
Abstract
Mutations of large conductance Ca2+- and voltage-activated K+ channels (BK) are associated with cognitive impairment. Here we report that CA1 pyramidal neuron-specific conditional BK knock-out (cKO) mice display normal locomotor and anxiety behavior. They do, however, exhibit impaired memory acquisition and retrieval in the Morris Water Maze (MWM) when compared to littermate controls (CTRL). In line with cognitive impairment in vivo, electrical and chemical long-term potentiation (LTP) in cKO brain slices were impaired in vitro. We further used a genetically encoded fluorescent K+ biosensor and a Ca2+-sensitive probe to observe cultured hippocampal neurons during chemical LTP (cLTP) induction. cLTP massively reduced intracellular K+ concentration ([K+]i) while elevating L-Type Ca2+ channel- and NMDA receptor-dependent Ca2+ oscillation frequencies. Both, [K+]i decrease and Ca2+ oscillation frequency increase were absent after pharmacological BK inhibition or in cells lacking BK. Our data suggest that L-Type- and NMDAR-dependent BK-mediated K+ outflow significantly contributes to hippocampal LTP, as well as learning and memory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mutations in KCNMA1, the human gene encoding the pore forming α subunit of the large-conductance calcium ion (Ca2+)- and voltage-activated potassium ion (K+) channel BK (maxi K+, KCa1.1, Slo1) are linked to a growing number of clinically relevant phenotypes. Besides movement disorders and epileptic seizures, these mutations are accompanied by cognitive impairments like developmental delay and intellectual disability [1], which are also linked to autism spectrum disorders and schizophrenia [2, 3].
Throughout the mammalian central nervous system [4], the ubiquitously expressed BK was found in pre- as well as postsynaptic locations [5, 6], where it associates closely with voltage-gated Ca2+ channels [7] and N-methyl-d-aspartate (NMDA)-type glutamate receptors (NMDAR; [8]). These act as Ca2+ sources to provide high local [Ca2+]i of more than 10 µM necessary for BK activation in the physiologically relevant voltage range of − 50 to 0 mV [7, 9, 10], reviewed in [11]. In presynaptic terminals, massive K+ outflow in response to BK activation and the ensuing membrane hyperpolarization limits synaptic transmission by reducing Ca2+ influx through voltage-gated Ca2+ channels [12]. In some postsynaptic cells, BK was described to decrease activity levels through hyperpolarization-mediated inhibition of voltage-gated cation channels [13]. In other cell populations like hippocampal CA1 pyramidal cells, however, BK increases firing rate by contributing to the fast repolarization after individual action potentials (AP) [14,15,16].
Brain development [17] as well as learning and memory aspects of cognition essentially depend on NMDAR-dependent plasticity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptor (AMPAR)-dependent glutamatergic transmission [18]. High frequency stimulation (HFS) of AMPAR provokes postsynaptic Ca2+ influx through NMDAR and voltage-activated L-Type Ca2+ channels (LTCC; [19, 20]). The resulting increase in [Ca2+]i strengthens synapses by increasing AMPAR conductance and exocytosis-mediated integration of additional postsynaptic AMPAR. This process of synaptic strengthening is called long-term potentiation (LTP). The converse mechanism termed long-term depression (LTD) reduces synaptic strength by AMPAR endocytosis [21].
Synaptic plasticity in the form of LTP and LTD has been most intensively studied in the hippocampus, a brain region involved in spatial and declarative learning and memory [22], where it is compromised in different models of cognitive impairment [23,24,25,26]. So far, however, little is known about the role of postsynaptic BK in hippocampal synaptic plasticity. Reportedly, BK-mediated afterhyperpolarization (fAHP) in hippocampal CA1 pyramidal cells is modulated in response to learning [27]. Additionally, global BK knock-out mice (BK KO or BK−/−) suffer from delayed acquisition of hippocampus-dependent spatial memory in the Morris Water Maze (MWM) [28].
Combining behavioral, electrophysiological and biochemical approaches, we found impaired hippocampal learning and LTP in hippocampus-specific conditional BK KO (cKO) mice as well as reduced Ca2+ oscillations and K+ loss in dissociated neurons lacking BK function during chemically induced LTP (cLTP) in vitro. Hence, we propose that BK-mediated hyperpolarizing K+-currents support postsynaptic Ca2+ activity necessary for hippocampal LTP expression.
Materials and methods
Animals
All experimental studies were approved by the local ethics Committee for Animal Research (Regierungspräsidium Tübingen, Germany) and performed according to the guidelines of the German Animal Welfare Act. Animals were kept in standardized cages on a 12/12 h light/dark cycle (lights on 6 a.m.–6 p.m.) under controlled temperature and humidity with access to food and water ad libitum.
8–12 weeks-old animals of both sexes used for characterization by PCR, Western blot and immunofluorescence as well as behavioral, electrophysiological and cLTP experiments were generated by mating the T29-1 transgenic subline expressing Cre recombinase under control of the CaMKII promoter in CA1 pyramidal neurons (B6.Cg-Tg(Camk2a-cre)T29-1Stl/J, JAX #005359) [29] with mice heterozygous for the floxed (BKfl/+; B6-Kcnma1tm2.1Ruth) or the BK KO allele (BK−/+; (B6-Kcnma1tm1Ruth) [30]. Subsequent intercrossing to floxed BK (BKfl/fl) mice produced age- and litter-matched CA1 pyramidal neuron-specific conditional BK knockouts (cKO, genotype: Cretg/+; BKfl/−) and respective controls (CTRL, genotype: Cretg/+; BKfl/+). Primary hippocampal neurons for cultures were obtained from homozygous P0–P1 wildtype (BK+/+) or KO (BK−/−) animals bred from either homozygous BK+/+ or heterozygous (BK−/+) breeders, respectively. Genetic status of neuronal cultures prepared from individual pups was determined by genotyping PCR of biopsies obtained from originating animals. Primer sequences are listed in Table S6.
Antibodies and reagents
All primary antibodies used in the present study are listed in Table S7. All secondary antibodies were isotype-specific and conjugated to Alexa Fluor 488- or Alexa Fluor 555 (Thermo Fisher Scientific). dl-2-Amino-5-phosphonopentanoic acid sodium salt (AP-5, #105), Forskolin (#1099), and Paxilline (#2006) were from Tocris. Nifedipine (#N7634), Picrotoxin (#P1675) and Rolipram (#R6520) were from Sigma-Aldrich. DNA oligonucleotide primers used in this study are listed in Table S6. If not specifically stated, all other reagents were of standard quality and from the usual vendors.
DNA Extraction and PCR
DNA was isolated from freshly dissected hippocampi of T29.1-Cretg; BKfl/+ using High Pure PCR Template Preparation Kit (Roche #11796828,001) according to manufacturer specifications. BKα-null allele (132 bp) was amplified using KAPA HotStart Mouse-Genotyping Kit (Roche #KK7352) and BK forward-1 5′-TGG TCT TCT TCA TCC TCG GG-3′; BK forward-2 5′-AAG GGC CAT TTT GAA GAC GTC-3′ and BK reverse 5′-CCA GCC ACG TGT TTG TTG G-3′ primers to confirm hippocampus specific recombination of BK.
Immunofluorescence
Mice were euthanized with CO2 and transcardially perfused with 20 mL Dulbecco's Balanced Salt Solution (DPBS, Thermo Scientific #14190144) followed by 20 mL 4% (wt/vol) paraformaldehyde in DPBS to fix the tissue. Brains were isolated, snap frozen in -40 to -60 °C cold isopentane and stored at − 80 °C. Brains were transferred to − 20 °C 2 h before 8 µm thick coronal sections were obtained. Slices from CTRL, cKO and BK−/− were collected on the same glass slides (epredia Superfrost™ Plus Adhesion Microscope Slides #J1800AMNZ) together with additional sections from BK+/+ that served as positive controls. Blocking buffer (BB) consisting of DPBS supplemented with 2% Glycerol, 5% NGS, 0.3% Triton-X-100, 2% (wt/vol) BSA and 50 mM NH4Cl was used for permeabilization and blocking of unspecific antibody binding. Primary antibodies against BK were diluted 1:1000 in BB and incubated overnight at 4 °C. Sections were then washed thrice with 0.01% Triton-X-100 in DPBS and blocked again with BB for 1 h before incubation with secondary antibody (1:2500) and Hoechst 33342 (1:1000) for 2 h. After three washes with 0.01% Triton-X-100 in DPBS, DPBS and water, sections were mounted with Permafluor (Fisher Scientific, #TA-030-FM). Primary antibodies are listed in Table S7.
Tissue lysis and western blot
Upon dissection, tissues were suspended in RIPA buffer (in mM: 50 Tris, 150 NaCl, 5 EGTA, 10 EDTA, final pH 7.4) containing protease inhibitors (in μg/ml; 1 phenylmethanesulfonyl fluoride, 1 pepstatin A, 10 leupeptin, 20 aprotinin), 1% NP-40, 10% glycerol, 0.05% sodium dodecyl sulfate (SDS), 0.4% deoxycholate and phosphatase inhibitors (Sigma Phosphatase Inhibitor Cocktail 2 and 3) and homogenized with a hand disperser (Polytron).
