
Abstract
Organ fibrosis caused by chronic allograft rejection is a major concern in the field of transplantation. Macrophage-to-myofibroblast transition plays a critical role in chronic allograft fibrosis. Adaptive immune cells (such as B and CD4+ T cells) and innate immune cells (such as neutrophils and innate lymphoid cells) participate in the occurrence of recipient-derived macrophages transformed to myofibroblasts by secreting cytokines, which eventually leads to fibrosis of the transplanted organ. This review provides an update on the latest progress in understanding the plasticity of recipient-derived macrophages in chronic allograft rejection. We discuss here the immune mechanisms of allograft fibrosis and review the reaction of immune cells in allograft. The interactions between immune cells and the process of myofibroblast formulation are being considered for the potential therapeutic targets of chronic allograft fibrosis. Therefore, research on this topic seems to provide novel clues for developing strategies for preventing and treating allograft fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although many patients with organ failure have been successfully treated since the development of allotransplantation, the 10-year survival rate after transplantation has not significantly improved [1, 2]. At present, allograft fibrosis caused by chronic allograft rejection is a major factor affecting the long-term survival of patients after transplantation [3, 4]. Persistent allogeneic immune-mediated inflammation under chronic dysfunction of transplanted organs leads to graft fibrosis [5,6,7]. However, the specific immunological mechanism of allograft fibrosis remains unclear.
It is traditionally believed that chronic allograft rejection is primarily mediated by indirect CD4+ T cell reaction [8]. Moreover, immunosuppressants currently used in clinical practice, which mainly act on the immune response of CD4+ T cells, have achieved some success; however, the long-term survival rate of grafts after organ transplantation must be improved. It has also been reported that innate immune cells can initiate the rejection of allogeneic nonself independent of lymphocytes [9,10,11,12], and an increasing number of recent and ongoing studies have focused on innate immune cells [13, 14]. Further, neutrophils, monocyte-derived dendritic cells (mo-DCs), plasmacytoid dendritic cells (pDCs), and innate lymphoid cells (ILCs) reportedly participate in chronic allograft rejection during mouse heart transplantation [15,16,17], mouse bone marrow plug transplantation [12], graft-versus-host disease (GVHD) [18], and mouse lung, skin, and human intestinal transplantation [19,20,21], respectively. It is reported that knocking out or inhibiting the function of recipient-derived macrophages after mouse lung, heart, and kidney allotransplantation reduce chronic allograft rejection [9, 22, 23].
Allograft immune rejection results from the coordinated response of several immune cell types as well as a series of complex processes in which the innate immune systems of the donor and recipient interact with the adaptive immune system. In this review, the latest progress of research on the interaction between recipient-derived macrophages and other immune cells, as well as their role in allograft fibrosis following chronic allograft rejection was reviewed. Furthermore, the molecular biological mechanism of recipient-derived macrophage plasticity in chronic allograft rejection was examined. This work provides a theoretical basis for developing targeted drugs to prevent and treat allograft fibrosis.
Plasticity of recipient-derived macrophages in allograft fibrosis
Peripheral blood monocytes can undergo epigenetic changes according to local tissue microenvironment changes [24]. When entering tissues, monocytes can differentiate into either monocyte-derived macrophages (mo-Macs) or mo-DCs [25]. The transcription factors involved in CD14+ monocyte differentiation are categorized as either mo-Mac-expressed (MafB, PU.1) or mo-DC-expressed (IRF4, AHR) [26]. mo-Macs express MafB and moderate levels of PU.1, which have been found to inhibit mo-DC differentiation [27]. In the presence of colony-stimulating factor 1 (CSF-1), CD14+ monocytes differentiate into mo-Macs unless they are exposed to certain cytokines (i.e., IL-4 and TNFα) in conjunction with AHR ligands, which promote mo-DC differentiation [26]. However, allograft immunity research has not yet elucidated the mechanism by which the recipient monocytes transform into macrophages and their various subsets in the allograft.
