Hellberg C, Bohmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6:307–320
Bollu LR, Mazumdar A, Savage MI et al (2017) Molecular pathways: targeting protein tyrosine phosphatases in cancer. Clin Cancer Res 23:2136–2142
Su PH, Lin YW, Huang RL et al (2013) Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene 32:15–26
Wang Y, Cao J, Liu W et al (2019) Protein tyrosine phosphatase receptor type R (PTPRR) antagonizes the Wnt signaling pathway in ovarian cancer by dephosphorylating and inactivating beta-catenin. J Biol Chem 294:18306–18323
Munkley J, Lafferty NP, Kalna G et al (2015) Androgen-regulation of the protein tyrosine phosphatase PTPRR activates ERK1/2 signalling in prostate cancer cells. BMC Cancer 15:9
Bunda S, Burrell K, Heir P et al (2015) Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 6:8859
Mainardi S, Mulero-Sánchez A, Prahallad A et al (2018) SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 24:961–967
Nichols RJ, Haderk F, Stahlhut C et al (2018) RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20:1064–1073
Gebregiworgis T, Kano Y, St-Germain J et al (2021) The Q61H mutation decouples KRAS from upstream regulation and renders cancer cells resistant to SHP2 inhibitors. Nat Commun 12:6274
Valencia-Sama I, Ladumor Y, Kee L et al (2020) NRAS status determines sensitivity to SHP2 inhibitor combination therapies targeting the RAS–MAPK pathway in neuroblastoma. Cancer Res 80:3413–3423
Chen K, Zhang YL, Qian L et al (2021) Emerging strategies to target RAS signaling in human cancer therapy. J Hematol Oncol 14:116
Bartha A, Gyorffy B (2021) TNMplot.com: a web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int J Mol Sci 22:2622
Lanczky A, Gyorffy B (2021) Web-based survival analysis tool tailored for medical research (KMplot): development and implementation. J Med Internet Res 23:27633
Rousseaux S, Debernardi A, Jacquiau B et al (2013) Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Trans Med 5:186166
Lavoie H, Gagnon J, Therrien M (2020) ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol 21:607–632
Gillies TE, Pargett M, Minguet M et al (2017) Linear integration of ERK activity predominates over persistence detection in Fra-1 regulation. Cell Syst 5:549-563.e545
Ropero S, Esteller M (2007) The role of histone deacetylases (HDACs) in human cancer. molecular oncology 1:19–25.24. Chou T-C (2010) drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446
Chou T-C (2010) Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res 70:440–446
Dus-Szachniewicz K, Wozniak M, Nelke K et al (2015) Protein tyrosine phosphatase receptor R and Z1 expression as independent prognostic indicators in oral squamous cell carcinoma. Head Neck 37:1816–1822
Majumdar G, Adris P, Bhargava N et al (2012) Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells. BMC Genomics 13:709
da Cunha JM, Ghisleni EC, Cardoso PS et al (2020) HDAC and MAPK/ERK inhibitors cooperate to reduce viability and stemness in medulloblastoma. J Mol Neurosci 70:981–992
Marks PA (2007) Discovery and development of SAHA as an anticancer agent. Oncogene 26:1351–1356
Narita T, Weinert BT, Choudhary C (2019) Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 20:156–174
Xu G, Wang J, Wu Z et al (2014) SAHA regulates histone acetylation, butyrylation, and protein expression in neuroblastoma. J Proteome Res 13:4211–4219
Liu J, Kang R, Tang D (2022) The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther 29:875–878
Ruess DA, Heynen GJ, Ciecielski KJ et al (2018) Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med 24:954–960
Zhou ZW, Ambrogio C, Bera AK et al (2020) KRAS(Q61H) preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer. Cancer Res 80:3719–3731
Wong GS, Zhou J, Liu JB et al (2018) Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med 24:1627–1627
Ahmed TA, Adamopoulos C, Karoulia Z et al (2019) Shp2 drives adaptive resistance to erk signaling inhibition in molecularly defined subsets of erk-dependent tumors. Cell Rep 26:65–78
Fedele C, Ran H, Diskin B et al (2018) SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov 8:1237–1249
Acknowledgements
We thank Ms. Yijing Wang for her great help in handing animals. We acknowledge TCGA and GEO databases for providing platforms and contributors for uploading datasets.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82104202) and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-028).
Author information
Authors and Affiliations
Contributions
TD: Conceptualization, Methodology, Investigation, Writing- Original draft preparation, Funding acquisition. XH: Investigation, Data curation, Writing-Original draft preparation. ZH: Investigation, Validation. WW: Software, Resources, data curation. SY: Investigation, visualization. MW: Methodology, Investigation. MJ: Methodology, Writing—Review & Editing, Supervision. NX: Supervision, Project administration. XC: Conceptualization, Supervision, Project administration. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared that no competing interest exists.
Ethical approval and consent to participate
The protocol for animal experiments was approved by the Ethics Committee for Animal Experiments of the Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College.
Consent to patient
All patient data were obtained from TCGA and GEO public databases and have been approved by the Ethical Committee of Chinese Academy of Medical Sciences & Peking Union Medical College.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Du, T., Hu, X., Hou, Z. et al. Re-expression of epigenetically silenced PTPRR by histone acetylation sensitizes RAS-mutant lung adenocarcinoma to SHP2 inhibition. Cell. Mol. Life Sci. 81, 64 (2024). https://doi.org/10.1007/s00018-023-05034-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05034-w