Protein concentration was measured using BCA assay (Thermo Scientific #23227). 80 µg total protein were incubated with SDS sample buffer at 95 °C for 5 min, separated by electrophoresis in 10% polyacrylamide gels, transferred to polyvinylidene fluoride (PVDF) membranes (Merck #IPFL00010) using a semi-dry blotting system (Carl Roth, 110 mA per membrane for 90 min), probed with the indicated primary antibodies (Table S7) and detected with fluorescently labelled secondary antibodies. Immunosignals were visualized using an Amersham Imager 600 (General Electrics). To simultaneously label multiple proteins of different molecular weight on the membrane using antibodies, the membranes were cut with a little safety margin, according to the size of the proteins of interest. The contrast of the displayed blots was increased to improve visibility of the bands. Importantly, however, densiometric quantification using ImageJ was performed with unprocessed raw images.
Blinding procedures
During the in vivo experiments (beam-walk, open-field and water-maze) as well as electrophysiology, the experimenter was unaware of the animals’ genotype. To mask the genotypes, a person other than the experimenter blinded genotypes prior to each experiment by assigning a randomized number to each mouse that did not provide the experimenter with any information about the genotype. Only after data analysis, the experimenter was informed about genotypes.
Beam-Walk
As previously described [31], mice were trained for 3 consecutive days with four trials per day on a rectangular beam of 1 m length with 12 mm edge length suspended in 0.5 m height. The area below the beam was cushioned to prevent fall injuries. On test day, latency to traverse rectangular (28, 12 and 5 mm edge length) and circular beams (28, 17 and 11 mm diameter) was recorded and analyzed offline by an observer unaware of the genotype for latency to cross beam, hind paw foot slips and number of falls.
Open-Field
Open-Field was performed as previously described [31]. Mice were recorded while freely exploring a circular arena with a diameter of 112 cm for 30 min using a suspended camera. Latency to enter border zone (15 cm from the edge), relative time spent in the border zone, total distance travelled, resting time and mean speed were quantified and analyzed using Smart 3.0 tracking software (Panlab). Additionally, the number of rearings during the first 5 min was quantified.
Spatial acquisition and reversal (Water-Maze)
As previously described [31], a 112 cm diameter pool was filled with 30 cm of water (25 °C) made opaque by milk powder. A transparent cylindrical escape platform of 12 cm diameter was submerged 0.5 cm beneath the water surface. The maze was virtually divided into 4 quadrants NE, SE, SW and NW. Swimming trials were recorded with a camera suspended above center of the pool and processed via Smart 3.0 tracking software (Panlab). Mice were trained for 5 consecutive days with four trials per day to find the platform submerged in the NW quadrant using visual cues. For every trial, mice were released into the pool from a different starting location. On day 6, the platform was removed for probe trial. Mice were released into SE quadrant opposite of the target and their movement was tracked for 60 s. After acquisition probe trial, a reversal phase was performed in which the cognitive flexibility was queried. For this purpose, the platform was positioned in the opposite SE quadrant and insertion locations were mirrored. Training days and number of trials as well as probe trial were otherwise performed identically to the initial acquisition phase.
Electrophysiology
Extracellular fEPSP were recorded as previously described [38]. In brief, brains were quickly isolated and immersed in ice-cold slicing buffer (in mM: 127 NaCl, 1.9 KCl, 26 NaHCO3, 1.2 KH2PO4, 10 D-glucose, 2 MgSO4, and 1.1 CaCl2, saturated with 5% CO2 and 95% O2, final pH 7.4). 400 µM thick forebrain sections were coronally cut on a vibrating microtome (Leica VT 1000S) and stored in artificial cerebrospinal fluid (ACSF, in mM: 127 NaCl, 1.9 KCl, 26 NaHCO3, 1.2 KH2PO4, 2.2 CaCl2, 1 MgSO4 and 10 D-glucose, oxygenated with 95% O2 plus 5% CO2, final pH 7.4) at 30 °C for at least 1 h before remaining at room temperature until measurement. Sections were transferred to a measurement chamber constantly perfused with oxygenated warm ACSF at 30 °C. Stimulation (bipolar concentric, TM53CCINS, WPI) and recording (ACSF-filled glass pipettes, 2–3 MΩ) electrodes were positioned in the stratum radiatum to measure Schaffer collateral fEPSPs. Signals were amplified using an Axopatch 200B amplifier (Molecular Devices), digitized by a LIH 8 + 8 (HEKA) at 5 kHz and recorded using WinWCP from the Strathclyde Electrophysiology Suite. Stimuli (100 µs) were applied through a stimulus isolator (WPI). For each individual slice the strength of the stimulation (typically between 50 and 100 μA) was chosen to evoke approximately 50% of the maximal response, defined by initial fEPSP slope. LTP was induced by high-frequency stimulation (HFS; 100 Hz, 1 s) at the same intensity as baseline. Baseline and LTP levels were determined by average fEPSP initial slopes from the period between −10 to 1 min before and 45 and 60 min after HFS, respectively. Paired Student's T-test was used to determine whether LTP was induced within a genotype (mean baseline vs. mean LTP levels). Two-way ANOVA was used to compare LTP levels (45 to 60 min after HFS) between CTRL and cKO. Before each LTP measurement, input–output ratio (IOR) was determined for stimulus intensities between 25–150 µA and paired-pulse facilitation (PPF) for inter-stimulus intervals of 10, 20, 50, 100, 200, and 500 ms (with the same intensity as for the LTP measurement) for each slice. The stimulation interval was 1/15 s, with 4 traces being averaged to one data point. Data were analyzed and processed using Clampfit 10 (Molecular Devices) and Microsoft Excel. Statistics and visualization were performed with GraphPad Prism. Two-way ANOVA with Sidak's multiple comparison was used to compare IOR and PPF between CTRL and cKO.
Chemically induced LTP (cLTP)
Freshly isolated brains were immediately transferred into ice cold sucrose dissection buffer (SACSF, in mM: 254 sucrose, 1.9 KCl, 1.2 KH2PO4, 26 NaHCO3, 10 D-glucose, 2 MgSO4 and 1.1 CaCl2, saturated with 5% CO2 and 95% O2, final pH 7.4). Hippocampi were isolated and cut into 350 µM transversal sections using a McIlwain Tissue chopper and incubated in oxygenated ACSF (see above) for 30 min at room temperature. cLTP was induced by adding 20 µM forskolin, 0.1 µM rolipram 50 µM picrotoxin for 10 min in the presence or absence of 5 µM paxilline [32]. This treatment increases cAMP levels [32] and subsequently increases network activity leading to tetanic-like bulk stimulation potentiating a majority of excitatory synapses [33]. Stimulation was terminated by subsequently storing slices in oxygenated ACSF for an additional 10 min.
cLTP induction was evaluated by Western immunoblot using the indicated antibodies (Table S7) as described above. After detection of phospho-specific GluA1 signals, membranes were stripped using Re-blot Plus Strong Antibody Stripping Solution (Millipore) before incubation with GluA1-specific antibodies and detection with secondary antibodies labelled with a fluorescent dye different from the one used for detection of phospho signals (all primary antibodies are listed in Table S7).
Primary dissociated hippocampal neurons
Primary hippocampal neurons were cultured from P0 BK+/+ and BK−/− as previously described [34]. In summary, hippocampi were isolated in dissection medium (DM) consisting of Hank’s Balanced Salt Solution (Invitrogen #14175095) supplemented with 1 mM sodium pyruvate, 0.1% (wt/vol) Glucose and 10 mM HEPES) and cleared of meninges. After washing thrice with DM, hippocampi were trypsinized (0.25% (wt/vol) in DM for 20 min at 37 °C) and subsequently incubated with 0.1% (wt/vol) Deoxyribonuclease I (Sigma #DN25) for 5 min at room temperature. After washing twice with DM, trypsin was deactivated by washing twice with plating medium (PM) consisting of BME medium (Invitrogen #21010046) supplemented with 10% FCS, 0.45% (wt/vol) glucose, 1 mM sodium pyruvate, 2 mM glutamine and 100 U/ml penicillin/streptomycin. Neurons were dissociated by triturating several times using a fire-polished glass pipette, counted and seeded (700,000 cells) onto 30 mm circular poly-l-lysine coated (Sigma #P2636) glass coverslips. After 2 h, PM was replaced with maintenance medium (MM; Neurobasal; Invitrogen #21103049) supplemented with B-27 (Invitrogen #17504044), 2 mM glutamine and 100 U/ml penicillin/streptomycin. Cells were maintained at 37 °C in a humidified environment with 5% CO2/95% air. Half of the MM was replaced every 3–4 days.