Allogeneic nonself-recognition by monocytes is necessary to initiate graft rejection. Dai et al. found for the first time that the paired immunoglobulin-like receptor-A (PIR-A) expressed on recipient Ly6Chigh monocytes and macrophages promote allograft rejection via direct binding to donor MHC I molecules, and inhibiting PIR-A recognition of MHC I using a specific antibody attenuates kidney and heart allograft rejection in mice [9]. Monocyte activation after allotransplantation can also occur independently of MHC mismatch between donors and recipients [10, 28]. Donor polymorphism in the gene encoding signal regulatory protein α (SIRPα) can regulate the affinity with CD47 on recipient Ly6Chigh monocytes to accumulate mature mo-DCs that produce interleukin-12 and present antigens to T cells [12, 29], which appears to aggravate organ rejection after allotransplantation. However, blocking the binding of the recipient monocyte SIRPα to CD47 from donor cells impairs transplant tolerance [30], which results in the differentiation of the donor myeloid-derived suppressor cells into M1 macrophages [31]. Therefore, it is possible that the strength of the interaction between SIRPα expressed by recipient or donor monocytes and CD47 expressed by donor or recipient cells determines whether monocyte differentiation favors mo-Macs or mo-DCs following allotransplantation.
The accumulation of macrophages in grafts has long been identified as a characteristic of organ transplantation rejection [32,33,34]. Immunological examination of grafts with chronic rejection showed that macrophages accounted for 74.2–86.6% of the total immune cells [6, 35]. Approximately 90% of macrophages in grafts were derived from monocytes obtained from recipient bone marrow [22, 23]. Recipient-derived macrophages have different characteristics from those of resident donor tissue macrophages [36]. These resident macrophages are sensitive to the stimulation of the transplantation environment and are prone to necrosis and apoptosis, while recipient-derived macrophages continuously replenish the lost resident macrophages in the graft [37,38,39]. Notably, this process persists after the acute surgical inflammation subsides [40]. Moreover, recipient-derived macrophages have considerable plasticity [41], and may thus play an important role in long-term immune rejection after allotransplantation.
Myofibroblasts [α smooth muscle actin+ (α-SMA+) fibroblasts] can produce extracellular matrix (ECM) components, which are the primary effectors of the organ fibrotic process in chronic allograft rejection [22, 23, 42, 43]. Myofibroblasts may arise from resident fibroblasts, pericytes, endothelial and epithelial cells, and circulating precursors [44,45,46,47]. Recent studies have primarily focused on myofibroblasts derived from recipient bone marrow progenitor cells [22, 23]. Macrophages derived from bone marrow cells can differentiate into α-SMA+ myofibroblasts within a fibrotic tissue in a process termed macrophage-to-myofibroblast transition (MMT) [22, 23, 41, 48,49,50,51]. To determine the origin of myofibroblasts in models of chronic allograft rejection, lineage tracing using LysMCreRosa26-tdTomato and CX3CR1creERRosa26-tdTomato mice has been employed, and to track myeloid cells and confirm the potential sources of α-SMA+ myofibroblasts, allografts have been analyzed by confocal microscopy, flow cytometry, and single-cell transcriptomic analysis to identify co-expression of macrophage makers (CX3CR1, CD68, or F4/80) and the myofibroblast marker (α-SMA) as well as collagen production [22, 23, 43]. MMT may serve as a key checkpoint for the progression of chronic allograft fibrosis.
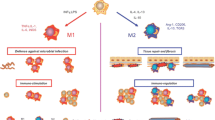
Recipient monocytes infiltrate into the allograft and are mainly polarized into M1 macrophages and M2 macrophages [52, 53]. In general, IFNγ/LPS/TNFα promote M1 macrophage activation (phenotypic markers: MHC II, CD86, Ly6chigh, and iNOS), while IL-4/IL-10/IL-13/TGFβ/CSF-1 stimulate M2 macrophage activation (phenotypic markers: CD206, Ly6clow, CX3CR1high, and Arg1) [17, 23, 54, 55]. M1 macrophages tend to produce IL-1, IL-6, IL-12, IL-23, CCL2, CXCL9, CXCL10, and CXCL11, which are involved in acute rejection [54, 56]. M2 macrophages produce fewer cytokines, such as IL-10 and TGFβ1, to regulate tissue inflammation resolution and produces pro-fibrotic mediators, such as CCL18 [57] and ECM, which promotes the progression of allograft fibrosis [3, 23, 43]. The infiltration of M2 macrophages is especially related to tissue fibrosis [3, 58]. Therefore, M2 macrophages likely play a key role in allograft fibrosis.