K+ imaging
FRET-based recordings of [K+]i were performed as previously described [35]. Neurons were transduced after 7 days in vitro (DIV) at a multiplicity of infection (MOI) of 100 with an adeno associated virus -DJ/8 vector system encoding a cytosol targeted K+ sensitive lc-LysM GEPII 1.0 FRET-based biosensor [36] under control of a CAG promoter. At 9 DIV, coverslips were mounted in a PC30 perfusion chamber (NGFI GmbH, Graz, Austria), perfused with imaging buffer (IB, in mM: 126.5 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 30 D-glucose, 10 sodium pyruvate, final pH 7.4) through a gravity-based perfusion system (NGFI GmbH, Graz, Austria) which was also used for drug application. Single cell live imaging was conducted on a Zeiss Observer Z.1 (Wetzlar, Germany) with an external light source (2200114 LED-Hub, Omicron Laserage, Rodgau-Dudendorf, Germany) and a Plan-Neofluar 40x/1.30 Oil immersion objective as previously described [37]. Emissions were simultaneously collected at 480 and 535 nm using an Optosplit II (Cairn Research, Faversham, UK). The LED hub (Omicron Laserage, Rodgau-Dudendorf, Germany) featured a 340 nm, 380 nm and 455 nm, LED with 340×, 380× and 427/10 bandpass filters, respectively (AHF Analysentechnik, Tübingen, Germany). Emissions were recorded using a 459/526/596 dichroic with a 475/543/702 emission filter (AHF Analysentechnik, Tübingen, Germany). Images were recorded using a pco.panda 4.2 bi sCMOS camera (PCO, Kelheim, Germany). VisiView software (Visitron Systems, Puchheim, Germany) was used to acquire images and to control the microscope. During live cell imaging, drugs were applied 10 min prior to the start of the recording. Background subtraction was performed using a background ROI. The ratio, which is proportional to [K+]i levels, was calculated from the corrected values of the YFP images to those of CFP. First 5 min of basal FRET ratio signal were used for normalization.
Ca2+ imaging
Ca2+-sensitive live cell imaging was conducted as described before [38]. 9 DIV neurons were loaded with 2.5 µM of the ratiometric dye Fura-2AM (AAT Bioquest, Sunnyvale, USA) in IB for 20 min at 37 °C followed by a washing step in IB for 10 min at room temperature. Excitation at 340 nm (Ca2+ bound to Fura-2) and 380 nm (Ca2+ free) induced emissions which were recorded on the same setup as K+ signals. Background subtraction was performed using a background ROI. The ratio, which is proportional to [Ca2+]i levels, was calculated from the corrected values of the images at 340 nm to those taken at 380 nm. Only spikes with at least 10% of the height of the glutamate peak were considered in the analysis.
Membrane potential imaging
Membrane potential was visualized by live cell fluorescence imaging using membrane potential sensitive dye Bis-(1,3-dibutylbarbituric acid)trimethine oxonol (DiBAC4(3)) [39]. 9 DIV neurons were loaded with a 1:40,000 dilution of a 10 mg/mL stock of DiBAC4(3) for 30 min at RT in IB. During recordings DiBAC4(3) in the indicated concentration was present in all buffers used. cLTP induction, recording of induced emissions at 516 nm and background subtraction by background ROI were performed as described for K+- and Ca2+ -sensitive recordings.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9.4.1. Data are expressed as mean ± standard error of mean (SEM). After data sets passed the Shapiro–Wilk test for Gaussian normal distribution, an unpaired (Figs. 1B; 2E and S3B, D) or paired (Figs. 2F, G) t-test was performed. Comparisons consisting of more than 2 data sets were tested with either one- or two-way ANOVA followed by a Dunnett’s (Fig. 4H), Sidak’s (Fig. 2A, B, F, G, 3; S1B-D and S2) or Tukey’s (Figs. 5F; S4K; S5C and S6F) multiple comparison test. The specific statistical tests used are explained in the respective legends. For an exact description, see also Tables S1–S5. P-values of ≤ 0.05 were represented with *, p ≤ 0.01 with **, and p ≤ 0.001 with ***, indicating comparisons between genotypes. Statistically significant differences to corresponding BK+/+ condition (Figs. 4H and 5F) were indicated by † = p ≤ 0.05; § = p ≤ 0.01; # = p ≤ 0.001.

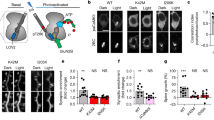
Characterization of conditional hippocampus-specific BK KO mice. A Representative immunoblot of whole hippocampal lysates from wildtype (BK+/+), conditional BK control (CTRL), conditional (cKO) and global BK KO (BK−/−) mice probed using BKα-specific antibodies revealed that BK protein levels were comparable between BK+/+ and CTRL, but significantly reduced in cKO. Compared to CTRL no BKα immunoreactivity was obtained in BK−/− samples. α-Tubulin immunoreactivity serves as loading control. B Quantification of BK protein band intensities normalized to α-tubulin. n = 3 independent samples from N = 3 mice. C Representative immunofluorescence labelling of cryosections from CTRL, cKO and BK−/− mice using specific antibodies against the pore forming α-subunit of BK (BKα, green). Nuclei were stained with Hoechst 33342 (blue). In CTRL, BKα immunosignals were detected in all hippocampal regions. In cKO, BK immunosignals were virtually absent in CA1, but were present in CA3 and DG. No BK immunosignals were detected in BK−/− mice. n = 3 protein samples obtained from N = 3 mice per genotype. Scale bar is 100 µm. D–G During 30 min of observation in the open field experiment, time spent in center of the arena (D), number of rearings (E), total distance travelled (F) and mean velocity (G) did not differ between cKO and CTRL (N = 14). H–J cKO and CTRL performed similar in the beam walk test as assessed by falls per trial (H), missteps per trial (I) and latency to cross the bar (J). Diagrams depict averages of all bar diameters of the square (□, left), round (○, middle) or both (□○, right) cross-sections indicated below (N = 10–14). Statistics: Unpaired Student's t-test (D–G), Two-way ANOVA with Sidak's multiple comparison test (H–J). All bar diagrams presented as means ± SEM. See also Fig. S1 and Table S1

Impaired MWM performance and hippocampal LTP in cKO. A–C Compared to CTRL (N = 20), cKO (N = 21) exhibit delayed learning and reduced memory in the MWM task. A Mean latencies to reach the hidden platform or original platform position over 5 training days (4 training sessions per day). Latency was similar on day 1 but decreased quicker in CTRL than cKO during the following trainings days. Since the hidden platform was removed, latency during probe trial on day 6 was measured for reaching the former platform position. The latency of the probe is offset, as it is not part of the acquisition phase. B CTRL but not cKO showed significant target quadrant (NE) preference during probe trial and also significantly avoided the opposing quadrant (SW). C Representative paths traveled by the CTRL and D cKO during acquisition probe trial. Start and finish indicated by dashed and solid red circle, respectively. E Swim speed of CTRL (N = 20) and cKO (N = 21) during acquisition probe trial were comparable. F Schaffer-collateral fEPSP initial slopes recorded from forebrain slices. 100 Hz 1 s high frequency stimulation (HFS) induced significantly more LTP in CTRL (n = 7 slices from N = 4 animals) than cKO (n = 8 slices from N = 4 animals). Top: Representative traces before (solid) and after (dashed) LTP induction. G Schaffer-collateral fEPSP initial slopes recorded from forebrain slices. 10 min perfusion of 20 µM forskolin, 50 µM picrotoxin and 0.1 µM rolipram (FRP) induced significantly more cLTP in CTRL (n = 10 slices from N = 4 animals) than cKO (n = 10 slices from N = 5 animals). Top: Representative traces before (solid) and after (dashed) LTP induction. Statistics: Two-way ANOVA (F, G) with Sidak's multiple comparison test (A, B), unpaired Student's t-test (E). All bar diagrams presented as means ± SEM. See also Fig. S2 and Table S2

BK deficiency precludes GluA1 S845 phosphorylation after LTP induction. Acute hippocampal slices from 8–12 weeks-old mice were treated with vehicle or 20 µM forskolin, 0.1 µM rolipram and 50 µM picrotoxin for 10 min to chemically elicit LTP in the presence or absence of the BK channel blocker PAX (5 µM). 10 min after cLTP induction, slices were lysed for immunodetection of phosphorylation at GluA1 residues S845 and S831. Blots were stripped and reprobed for total GluA1. Loading control: α-Tubulin. Representative blots on the left, densitometric quantification on the right. Relative expression was normalized to the mean of the respective untreated CTRL group not stimulated by cLTP to better represent the ratios. A cLTP significantly increased GluA1 S845 phosphorylation in CTRL but not cKO (N = 4 animals per genotype), B while S831 phosphorylation levels were not altered. C Presence of PAX precludes changes in S845 phosphorylation levels. D S831 phosphorylation remains unchanged after cLTP in both CTRL (N = 3) and cKO (N = 3) also in the presence of PAX. Statistics: Two-way ANOVA with Sidak's multiple comparison test. All bar diagrams presented as means ± SEM. See also Table S3

cLTP induction causes massive K+ outflow depending on BK, NMDAR and LTCC. Live imaging of 9 DIV hippocampal neuronal cultures (representative cells in A), virally transduced at 7 DIV with the FRET-based K+-sensitive sensor (GEPII), allowed single-cell live recording of [K+]i in response to external stimuli. (B) Representative time course of ratio between single fluorescence intensities (YFP/CFP, black) highlights reduction of [K+]i after repeated application of 20 µM glutamate. Single fluorescence intensities shown in yellow and blue as indicated. C–G Individual (gray) and averaged (black or red in (D)) YFP/CFP in response to cLTP induction in neurons. Glutamate (20 µM) application at the end of each measurement verified cell viability (inset on lower right of each panel). C cLTP strongly and persistently decreased [K+]i in BK+/+ (n = 11 independent experiments from a total of n = 54 neurons obtained from N = 6 preparations). D cLTP induction failed to decrease [K+]i in BK−/− (n = 9 independent experiments with a total of n = 48 neurons obtained from N = 5 preparations). E BK inhibition by PAX (5 µM) prevented decrease of [K+]i during cLTP (n = 10) independent experiments from a total of n = 43 neurons obtained from from N = 5 preparations). F Compared to vehicle, NMDAR inhibition by AP5 (100 µM) significantly reduced K+ efflux during cLTP (n = 9 independent experiments with a total of n = 37 neurons obtained from N = 5 preparations). G Compared to vehicle, LTCC inhibition by NIFE (5 µM) significantly reduced K+ efflux during cLTP (n = 6 independent experiments with a total of n = 52 neurons obtained from N = 4 preparations). H Maximum differences between average baseline and minimum YFP/CFP during cLTP recordings in panels C-G to corresponding BK+/+ condition: § = p ≤ 0.01; # = p ≤ 0.001. Statistics: One-way ANOVA with Dunnett's multiple comparison test. All bar diagrams presented as means ± SEM. See also Fig. S4 and Table S4