In vitro and in vivo experiments have demonstrated that the Notch2 signaling pathway controls the transformation of Ly6Chigh monocytes into Ly6Clow CX3CR1high monocytes under homeostasis [59]. The induction of the colony-stimulating factor receptor-1(CSF-1R) signaling axis is necessary for the differentiation of Ly6Chigh monocytes into Ly6Clow CX3CR1high monocytes under pathological conditions [60, 61], and this is the main mechanism by which M2 macrophages proliferate and accumulate following allotransplantation [22, 62]. Smooth muscle cell-related gene expression in CD16high CX3CR1high monocytes is increased during chronic rejection of transplanted human kidney [63]. It has been reported that increased levels of Ly6Clow CX3CR1high macrophages are consistent with fibrosis or gene-related M2 macrophage function [64]. The recipient-derived CX3CR1high population, which is derived from infiltrating Ly6Chigh(mouse) /CD14high(human) monocytes, has a profibrotic function and is composed of different continuously differentiated subsets of cells [22, 41]. Recipient-derived CX3CR1high macrophages may be mainly CD206+ M2 macrophages in chronic allograft fibrosis, and it has been reported that CD206+ M2 macrophages are the precursors of MMT [23, 41, 43, 50, 65, 66]. However, whether early M1 macrophages switch phenotype to become M2 macrophages remains unclear.
The findings outlined above indicate that recipient-derived CX3CR1high macrophages transformed to myofibroblasts via MMT play a critical role in chronic allograft fibrosis (Fig. 1). Understanding the dynamic reprogramming landscapes of recipient-derived macrophages is important to exploit the mechanisms of chronic allograft fibrosis. However, chronic allograft rejection does not arise from recipient-derived macrophage activity alone but from a coordinated immune cell response, and the role of recipient-derived macrophages and other immune cells in allograft fibrosis has not yet been systematically reviewed; relevant studies are discussed in the next section.

The plasticity of recipient-derived macrophages in chronic allograft rejection. Peripheral blood monocytes can undergo epigenetic changes according to the allograft microenvironment. In the allograft with chronic rejection, recipient monocytes can differentiate into CX3CR1+ CD68+ macrophages. MMT (recipient-derived CX3CR1+ CD68+ CD206+ M2 macrophages to α-SMA+ myofibroblasts) can result in the production of ECM components, which promote chronic allograft fibrosis.
Interaction of recipient-derived macrophages with adaptive immune cells in allograft fibrosis
Interaction with B cells
The population of B cells has been found to increase gradually during chronic transplant rejection [2, 67, 68]. Antibody-mediated rejection (AMR) is a major determinant of chronic allograft failure [69,70,71,72]. HLA-donor-specific antibody (DSA) binding to vascular endothelial cells of the allograft triggers inflammation, vessel injury, and AMR. The histological characteristic of AMR is the accumulation of recipient-derived macrophages [73, 74]. Therefore, the interaction between macrophages and B cells likely plays an important role in chronic graft rejection.
During human chronic lung allograft dysfunction, macrophages and B cells coexist in the luminal tissue of the bronchial tub [75, 76], and the existence of DSA may promote the development of bronchiolitis obliterans syndrome after human lung allotransplantation [77]. The accumulation of CD68+ macrophages in endomyocardial tissue following human cardiac transplantation correlates well with the increased numbers of alloantibodies [74]. The mechanism underlying the interaction between B cells and macrophages in chronic transplant rejection requires further investigation.