Ca2+ oscillations during cLTP induction depend on BK, NMDAR and LTCC. A–E Time course of ratio between fluorescence intensities at 340 nm and 380 nm (RF340/F380) in 9 DIV hippocampal neuronal cultures loaded with the Ca2+-sensitive dye Fura-2-AM. Traces recorded from individual neurons during multiple measurements are plotted in grey, representative traces in black except for (B) which is in red. Glutamate (20 µM) application at the end of each measurement verified cell viability and served as positive control. A, B Neuronal Ca2+ oscillations spontaneously occurred under control conditions in BK+/+ (A; n = 4 independent experiments with a total of n = 31 neurons obtained from 4 preparations) but not BK−/− neurons (B; n = 4 independent experiments with a total of n = 35 neurons obtained from 3 preparations). In BK+/+, cLTP induction increased oscillation frequency to a significantly higher degree than in BK−/−. C–E Inhibition of BK, NMDAR, and LTCC by 10 min pre-incubation with 5 µM PAX (C; n = 4 independent experiments with a total of n = 29 neurons obtained from 3 preparations), 100 µM AP5 (D; n = 4 independent experiments with a total of n = 29 neurons obtained from 3 preparations) and 5 µM NIFE (E; n = 4 independent experiments with a total of n = 69 neurons obtained from 5 preparations), respectively, reduced spontaneous Ca2+ oscillations and prevented the cLTP-induced rise in frequency. F Spiking frequency before and after cLTP induction in indicated conditions. cLTP increased Ca2+ oscillation frequency in BK+/+ neurons. Genetically or pharmacological BK inhibition as well as AP5 and NIFE prevented frequency increase. Compared with corresponding BK+/+ condition: # = p ≤ 0.001. Statistics: Two-way ANOVA with Tukey's multiple comparison test. All bar diagrams presented as means ± SEM. See also Table S5
Results
Characterization of conditional CA1-specific BK KO mice
Impaired MWM memory acquisition in BK−/− was previously reported [28]. BK−/− were also observed to suffer from cerebellar ataxia [30, 40], which could impact their MWM swimming performance. In order to determine whether memory performance is affected independently of motor performance, we generated a conditional KO line lacking the pore-forming BKα subunit in hippocampal CA1 pyramidal cells (cKO), the postsynaptic neurons of hippocampal Schaffer-collateral CA3 to CA1 synapses. Plasticity of these synapses is generally agreed to correlate with the animal’s performance in spatial learning tasks [41]. Ablation of the floxed BKα (BKfl/+) pore exon in CA1 pyramidal cells was achieved by intercrossing mice carrying the floxed allele to a transgenic subline (T29-1) expressing Cre recombinase under control of CaMKII (Cretg/+) in CA1 pyramidal cells [29] to generate age- and litter-matched cKO (genotype: Cretg/+; BKfl/−) and the corresponding controls (CTRL, genotype: Cretg/+; BKfl/+). T29-1 was used in many studies to achieve recombination in CA1 pyramidal cells [42, 43]. It is one of several Cre-expressing sublines originally created by the Tonegawa lab under control of the CamKIIα promoter. All original founder lines display different expression patterns [29], with T29-1 causing very high recombination efficiency and also a remarkable selectivity in CA1 pyramidal cells. First, specific hippocampal recombination in cKO was confirmed by polymerase chain reaction (PCR) (Fig. S1A). Additionally, Western blots demonstrated significantly reduced immunoreactivity to a validated BKα antibody [44] in the hippocampus of cKO compared to CTRL (Fig. 1A, B) with tissue from wildtype (BK+/+) and BK−/− serving as positive and negative controls, respectively. Consistent with the Cre expression pattern [29], BKα immunofluorescence was consistently reduced in CA1 but not CA3 or the dentate gyrus (DG) of cKO compared to CTRL as shown in hippocampal sections of adult mice. Again, absence of BKα immunoreactivity in slices from global BK−/− confirmed antibody specificity (Fig. 1C). While Cre-mediated recombination is not completely restricted to CA1 pyramidal cells, cKO mice do not seem to express any BK in these cells. Therefore, these mice are well suited to test BK’s role in hippocampal synaptic plasticity and hippocampus-dependent learning paradigms [41].
Anxiety behavior and motor abilities are normal in cKO
Comparison of learning behavior between genotypes in the MWM can be perturbed by differences in anxiety and motor abilities. Thus, open field and beam walk tests were performed to assess if cKO suffer from corresponding impediments. Both paradigms examine test subjects’ motor abilities [45, 46], while mouse behavior in the open field additionally represents an important indicator of anxiety levels [47, 48]. cKO and CTRL did not differ in anxiety and exploratory behavior as evidenced by similar amounts of time spent in the center of the open field arena (Fig. 1D) and similar number of rearings (Fig. 1E). During their time in the open field, animals from both genotypes did not differ in total distance travelled (Fig. 1F) and mean movement speed (Fig. 1G). These equivalent motor abilities of cKO and CTRL were further confirmed by the more complex beam walk test in which animals walk over round- and square-shaped beams of decreasing diameter and increasing difficulty. Neither falls or missteps nor the time to cross the beam differed between genotypes (Fig. 1H–J and Fig. S1B–D). Additionally, results from the beam walk test do not suggest any defects in the visual abilities of cKO. This is expected, as in T29.1 Cre activity is only sparsely detected in retinal bipolar, amacrine, and ganglion cells [49] and global BK knockout mice display unaltered electroretinography results under normal lighting conditions [50]. In summary we conclude that BKα is specifically depleted from hippocampal CA1 pyramidal cells of cKO mice, which do not display altered anxiety levels or locomotor abilities in comparison to their age- and litter-matched CTRLs.
Hippocampal learning, memory and Schaffer collateral LTP is impaired in cKO
To test hippocampus-dependent spatial learning and cognitive performance, cKO and CTRL mice were subjected to a MWM task [51]. Over the course of five training days with four training sessions per day, mice learned to remember the hidden platform’s position in the NE quadrant. On the first day, CTRL and cKO exhibit similar latencies (Fig. 2A). On the second day already, performance in CTRL improved more quickly than in cKO. As evidenced by continuous shortening of the latency, cKO also learned the platform location over the course of 5 training days, but never reached the performance of CTRL (Fig. 2A). During probe trial on day 6, cKO latency to the platform location was similar to CTRL, but cKO showed significantly less target quadrant preference while spending significantly more time in the opposite SW quadrant than CTRL (Fig. 2B). The lack of target quadrant preference was evident when observing the animals’ swimming path. While CTRL focused on examining the target quadrant (Fig. 2C), cKO kept randomly scanning the arena (Fig. 2D). Swim speed as evaluated at the acquisition probe trial was similar between CTRL and cKO (Fig. 2E), ruling out swimming abilities as source for different latencies to reach the platform as well as quadrant preferences.
Schaffer collateral LTP is defective in cKO
Acquisition of spatial memory in the MWM was repeatedly shown to correlate with NMDAR- dependent LTP of hippocampal Schaffer collateral synapses [41, 52]. Therefore, we measured hippocampal LTP by recording extracellular field potentials (fEPSP) in acute hippocampal slices. In accordance with MWM observations, HFS elicited stable LTP in CTRL, while initially potentiated fEPSP slopes dropped to baseline levels in cKO within 60 min (Fig. 2F, Table S2F). We could not find any indications that altered LTP is due to changes in basal synaptic transmission, as both genotypes displayed similar fEPSP amplitudes in response to a range of input strengths (Fig. S2D). Importantly, similar levels of paired-pulse facilitation (PPF) also indicated unaltered presynaptic function between CTRL and cKO (Fig. S2C).
Additionally, we also observed reduced LTP in a chemically induced LTP (cLTP) paradigm, in which synapses are not potentiated electrically, but chemically. We tested this key mechanism in cKO by applying cLTP, a model in which LTP is induced by application of 20 µM forskolin, to activate adenylyl cyclases, 0.1 µM rolipram to inhibit phosphodiesterase 4 (PDE4) and 50 µM picrotoxin, to block GABAA receptors [32, 53, 54]. Interestingly, forskolin/rolipram/picrotoxin (FRP)-induced LTP was previously demonstrated to depend on NMDAR [32]. 10 min FRP perfusion of hippocampal slices sustainably increased initial fEPSP slopes in both genotypes to levels significantly higher than the respective baselines. This potentiation, however, was significantly less pronounced in cKO than CTRL (Fig. 2G, Table S2G). In both genotypes, FRP-induced comparatively more potentiation than HFS. This is probably the result of a more complete activation of the total neuronal population by the cLTP cocktail, while electrical stimulation is restricted to tissue directly adjacent to the stimulation electrode.