In recipient mice with B cell loss, macrophage infiltration and ECM accumulation were significantly reduced and graft fibrosis was alleviated [67, 78, 79]. Thus, B cells have been demonstrated to promote allograft fibrosis. In a mouse skin graft model, activated MHC IIhigh macrophages produce B cell activating factor (BAFF), which activates B cells to secrete DSA, leading to graft damage in AMR [80]. Moreover, cardiac allografts from murine recipients treated with MHC I DSA promoted monocyte differentiation into CD68+ CD206+ M2 macrophages in the endothelium of grafts [81]. Therefore, macrophages recruit B cells in the early stage of graft rejection, while B cells secrete DSA to promote the accumulation of M2 macrophages, promoting allograft fibrosis.
CSF-1R+ macrophages secrete CXCL13 to recruit CXCR5+ B cells to promote implant fibrosis, but the reduction in fibrosis after CSF-1R inhibitor administration was more significant than that after B cell knockout [67]. In a mouse lung transplant experiment, CXCL13 neutralization concurrent with Foxp3+ T cell depletion prevented AMR and preserved the airway epithelium [69]. Therefore, during chronic transplantation rejection, MHC IIhigh macrophages are responsible for CXCR5+ B cell recruitment and activation by secreting CXCL13 and BAFF, and activated CXCR5+ B cells secrete DSA, which promotes the emergence of a large number of CD68+ CD206+ M2 macrophages in the grafts (Fig. 2A). Macrophages may play a leading role in this process.

The diverse immune reactions of recipient-derived macrophages in chronic allograft rejection. Recipient-derived macrophages transformed to myofibroblasts via MMT play a critical role in chronic allograft fibrosis. During the occurrence of MMT, A CXCL13 and BAFF secreted by macrophages recruit and activate CXCR5+ B cells, which secrete DSA, which promotes the accumulation of CD68+ CD206+ M2 macrophages in grafts, while B IFN-γ secreted by Th1 cells inhibits the occurrence of MMT, and Th2 cells can induce the accumulation of M2 macrophages in grafts by secreting IL-4 and IL-13. C Neutrophils secrete CSF-1 or extracellular ATP, resulting in high M2 macrophage accumulation in the allograft. D IFN-γ secreted by ILC1s inhibits the occurrence of MMT, and ILC2s can induce the accumulation of M2 macrophages in grafts by secreting IL-4, IL-13, and G-CSF.
Interaction with CD4+ T cells
Recipient-derived macrophages are the dominant cell population after allotransplantation [9, 10, 73]. However, human chronic lung allograft dysfunction is associated with an increase in allo-reactive CD4+ T cells and mononuclear phagocytic cells [82]. In fact, the fibrosis degree and M2 macrophage count could be regulated by the T cell response [83]. The ability of primed macrophages to reject allografts requires the help of CD4+ T cells but not of CD8+ T cells [84]. Therefore, the interaction between recipient-derived macrophages and CD4+ T cells is considered crucial in chronic allograft rejection.
The Th1 cell surface protein Tim-3 regulates macrophage activation and the severity of autoimmune diseases [85]. However, direct interaction of recipient-derived macrophages with Th1 cells has not been detected in chronic allograft rejection. Donor-derived VIP-expressing pDCs limit IFN-γ+CD4+ T cell proliferation and polarization in vitro, thereby preventing GVHD [18]. It is suggested that IFN-γ+CD4+ T cells promote chronic allograft rejection. However, Th1 cells directly promote collagen degradation by releasing IFN-γ, which is an antifibrotic cytokine [86]. Moreover, IFN-γ secreted by Th1 cells disrupts fibrosis mediated by the TGFβ/Smad3 signaling pathway [87, 88]. The transformation of recipient-derived M2 macrophages into myofibroblasts is regulated by the TGFβ/Smad3 signaling pathway [23, 51, 89]. Therefore, Th1 cells may inhibit the development of allograft fibrosis by secreting IFN-γ to inhibit MMT (Fig. 2B). This may be one of the reasons why immunosuppressants targeting CD4+ T cells have not been effective in preventing and treating allograft fibrosis.
Activated Th2 cells secrete IL-4 and IL-13, which can induce the accumulation of M2 macrophages and promote graft fibrosis [7, 19, 90] (Fig. 2B), and promote MMT through activation of the JAK/STAT pathway [41]. However, immunosuppressants targeting CD4+ T cells fail to prevent and treat allograft fibrosis. Th2 cells may not play a major role in the transformation of recipient monocytes into M2 macrophages in the allograft microenvironment. Moreover, the number and function of innate lymphoid cells (ILCs) in the recipient circulation are reportedly unaffected by immunosuppressive drugs [91]. Type 2 ILCs (ILC2s) also secrete IL-4 and IL-13 [19], which may not be affected by immunosuppressants targeting CD4+ T cells.
It has been reported that inhibiting proliferation and accumulation of macrophages in the graft can reduce the risk of rejection without changing the number of peripheral leukocytes or altering T cell function [62, 92]. T cell knockout did not improve the degree of fibrosis on the implant surface, while macrophage knockout almost completely improved fibrosis on the surface [67]. It has been reported that CD4+ T cells also promote macrophages to acquire long-term specific memory of mouse skin grafts [93]. In the process of chronic rejection, the progressive accumulation of recipient-derived macrophages may be the main mechanism leading to allograft fibrosis, and CD4+ T cells play an auxiliary role in this process.
Interaction of recipient-derived macrophages with innate immune cells in allograft fibrosis
Interaction with neutrophils
Neutrophils can promote immune rejection of allogeneic transplants. In the early stages of rejection after transplantation, activated neutrophils cause tissue damage by releasing neutrophil extracellular traps (NETs) and proteases [94, 95]. Extensive NETs were formed in the grafts of acute renal tubular necrosis after human kidney transplantation and primary graft dysfunction (PGD) after human lung transplantation [95, 96]. In addition, neutrophils can initiate early rejection by interacting with monocytes and macrophages. In transplant-mediated ischemia reperfusion injury, intravital 2P microscopy revealed that neutrophils migrate across the endothelium immediately following inflammatory Ly6Chigh monocyte migration [97]. Neutrophils can damage cells or the ECM and release danger-associated molecular patterns (DAMPs) [98]. DAMPs then stimulate tissue-resident macrophages to secrete inflammatory mediators (such as ELR+-CXC and IL-1β) that activate the vascular endothelium and recruit circulating neutrophils, which adhere to the vascular endothelium of the graft [99].
Neutrophils have an important role in chronic allograft rejection [99]. A report showed that the accumulation of neutrophil-derived defensin peptides influenced the induction of airway inflammation and fibrosis in human lung allografts [100]. Christoffersson et al. found a unique vascular endothelial growth factor-A-induced subpopulation of CXCR4high MMP-9high neutrophils with vascular remodeling ability in mouse islet graft [101]. Macrophages, lymphocytes, and neutrophils highly infiltrated chronic rejection grafts [6, 102]. In a study of mouse heart transplantation, it was found that neutrophils were the main source of CSF-1 after co-stimulation block, which promoted the transformation of Ly6chigh monocytes into M2 macrophages[16] (Fig. 2C). Therefore, neutrophils may indirectly play a role in promoting fibrosis by promoting the transformation of recipient-derived macrophages.
Neutrophils in inflammatory tissues can produce elevated levels of extracellular adenosine triphosphate (ATP) [103]. The purinergic receptor P2X7 (P2x7r), an ATP-gated ion channel protein, and its main ligand, extracellular ATP, have recently been found to play an important role in promoting CD11b+ CD206+ P2x7r+ macrophage-mediated myocardial fibrosis during mouse chronic cardiac transplantation rejection [17] (Fig. 2C). In inflammatory tissues at the chronic graft rejection site, it remains unclear whether neutrophils produce a large number of extracellular ATP and promote the accumulation of M2 macrophages in allografts through the ATP/P2x7r axis.