Hippocampal LTP is generally associated with MWM learning ability, while hippocampal LTD is linked to cognitive flexibility as observed during MWM reversal training [55, 56]. Recently, defective hippocampal LTD and an associated reduction in reversal learning and cognitive flexibility were described in mice lacking the Na+-activated K+ channel Slack (Sequence Like a Ca2+-activated K+ Channel, KNa1.1, Slo2.2) which is highly homologous to BK [31, 38]. We therefore conducted reversal training subsequent to the acquisition phase of the MWM by placing the platform on the opposite SW side and by mirroring the animals' insertion points. As compared to the last acquisition trial(s) and the acquisition probe trials, both genotypes took longer to locate the new platform position in the reversal phase (Fig. S2A), but latencies were comparable between CTRL and cKO throughout of the reversal phase. Apparently, both cKO and CTRL were able to effectively update their memory of the new platform location (Fig. S2B), indicating comparable memory flexibility in both genotypes.
Together, our behavioral and electrophysiological data suggest that BK is necessary for normal hippocampal synaptic plasticity in the form of MWM learning and Schaffer collateral LTP.
BK deletion does not alter glutamate receptor subunit composition
Function of heterotetrameric NMDA- [57] and AMPA- [21] receptor complexes necessary for synaptic transmission and plasticity is crucially determined by their respective subunit composition. It was previously reported that animals lacking the closely related Na+-activated K+ channel Slack show decreased hippocampal GluN2B expression and impaired hippocampal synaptic plasticity [38]. We therefore examined protein levels of the most prevalent hippocampal NMDAR and AMPAR subunits [58, 59] in adult cKO and CTRL prior to or after completion of the MWM task. Expression levels were not different between cKO and CTRL for GluN1, GluN2A, GluN2B, GluA1 and GluA2 in the naïve state (Fig. S3A) nor for GluN2A, GluN2B and GluA1 after completion of the MWM task (Fig. S3B). GluN1 and GluA2 could not be analyzed due to low sample availability. Taken together, this suggests that defective hippocampal learning, memory and LTP in cKO is not due to altered glutamate receptor subunit expression.
Reduced GluA1-S845 phosphorylation in cKO after LTP induction
Induction of LTP by NMDAR stimulation initiates a signaling cascade resulting in GluA1 phosphorylation at S845, which is a crucial step to allow LTP expression by regulated AMPAR exocytosis [60,61,62]. We used cLTP to test this key mechanism in cKO. Compared to vehicle-treated controls, cLTP induction for 10 min with FRP (20 µM/0.1 µM/50 µM) significantly increased S845 phosphorylation in hippocampal slices of CTRL but not cKO (Fig. 3A). As shown earlier [54], cLTP induction by FRP did not influence GluA1 S831 phosphorylation confirming specificity of the elicited signaling (Fig. 3B). Interestingly, however, acute pharmacological blockade of BK channels with paxilline (PAX; 5 µM) effectively prevented cLTP-induced GluA1 S845 phosphorylation in CTRL, while PAX was less effective in cKO (Fig. 3C). In both genotypes, pharmacological BK blockade did not influence GluA1 S831 phosphorylation levels (Fig. 3D). This confirms our electrophysiological and MWM studies that suggest a pivotal role for BK in the cellular signaling mechanisms necessary for the expression of hippocampal LTP as well as hippocampus-dependent spatial learning and memory.
BK-mediated K+ efflux sustains neuronal Ca2+ oscillations during cLTP
To examine in detail how BK supports hippocampal LTP and, in turn, hippocampus-dependent memory formation, we next performed live cell imaging of dissociated hippocampal neuronal cultures generated from homozygous BK+/+ and BK−/− pups. Neurons were virally transduced with the genetically encoded potassium ion indicator (GEPII) NES lc-lysM GEPII 1.0 [35, 36] (Fig. 4A) or loaded with the Ca2+-sensitive dye Fura-2-AM (Fig. 5). For NES lc-LysM GEPII 1.0, the ratio of Förster resonance energy transfer (FRET) fluorescence divided by cyan fluorescent protein (CFP) fluorescence is directly proportional to [K+]i. GEPII sensitivity to [K+]i changes was verified by reproducible rapid decrease of the FRET/CFP ratio after repeated perfusion of 20 µM glutamate for 10 s presumably in response to neuronal depolarization (Fig. 4B). Additionally, we observed antiparallel progression of single fluorescent protein fluorescence intensity traces recorded over time, which indicates a valid FRET signal (Figs. 4B and S4A–E). cLTP induction by FRP application massively reduced [K+]i in BK+/+ (Fig. 4C and H). This drop in [K+]i was not observed after cLTP induction in BK−/− neurons (Fig. 4D and H) or after BK inhibition by PAX in BK+/+ (Fig. 4E and H). Neuron viability and physiological functionality was verified by glutamate application after each measurement (insets in Fig. 4C–G). This suggests that BK channels are necessary for sustained [K+]i depression during and after cLTP induction. Surprisingly, inhibition of both, NMDAR (Fig. 4F) as well as LTCC (Fig. 4G) prevented [K+]i reduction during cLTP by about 50% (Fig. 4H). This indicates that K+ outflow during cLTP depends on LTCC and NMDAR as sources for the high local [Ca2+]i required for BK activation. Comparison of the mean baseline between non-normalized curves of all K+ sensitive measurements (Fig. S4F–J) revealed a comparable basal [K+]i in both genotypes (Fig. S4K).
The sustained reduction of [K+]i after cLTP induction (Fig. 4C) was surprising. To evaluate whether it might affect the neuronal resting membrane potential, we elicited cLTP in the presence of the membrane potential-sensitive dye DiBAC4(3) [39]. The dye enters depolarized cells leading to increased fluorescence. Upon cLTP induction by FRP, DiBAC4(3) fluorescence was not significantly affected in either BK+/+ and PAX-inhibited BK+/+, while glutamate application provoked a rapid and profound increase in DiBAC4(3) fluorescence (Fig. S5A–C). The fact that the membrane potential hardly changes despite a sustained K+ efflux (Fig. 4C) might be a consequence of sustained Ca2+ influx through Ca2+ oscillations during cLTP and washout (Fig. 5A), which could offset the increased K+ permeability to result in a constant net membrane potential.
BK supports Ca2+ oscillations during LTP
Although high local [Ca2+]i is necessary for BK activation, BK channel activity also supports Ca2+ entry by rapidly repolarizing the plasma membrane to return voltage-gated Na+ or Ca2+ channels into an activatable state to support fast AP firing rates [14,15,16]. To test this reciprocal relationship, we induced cLTP in hippocampal neurons loaded with the Ca2+-sensitive dye Fura-2-AM (Fig. 5). In line with previous observations [63], spontaneous Ca2+ spikes were observed under basal conditions in 86% of all BK+/+ neurons (Fig. 5A). In all BK+/+ neurons, however, cLTP led to a significant increase in Ca2+ oscillation frequency that was preserved after wash-out (Fig. 5A and F). Interestingly, no spontaneous basal activity was observed in neurons lacking BK (Fig. 5B) while spiking activity in these cells was also increased by cLTP albeit at a significantly lower frequency than in BK+/+ (Fig. 5F). When BK activity was pharmacologically blocked in BK+/+, basal Ca2+ oscillations were only observed in 45% of these neurons and their frequency did not increase during or after cLTP induction (Fig. 5C and F). As expected, blocking neuronal Ca2+ entry through NMDAR and LTCC, by AP5 and NIFE, respectively, prevented FRP cLTP-induced increases in Ca2+ spiking frequency (Fig. 5D–F). Spontaneous activity was observed under NIFE, but not AP5. Comparison of mean baselines from non-normalized curves of all Ca2+ sensitive measurements (Fig. S6A–E) indicates equivalent basal [Ca2+]i in both genotypes (Fig. S6F).
In conclusion, cLTP-provoked Ca2+ spikes require NMDAR and LTCC in addition to functional BK expression. Hence, the low-frequency activity of hippocampal neurons lacking BK lays basis for the impaired expression of hippocampal LTP and deficits in spatial learning and memory performances in cKO.
Discussion
Here we report efficient hippocampus-specific conditional BK deletion (cKO. Figs. 1A–C and S1A) by Cre-mediated recombination under control of the CA1-selective T29-1 Cre subline [29]. Impairment of spatial learning and memory performance in the MWM as well as reduced electrically and chemically induced hippocampal LTP in cKO (Fig. 2) confirms an earlier report of restricted MWM learning in BK−/− global BK KO mice [28]. Unlike BK−/−, however, cKO do not display motor deficits like tremor and ataxia (Fig. 1D–J and S1B–D) previously reported to affect BK−/− [30, 40] that might interfere with their performance in behavioral assays.
LTP expression relies on activation of postsynaptic signaling mechanisms by Ca2+ entry through NMDAR and LTCC [64]. This increased postsynaptic [Ca2+]i, in turn, promotes insertion of additional AMPAR into postsynaptic sites after phosphorylation of the GluA1 subunit at either S831 or S845 to ultimately enhance synaptic transmission efficiency [19,20,21, 52]. In accordance with lack of hippocampal LTP, cLTP induction in cKO failed to increase phosphorylation levels at S845 of the AMPAR subunit GluA1, while phosphorylation at S831 remained unaffected (Fig. 3A, B). Additionally, S845 phosphorylation during cLTP was sensitive to acute BK inhibition by PAX (Fig. 3C). This suggests that BK activity during LTP induction is necessary for LTP expression through GluA1 phosphorylation at S845 that, in turn, increases AMPAR conductance and exocytosis of AMPAR from intracellular compartments [21]. BK might affect protein kinase A (PKA)-mediated S845 phosphorylation by supporting postsynaptic Ca2+ entry through NMDAR or LTCC to activate Ca2+-dependent adenylyl cyclase [65]. Alternative explanations for impaired hippocampal LTP were ruled out by confirming normal synaptic function (Fig. S2C, D) as well as AMPAR and NMDAR subunit composition in cKO (Fig. S3). Interestingly, cKO performed equal to CTRL in the reversal phase of the MWM (Fig. S2A), which tests the memory flexibility of the experimental animals. Many studies suggest that physiological LTD may be the base for proper memory flexibility [55, 56]. This stands in contrast to adult Slack KO, which are deficient in MWM reversal learning [31] and display normal LTP [38]. Thus, even as BK and Slack share high sequence homology and several topological features, both channels seem to serve very different functions in hippocampal synaptic plasticity. In addition to BK and Slack, small-conductance Ca2+-activated K+ channels (SK), which are also gated by Ca2+ to cause an efflux of K+ yielding a single channel conductance of 10–20 pS, are widely expressed in central neurons including the postsynaptic membrane of glutamatergic synapses [66]. Blocking SK channels with apamin promoted hippocampal synaptic plasticity and improved spatial learning in the MWM task by reducing SK-mediated AHP to disinhibit NMDAR [67,68,69]. In contrast to post-synaptic BK function, these findings suggest that the repolarizing conductance provided by SK activation decreases hippocampal glutamate receptor-mediated depolarization and thereby synaptic plasticity, neuronal excitability, and memory formation [66, 70, 71]. However, apamin-sensitive SK channels are also present at intracellular sites, and they are widely distributed throughout the surface of hippocampal neurons as well as in interneurons. Thus, multiple SK channel pools may indirectly contribute to LTP expression and thereby to learning performance [72,73,74].
Patients carrying both gain- and loss-of-function (LOF) mutations of BK suffer from multifaceted combinations of movement and seizure disorders as well as developmental delay and intellectual disability [1]. Cognitive development and neuronal functions like learning and memory are associated with synaptic plasticity [17, 18]. Thus, our finding of impaired hippocampal LTP might explain cognitive impairment in these patients, particularly in the case of LOF mutations. Additionally interesting in this context is that reduced BK expression was associated with Alzheimer's disease (AD), while reduced LTP was demonstrated in synapses from AD patients and related mouse models [75, 76]. Accordingly, pharmacological BK activation not only rescues hippocampal LTP in a mouse model of AD but also recovers cognitive function as assessed by behavioral assays [76].
How could BK influence hippocampal synaptic plasticity in the form of LTP? LTP induction depends on postsynaptic Ca2+ entry. Accordingly, we observe that the frequency of spontaneous Ca2+ oscillations in dissociated hippocampal neuronal cultures was greatly raised by cLTP induced signaling (Fig. 5A). Concomitant to this increased Ca2+ spike activity, we observed a massive BK-dependent reduction in [K+]i (Fig. 4C). We assume that this outflow of K+ through BK is triggered by Ca2+ entry through NMDAR and LTCC (Fig. 4F and G). Both of these channels were previously demonstrated to physically interact with postsynaptic BK channels [7, 8] and to provide elevated local [Ca2+]i necessary for BK activation [7, 9,10,11]. It is therefore tempting to speculate that BK activation is one of the mechanisms by which both NMDAR and LTCC exert their essential contribution to the expression of hippocampal LTP [19, 20]. In contrast, inhibition of each NMDAR and LTCC substantially reduced Ca2+ during cLTP (Fig. 5D and E), while the reduction of [K+]i was still at about half the amplitude of the control (Fig. 4F–H). It is conceivable that this drop in [K+]i is due to Ca2+-independent BK activation e.g. by direct phosphorylation of BK [77] or due to altered assembly with auxiliary subunits [78]. Alternatively, each Ca2+ source (NMDAR and LTCC) might activate a locally associated BK population. This would slightly reduce cellular [K+]i, but not sufficiently to support LTP. Only the simultaneous activity of both Ca2+ sources raises global [Ca2+]i sufficiently to trigger LTP and to efficiently activate BK channels.
While Ca2+ entry activates BK channels, we were surprised to discover that BK activity, in contrast to SK activity [70, 79], seems to support neuronal Ca2+ oscillations (Fig. 4C and D). This seems contradictory, as neuronal hyperpolarization by K+ channels in general and BK in particular is thought to inhibit NMDAR as well as voltage-gated cation channels [8, 80]. It has, however, also been proposed that BK is involved in fast repolarization after APs [14,15,16]. Consistent with our observations, BK inhibition was previously shown to slow the firing rate of hippocampal CA1 pyramidal cells, GABAergic neurons, and intracardiac autonomic neurons [15, 81, 82]. Others, however, reported increased firing frequencies after BK blockade in CA1 pyramidal neurons of the rat [83].
So far, we assumed neurons to act as single functional units despite their well-known morphological subdivision into different compartments. An increasing body of evidence, however, suggests specialized functions of dendritic compartments, particularly in respect to synaptic plasticity [84,85,86,87]. Fear learning-induced increases in local Ca2+, for example, were found in apical but not basal dendrites of layer 2/3 (L2/3) pyramidal neurons in the mouse auditory cortex [88]. Paradoxically, just in those apical dendrites an inactivating function of BK could be observed [89], which consequently leads to decrease in neuronal excitation as BK is activated by Ca2+ spikes mediated by NMDAR [90] and voltage-gated Ca2+ channels [91]. This contrasts with our findings suggesting positive feedback involvement of somatic BK in LTP through fAHP (Figs. 4C and 5A). While this seems like a contradiction at first glance, dendritic BK may nevertheless support increased plasticity through three factors. First, by hyperpolarizing the membrane potential, BK could impede the triggering of action potentials by synaptic inputs [89]. This could increase the specificity of synaptic potentiation, as only closely tuned inputs can cross the increased threshold [92]. Next, excessive and thus neurotoxic Ca2+ accumulation may be prevented, so that fewer synapses or dendrites perish [12]. Lastly, hyperpolarization in apical dendrites by BK may influence the timing of postsynaptic potentials and Ca2+ spikes to optimize temporal coding and signal summation, which might facilitate coincidence detection and thus also the occurrence of LTP [93].
Based on the presented data we propose a mechanism by which BK is activated by Ca2+ entry through both NMDAR and LTCC in postsynaptic membranes. BK-mediated K+ outflow, in turn, amplifies Ca2+ spiking frequency, which facilitates Ca2+ entry in support of the Ca2+-mediated mechanisms ultimately leading to AMPAR phosphorylation and LTP expression.
Availability of data and materials
All data generated or analyzed during this study that are not included in this published article and its supplementary information files are available from the corresponding authors on reasonable request. Further information and requests for resources and reagents should be directed to and will be fulfilled either by the Lead Contact, Lucas Matt (lucas.matt@uni-tuebingen.de) or by Robert Lukowski (robert.lukowski@uni-tuebingen.de). This study includes no data deposited in external repositories.