Neutrophil knockout alone does not significantly improve fibrosis caused by chronic rejection [67]. In a mouse acellular nerve allograft (ANA) model, systemic depletion of recipient-derived macrophages (but not neutrophils) severely hindered angiogenesis and subsequent ANA nerve regeneration, suggesting that blood-derived macrophages were the main contributors to angiogenesis in ANAs [104]. Further investigation is required to determine whether neutrophils secrete a large amount of CSF-1 or extracellular ATP for long periods, resulting in high M2 macrophage accumulation in the allograft in chronic organ transplant rejection, thus aggravating organ fibrosis.
Interaction with ILCs
In recent years, ILCs have been newly identified with innate cell-like characteristics [105,106,107] and found to play several important roles in the innate immune system [108]. ILCs are classified into three main groups: group 1 (ILC1s and NK cells), group 2 (ILC2s), and group 3 (ILC3s), which correspond to Th1 (NK cells correspond to CD8+ cytotoxic T cells), Th2, and Th17 cells, respectively [106]. They lack conventional antigen receptors, instead identifying non-specific danger signals and cytokines [105].
ILCs reside in a variety of tissues and can proliferate and enrich regionally[109, 110], which helps the innate immune system to quickly initiate defensive responses and participate in tissue repair and homeostasis [111]. In pathological settings, ILCs can be supplemented from bone marrow or lymphoid organ precursors, in addition to local proliferation [109, 112].
ILCs are implicated in chronic inflammation, autoimmunity, and cancer [113,114,115,116]. In the field of organ transplantation, ILCs are categorized as donor-derived or recipient-derived ILCs. In human bone marrow transplantation, a high number of donor-derived ILCs tend to proliferate and are related to GVHD development [117]. In mouse liver and heart allograft, the infiltration of recipient cells increased, and the depletion of donor lymphocytes occurred on the 28th day after transplantation. Moreover, conventional NK cells and ILC1s in grafts showed an increasing trend and were mainly derived from the recipient; however, changes in ILC2s and ILC3s have not been verified further at the late transplantation period [2]. The number and the function of ILCs in the recipient’s circulation are reportedly unaffected by immunosuppressive drugs [91]. Although ILCs only account for a small proportion of graft immune cells, they likely play an important role in chronic allograft rejection.
Interaction with NK cells and ILC1s
Together with ILC1s, NK cells constitute group 1 ILCs, which are characterized by their capacity to produce IFN-γ and their functional dependence on the transcription factor T-bet. Human donor lung tissue with a high level of ILC1s before transplantation showed no occurrence of PGD after transplantation [118], suggesting that ILC1s may promote early transplantation tolerance. The number of ILC1s and NK cells was reportedly decreased in the early stages of mouse heart and liver allotransplantation, but that from D28 recipients was increased [2], suggesting that recipient-derived group 1 ILCs may be involved in the chronic rejection process. The role of ILC1s and NK cells in chronic allograft fibrosis, particularly with respect to macrophages, is still not well understood.
In a human bronchial asthma model experiment, ILC1s co-cultured with alveolar macrophages (AMs) induced M1 macrophage-related gene expression [119], which may be related to the secretion of IFN-γ by ILC1s. The mechanism underlying the interaction between macrophages and ILC1s during allograft fibrosis has not been reported. ILC1s may inhibit allograft fibrosis by secreting IFN-γ to inhibit MMT (Fig. 2D). NK cells have dual immunomodulatory effects after lung, kidney, skin, and heart transplantation [120,121,122,123,124]. After 3 to 4 days of mouse allogeneic skin grafts, the mRNA expression of monocyte chemotactic cytokines increased, and after treatment with anti-NK1.1 antibody, the mRNA levels of these cytokines in the grafts were downregulated by nearly 70% [123]. Hence, the presence of NK cells in the graft is considered to contribute to recipient-derived macrophage accumulation.
Interaction with ILC2s
ILC2s express the transcription factor GATA3, mainly secrete cytokines, such as IL-4, IL-5, and IL-13, and play an important role in tissue repair [124]. ILC2s were found to decrease in the early post-transplant period after lung, heart, and liver transplantation [2, 118], which may impair the development of immune tolerance. In human islet cell transplantation, IL-10 secreted by ILC2s promoted graft tolerance [125]. At present, the mechanism underlying the role of ILC2s in chronic graft rejection is not clear.
In a human bronchial asthma model experiment, ILC2s co-cultured with AMs induced M2 macrophage-related gene expression [119], which may be related to the secretion of IL-4 and IL-13 by ILC2s. It has been reported that ILC2s secrete G-CSF in addition to IL-5, IL-13, and Areg [126]. Further, G-CSF can reportedly increase the M2 macrophage ratio during donor bone marrow transplantation and prevent the occurrence of GVHD [127]. Mast cells interact with ILC2s via the IL-33/IL-13 axis after lung transplantation to promote M2 macrophage accumulation during allograft fibrosis [19]. Hence, the secretion of IL-4, IL-13, and G-CSF by ILC2s in the allograft is considered to contribute to the massive accumulation of recipient M2 macrophages and MMT (Fig. 2D).
Interaction with ILC3s
ILC3s express the transcription factor RORγt; secrete IL-17, IL-22, and GM-CSF under the induction of IL-2, IL-7, IL-1β, and IL-23; induce the formation of tertiary lymphoid tissue; and influence tissue defense and inflammation responses [128,129,130]. ILC3s consist of two distinct subsets based on cell surface expression of natural cytotoxicity receptors (NCRs), such as NKp44 in humans and NKp46 in mice [131]. NCR+ ILC3s produce mainly IL-22, whereas NCR− ILC3s produce IL-17 and limited amounts of IL-22 [132]. In recent years, ILC3s have been reported to promote chronic inflammation and fibrosis [105, 110, 133, 134]. Nevertheless, the role of ILC3s in chronic graft rejection remains unclear.
IL-22 is a key molecule involved in tissue repair and mucosal defense. Because IL-22R is expressed in fibroblasts and myofibroblasts, IL-22 signal transduction can promote the process of fibrosis [134, 135]. IL-22 overexpression promotes the fibroblast response to TNF and the activation of pro-inflammatory fibroblasts [136]. IL-22 promotes fibrosis by enhancing the role of TGFβ [134, 137]. In a study of mouse lung transplantation model, ILC3s were the main cell group secreting IL-22, which leads to the formation of tertiary lymphoid tissue in the allograft [138]. Increased ILC3s after human skin grafting can induce psoriasis, with 78% of ILC3s expressing IL-22 and 7–13% ILC3s expressing both IL-17 and IL-22 [20]. After human intestinal transplantation, ILC3 decreased in the early stage, and the secretion of IL-22 by NKp44+ ILC3s in the graft showed an increasing trend one month later [21, 139]. In particular, the proportion of ILC3s in grafts with chronic rejection was relatively high over a long period after intestinal transplantation [140]. Therefore, IL22+ ILC3s may promote graft fibrosis during chronic allograft rejection.
The main effector cells of chronic allograft rejection are recipient-derived macrophages. It has been reported that CX3CR1+ mononuclear phagocytes maintain the proliferation and survival of ILC3s by secreting IL-23 and IL-1β and promote the secretion of IL-22 by ILC3s, thus accelerating fibroblast proliferation and fibrosis [134, 141,142,143]. M1 macrophages have been reported to secrete IL-23 and IL-1β to promote CXCR5 expression by ILC3s, which migrate to the lungs along the CXCR5/CXCL13 axis and promote the formation of pulmonary granuloma by secreting IL-22 and IL-17 [112]. However, the mechanism underlying the interaction between macrophages and ILC3s in the allograft microenvironment has not been reported. Although the proportion of ILC3s in immune cells is low, its function cannot be underestimated. ILC3s induce the formation of a so-called battlefield for graft rejection by promoting the formation of tertiary lymphoid tissue, and their survival is supported by macrophages.