References
Miller JP, Moldenhauer HJ, Keros S, Meredith AL (2021) An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy. Channels (Austin) 15(1):447–464. https://doi.org/10.1080/19336950.2021.1938852
Laumonnier F, Roger S, Guerin P, Molinari F, M’Rad R, Cahard D et al (2006) Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry 163(9):1622–1629. https://doi.org/10.1176/ajp.2006.163.9.1622
Zhang L, Li X, Zhou R, Xing G (2006) Possible role of potassium channel, big K in etiology of schizophrenia. Med Hypotheses 67(1):41–43. https://doi.org/10.1016/j.mehy.2005.09.055
Sausbier U, Sausbier M, Sailer CA, Arntz C, Knaus H-G, Neuhuber W et al (2005) Ca2+-activated K+ channels of the BK-type in the mouse brain. Histochem Cell Biol 125(6):725. https://doi.org/10.1007/s00418-005-0124-7
Misonou H, Menegola M, Buchwalder L, Park EW, Meredith A, Rhodes KJ et al (2006) Immunolocalization of the Ca2+-activated K+ channel Slo1 in axons and nerve terminals of mammalian brain and cultured neurons. J Compar Neurol 496(3):289–302. https://doi.org/10.1002/cne.20931
Sailer CA, Kaufmann WA, Kogler M, Chen L, Sausbier U, Ottersen OP et al (2006) Immunolocalization of BK channels in hippocampal pyramidal neurons. Eur J Neurosci 24(2):442–454. https://doi.org/10.1111/j.1460-9568.2006.04936.x
Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S et al (2006) BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314(5799):615–620. https://doi.org/10.1126/science.1132915
Zhang J, Guan X, Li Q, Meredith AL, Pan HL, Yan J (2018) Glutamate-activated BK channel complexes formed with NMDA receptors. Proc Natl Acad Sci USA 115(38):E9006–E9014. https://doi.org/10.1073/pnas.1802567115
Berkefeld H, Fakler B (2013) Ligand-gating by Ca2+ is rate limiting for physiological operation of BK(Ca) channels. J Neurosci 33(17):7358–7367. https://doi.org/10.1523/JNEUROSCI.5443-12.2013
Perez GJ, Bonev AD, Nelson MT (2001) Micromolar Ca(2+) from sparks activates Ca(2+)-sensitive K(+) channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol 281(6):C1769–C1775. https://doi.org/10.1152/ajpcell.2001.281.6.C1769
Shah KR, Guan X, Yan J (2021) Structural and functional coupling of calcium-activated BK channels and calcium-permeable channels within nanodomain signaling complexes. Front Physiol 12:796540. https://doi.org/10.3389/fphys.2021.796540
Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R et al (2001) Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci 21(24):9585–9597. https://doi.org/10.1523/JNEUROSCI.21-24-09585.2001
Contet C, Goulding SP, Kuljis DA, Barth AL (2016) BK channels in the central nervous system. Int Rev Neurobiol 128:281–342. https://doi.org/10.1016/bs.irn.2016.04.001
Storm JF (1987) Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol 385:733–759. https://doi.org/10.1113/jphysiol.1987.sp016517
Gu N, Vervaeke K, Storm JF (2007) BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol 580(Pt.3):859–882. https://doi.org/10.1113/jphysiol.2006.126367
Jin W, Sugaya A, Tsuda T, Ohguchi H, Sugaya E (2000) Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res 860(1–2):21–28. https://doi.org/10.1016/s0006-8993(00)01943-0
Volk L, Chiu SL, Sharma K, Huganir RL (2015) Glutamate synapses in human cognitive disorders. Annu Rev Neurosci 38:127–149. https://doi.org/10.1146/annurev-neuro-071714-033821
Takeuchi T, Duszkiewicz AJ, Morris RG (2014) The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369(1633):20130288. https://doi.org/10.1098/rstb.2013.0288
Freir DB, Herron CE (2003) Inhibition of l-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo. Brain Res 967(1–2):27–36. https://doi.org/10.1016/s0006-8993(02)04190-2
Malenka RC (1991) Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6(1):53–60. https://doi.org/10.1016/0896-6273(91)90121-f
Diering GH, Huganir RL (2018) The AMPA receptor code of synaptic plasticity. Neuron 100(2):314–329. https://doi.org/10.1016/j.neuron.2018.10.018
Lisman J, Buzsaki G, Eichenbaum H, Nadel L, Ranganath C, Redish AD (2017) Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat Neurosci 20(11):1434–1447. https://doi.org/10.1038/nn.4661
Kim JH, Lee HK, Takamiya K, Huganir RL (2003) The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 23(4):1119–1124
Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J et al (2002) SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22(22):9721–9732
Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L (2009) The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev 23(11):1289–1302. https://doi.org/10.1101/gad.1783809
Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L (2011) Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron 72(2):300–315. https://doi.org/10.1016/j.neuron.2011.09.001
Matthews EA, Disterhoft JF (2009) Blocking the BK channel impedes acquisition of trace eyeblink conditioning. Learn Mem 16(2):106–109. https://doi.org/10.1101/lm.1289809
Typlt M, Mirkowski M, Azzopardi E, Ruettiger L, Ruth P, Schmid S (2013) Mice with deficient BK channel function show impaired prepulse inhibition and spatial learning, but normal working and spatial reference memory. PLoS One 8(11):e81270. https://doi.org/10.1371/journal.pone.0081270
Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ et al (1996) Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87(7):1317–1326. https://doi.org/10.1016/s0092-8674(00)81826-7
Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H et al (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA 101(25):9474–9478. https://doi.org/10.1073/pnas.0401702101
Bausch AE, Dieter R, Nann Y, Hausmann M, Meyerdierks N, Kaczmarek LK et al (2015) The sodium-activated potassium channel Slack is required for optimal cognitive flexibility in mice. Learn Mem 22(7):323–335. https://doi.org/10.1101/lm.037820.114
Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B et al (2004) Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol 91(5):1955–1962. https://doi.org/10.1152/jn.00941.2003
Kopec CD, Li B, Wei W, Boehm J, Malinow R (2006) Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 26(7):2000–2009. https://doi.org/10.1523/JNEUROSCI.3918-05.2006
Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF et al (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7(9):1741–1754. https://doi.org/10.1038/nprot.2012.099
Ehinger R, Kuret A, Matt L, Frank N, Wild K, Kabagema-Bilan C et al (2021) Slack K(+) channels attenuate NMDA-induced excitotoxic brain damage and neuronal cell death. FASEB J 35(5):e21568. https://doi.org/10.1096/fj.202002308RR
Bischof H, Rehberg M, Stryeck S, Artinger K, Eroglu E, Waldeck-Weiermair M et al (2017) Novel genetically encoded fluorescent probes enable real-time detection of potassium in vitro and in vivo. Nat Commun 8(1):1422. https://doi.org/10.1038/s41467-017-01615-z
Bischof H, Burgstaller S, Springer A, Matt L, Rauter T, Bachkonig OA et al (2021) Potassium ions promote hexokinase-II dependent glycolysis. iScience 24(4):102346. https://doi.org/10.1016/j.isci.2021.102346
Matt L, Pham T, Skrabak D, Hoffmann F, Eckert P, Yin J et al (2021) The Na+-activated K+ channel Slack contributes to synaptic development and plasticity. Cell Mol Life Sci 78(23):7569–7587. https://doi.org/10.1007/s00018-021-03953-0
Bräuner T, Hülser DF, Strasser RJ (1984) Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochim Biophys Acta 771(2):208–216. https://doi.org/10.1016/0005-2736(84)90535-2
Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G et al (2010) Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci USA 107(27):12323–12328. https://doi.org/10.1073/pnas.1001745107
Morris RG, Anderson E, Lynch GS, Baudry M (1986) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 319(6056):774–776. https://doi.org/10.1038/319774a0
Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87(7):1327–1338. https://doi.org/10.1016/s0092-8674(00)81827-9
Goto A, Bota A, Miya K, Wang J, Tsukamoto S, Jiang X et al (2021) Stepwise synaptic plasticity events drive the early phase of memory consolidation. Science 374(6569):857–863. https://doi.org/10.1126/science.abj9195
Lu R, Lukowski R, Sausbier M, Zhang DD, Sisignano M, Schuh CD et al (2014) BKCa channels expressed in sensory neurons modulate inflammatory pain in mice. Pain 155(3):556–565. https://doi.org/10.1016/j.pain.2013.12.005
Carter RJ, Morton J, Dunnett SB (2001) Motor coordination and balance in rodents. Curr Protoc Neurosci. https://doi.org/10.1002/0471142301.ns0812s15
Gould TD, Dao DT, Kovacsics CE. The open field test. Mood and anxiety related phenotypes in mice: characterization using behavioral tests. 2009;42:1–20.https://doi.org/10.1007/978-1-60761-303-9_1
Prut L, Belzung C (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463(1–3):3–33. https://doi.org/10.1016/s0014-2999(03)01272-x
Choleris E, Thomas AW, Kavaliers M, Prato FS (2001) A detailed ethological analysis of the mouse open field test: effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci Biobehav Rev 25(3):235–260. https://doi.org/10.1016/s0149-7634(01)00011-2
Ivanova E, Hwang GS, Pan ZH (2010) Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience 165(1):233–243. https://doi.org/10.1016/j.neuroscience.2009.10.021
Tanimoto N, Sothilingam V, Euler T, Ruth P, Seeliger MW, Schubert T (2012) BK channels mediate pathway-specific modulation of visual signals in the in vivo mouse retina. J Neurosci 32(14):4861–4866. https://doi.org/10.1523/jneurosci.4654-11.2012
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1(2):848–858. https://doi.org/10.1038/nprot.2006.116
Citri A, Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33(1):18–41. https://doi.org/10.1038/sj.npp.1301559
Nelissen E, Argyrousi EK, Van Goethem NP, Zhao F, Hines CDG, Swaminath G et al (2021) Soluble guanylate cyclase stimulator vericiguat enhances long-term memory in rats without altering cerebral blood volume. Biomedicines. https://doi.org/10.3390/biomedicines9081047
Diering GH, Heo S, Hussain NK, Liu B, Huganir RL (2016) Extensive phosphorylation of AMPA receptors in neurons. Proc Natl Acad Sci USA 113(33):E4920–E4927. https://doi.org/10.1073/pnas.1610631113
Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S et al (2008) Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58(1):104–117. https://doi.org/10.1016/j.neuron.2008.01.039
Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11(7):459–473. https://doi.org/10.1038/nrn2867
Hansen KB, Yi F, Perszyk RE, Menniti FS, Traynelis SF (2017) NMDA receptors in the central nervous system. Methods Mol Biol 1677:1–80. https://doi.org/10.1007/978-1-4939-7321-7_1
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62(3):405–496. https://doi.org/10.1124/pr.109.002451
Zhao Y, Chen S, Swensen AC, Qian WJ, Gouaux E (2019) Architecture and subunit arrangement of native AMPA receptors elucidated by cryo-EM. Science 364(6438):355–362. https://doi.org/10.1126/science.aaw8250
Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G et al (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112(5):631–643. https://doi.org/10.1016/s0092-8674(03)00122-3
Oh MC, Derkach VA, Guire ES, Soderling TR (2006) Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281(2):752–758. https://doi.org/10.1074/jbc.M509677200
Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6(2):136–143. https://doi.org/10.1038/nn997
Dravid SM, Murray TF (2004) Spontaneous synchronized calcium oscillations in neocortical neurons in the presence of physiological [Mg(2+)]: involvement of AMPA/kainate and metabotropic glutamate receptors. Brain Res 1006(1):8–17. https://doi.org/10.1016/j.brainres.2004.01.059
Malenka RC (1991) The role of postsynaptic calcium in the induction of long-term potentiation. Mol Neurobiol 5(2–4):289–295. https://doi.org/10.1007/BF02935552
Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC (2003) Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical ‘barrel’ map development. Nat Neurosci 6(9):939–947. https://doi.org/10.1038/nn1106
Adelman JP, Maylie J, Sah P (2012) Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol 74(1):245–269. https://doi.org/10.1146/annurev-physiol-020911-153336
Norris CM, Halpain S, Foster TC (1998) Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci 18(9):3171–3179. https://doi.org/10.1523/JNEUROSCI.18-09-03171.1998
Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP et al (2002) Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci 22(23):10163–10171. https://doi.org/10.1523/JNEUROSCI.22-23-10163.2002
Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G (2004) A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci 24(22):5151–5161. https://doi.org/10.1523/JNEUROSCI.0800-04.2004
Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP (2005) SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci 8(5):642–649. https://doi.org/10.1038/nn1449
Tigaret CM, Olivo V, Sadowski J, Ashby MC, Mellor JR (2016) Coordinated activation of distinct Ca(2+) sources and metabotropic glutamate receptors encodes Hebbian synaptic plasticity. Nat Commun 7:10289. https://doi.org/10.1038/ncomms10289
Behnisch T, Reymann KG (1998) Inhibition of apamin-sensitive calcium dependent potassium channels facilitate the induction of long-term potentiation in the CA1 region of rat hippocampus in vitro. Neurosci Lett 253(2):91–94. https://doi.org/10.1016/s0304-3940(98)00612-0
McKay BM, Oh MM, Disterhoft JF (2013) Learning increases intrinsic excitability of hippocampal interneurons. J Neurosci 33(13):5499–5506. https://doi.org/10.1523/JNEUROSCI.4068-12.2013
Lujan R, Merchan-Perez A, Soriano J, Martin-Belmonte A, Aguado C, Alfaro-Ruiz R et al (2021) Neuron class and target variability in the three-dimensional localization of SK2 channels in hippocampal neurons as detected by immunogold FIB-SEM. Front Neuroanat 15:781314. https://doi.org/10.3389/fnana.2021.781314
Prieto GA, Trieu BH, Dang CT, Bilousova T, Gylys KH, Berchtold NC et al (2017) Pharmacological rescue of long-term potentiation in Alzheimer diseased synapses. J Neurosci 37(5):1197–1212. https://doi.org/10.1523/JNEUROSCI.2774-16.2016
Wang L, Kang H, Li Y, Shui Y, Yamamoto R, Sugai T et al (2015) Cognitive recovery by chronic activation of the large-conductance calcium-activated potassium channel in a mouse model of Alzheimer’s disease. Neuropharmacology 92:8–15. https://doi.org/10.1016/j.neuropharm.2014.12.033
Reinhart P, Chung S, Martin B, Brautigan D, Levitan I (1991) Modulation of calcium-activated potassium channels from rat brain by protein kinase A and phosphatase 2A. J Neurosci 11(6):1627–1635. https://doi.org/10.1523/jneurosci.11-06-01627.1991
Li Q, Yan J (2016) Modulation of BK channel function by auxiliary beta and gamma subunits. Int Rev Neurobiol 128:51–90. https://doi.org/10.1016/bs.irn.2016.03.015
Titley HK, Brunel N, Hansel C (2017) Toward a neurocentric view of learning. Neuron 95(1):19–32. https://doi.org/10.1016/j.neuron.2017.05.021
Gomez R, Maglio LE, Gonzalez-Hernandez AJ, Rivero-Perez B, Bartolome-Martin D, Giraldez T (2021) NMDA receptor-BK channel coupling regulates synaptic plasticity in the barrel cortex. Proc Natl Acad Sci USA 118(35):e2107026118. https://doi.org/10.1073/pnas.2107026118
Gittis AH, Moghadam SH, du Lac S (2010) Mechanisms of sustained high firing rates in two classes of vestibular nucleus neurons: differential contributions of resurgent Na, Kv3, and BK currents. J Neurophysiol 104(3):1625–1634. https://doi.org/10.1152/jn.00378.2010
Perez GJ, Desai M, Anderson S, Scornik FS (2013) Large-conductance calcium-activated potassium current modulates excitability in isolated canine intracardiac neurons. Am J Physiol Cell Physiol 304(3):C280–C286. https://doi.org/10.1152/ajpcell.00148.2012
Matthews EA, Weible AP, Shah S, Disterhoft JF (2008) The BK-mediated fAHP is modulated by learning a hippocampus-dependent task. Proc Natl Acad Sci USA 105(39):15154–15159. https://doi.org/10.1073/pnas.0805855105
Beaulieu-Laroche L, Toloza EHS, van der Goes M-S, Lafourcade M, Barnagian D, Williams ZM et al (2018) Enhanced dendritic compartmentalization in human cortical neurons. Cell 175(3):643–51.e14. https://doi.org/10.1016/j.cell.2018.08.045
Poirazi P, Brannon T, Mel BW (2003) Pyramidal neuron as two-layer neural network. Neuron 37(6):989–999. https://doi.org/10.1016/S0896-6273(03)00149-1
Petreanu L, Mao T, Sternson SM, Svoboda K (2009) The subcellular organization of neocortical excitatory connections. Nature 457(7233):1142–1145. https://doi.org/10.1038/nature07709
Makino H, Komiyama T (2015) Learning enhances the relative impact of top-down processing in the visual cortex. Nat Neurosci 18(8):1116–1122. https://doi.org/10.1038/nn.4061
Godenzini L, Shai AS, Palmer LM (2022) Dendritic compartmentalization of learning-related plasticity. Eneuro 9(3):ENEURO.0060-22.2022. https://doi.org/10.1523/eneuro.0060-22.2022
Bock T, Stuart GJ (2016) The impact of BK channels on cellular excitability depends on their subcellular location. Front Cell Neurosci 10:206. https://doi.org/10.3389/fncel.2016.00206
Antic SD, Zhou WL, Moore AR, Short SM, Ikonomu KD (2010) The decade of the dendritic NMDA spike. J Neurosci Res 88(14):2991–3001. https://doi.org/10.1002/jnr.22444
Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA (1992) Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 9(6):1099–1115. https://doi.org/10.1016/0896-6273(92)90069-p
Sjöström PJ, Rancz EA, Roth A, Häusser M (2008) Dendritic excitability and synaptic plasticity. Physiol Rev 88(2):769–840. https://doi.org/10.1152/physrev.00016.2007
Xu N-l, Ye C-q, Poo M-m, Zhang X-h (2006) Coincidence detection of synaptic inputs is facilitated at the distal dendrites after long-term potentiation induction. J Neurosci 26(11):3002–3009. https://doi.org/10.1523/jneurosci.5220-05.2006
Acknowledgements
We thank John Dempster for WinWCP as well as Clement Kabagema-Bilan and Michael Glaser for excellent technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) with individual grants to LM and RL. TP, TH, DC, MCS, MK, PR and RL were supported by GRK2381: “cGMP: From Bedside to Bench”, DFG Grant Number 335549539. HB is a fellow of the Austrian Science Fund (FWF) funded Erwin-Schrödinger-Program, project number J4457. LM received support from the Wilhelm Schuler-Stiftung of the University of Tübingen.
Author information
Authors and Affiliations
Contributions
TP, HB, LM and RL designed experiments, TP, TH, DC, DSp, DK and SS performed experiments. TP, TH, DC, DSp, DK, SS and LM analyzed data. HB, DSk, MCS, SM, MK, BG, RE and NP contributed resources, materials and protocols. PR, RL and LM contributed to discussions. TP and LM wrote the manuscript. TP, RL and LM edited the manuscript. PR, RL and LM obtained funding. RL and LM supervised the project. All authors approved the content and submission of the paper.
Corresponding author
Ethics declarations
Conflict of interest
RL has a cooperation with Cyclerion Therapeutics Inc. on a topic not directly related to this study. All other authors authors have stated explicitly that there are no conflicts of interest in connection with this article.
Ethical approval
All experimental procedures were conducted in accordance with the animal protection laws in Germany and were approved by the local Ethics Committee for Animal Research (Regierungspräsidium Tübingen).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lucas Matt: Lead contact.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pham, T., Hussein, T., Calis, D. et al. BK channels sustain neuronal Ca2+ oscillations to support hippocampal long-term potentiation and memory formation. Cell. Mol. Life Sci. 80, 369 (2023). https://doi.org/10.1007/s00018-023-05016-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05016-y