Conclusion and perspectives
The human immune system has not evolved to account for organ transplantation. As organ transplants constitute an artificial intervention for treating patients with end-stage diseases, immune networks do not follow a natural disease response course and instead exhibit highly specific response mechanisms. In this unique pathological microenvironment, source immune cells respond to the invasion of external cells, while recipient-derived immune cells are reprogrammed [2, 144,145,146]. For example, MMT occurs in the allograft, and cytokines that should not be secreted by cells are secreted in the transplant microenvironment, for example, the secretion of large amounts of CSF-1 by neutrophils after allotransplantation [16]. ILC3s secrete high levels of IL-22 after human intestinal and mouse lung allotransplantation [21, 138, 139]. Such distinct and critical immune responses constitute an interesting obstacle in organ transplant immunology.
Immunosuppressants currently used in clinical practice mainly act on the molecular before and after the immune response of CD4+ T cells, but the long-term survival rate of grafts after organ transplantation is still disappointing. There is currently a lack of clinical inhibitors against innate immune rejection. This review focused on chronic rejection after organ transplantation. The findings outlined above indicate that recipient-derived CX3CR1high macrophages transformed to myofibroblasts via MMT play a critical role in chronic allograft fibrosis. B cells, CD4+ T cells, neutrophils, and ILC2s participate in the occurrence of MMT by secreting cytokines (Fig. 2). Potential strategies for inhibiting the accumulation of CX3CR1high macrophages in allografts should be developed to prevent and treat allograft fibrosis. According to this review, CSF-1 is the key cytokine promoting the transformation of monocytes into recipient-derived CX3CR1high macrophages in grafts. Moreover, small-molecule kinase inhibitors and neutralizing antibodies against CSF-1R suppressed the accumulation of M2 macrophages and inhibited organ fibrosis, without affecting the function of other immune cells [62, 67, 147,148,149]. Targeting CSF-1/CSF-1R axis may allow for a more selective method for inhibiting allograft fibrosis.
However, the phenomenon of the secretion of a large amount of IL-22 by ILC3s during chronic transplant rejection cannot be ignored. IL-22 promotes fibrosis by enhancing the role of TGFβ [134] and may be involved in the proliferation of myofibroblasts and the occurrence of MMT. It has been reported that IL-22 can inhibit rejection in the early stage after transplantation [138]. However, in the late stage of chronic transplantation rejection, IL-22+ ILC3 may have a fibrotic effect on transplanted organs, which needs to be proved through a large number of studies. In the future, the proportion of ILC3s or levels of IL-22 in the peripheral blood or near allografts can be measured to determine the time of initiation of organ fibrosis and the time to administer anti-IL-22 drugs. This holds promise for the prevention and treatment of fibrosis after organ transplantation.
Availability of data and materials
Not applicable.
References
Chambers DC, Cherikh WS, Harhay MO, Hayes D Jr, Hsich E, Khush KK et al (2019) The international thoracic organ transplant registry of the international society for heart and lung transplantation: thirty-sixth adult lung and heart-lung transplantation report-2019; focus theme: donor and recipient size match. J Heart Lung Transplant 38:1042–1055. https://doi.org/10.1016/j.healun.2019.08.001
Prosser A, Huang WH, Liu L, Dart S, Watson M, de Boer B et al (2021) Dynamic changes to tissue-resident immunity after MHC-matched and MHC-mismatched solid organ transplantation. Cell Rep 35:109141. https://doi.org/10.1016/j.celrep.2021.109141
Zhang H, Li Z, Li W (2021) M2 macrophages serve as critical executor of innate immunity in chronic allograft rejection. Front Immunol 12:648539. https://doi.org/10.3389/fimmu.2021.648539
van den Bosch TP, Kannegieter NM, Hesselink DA, Baan CC, Rowshani AT (2017) Targeting the monocyte-macrophage lineage in solid organ transplantation. Front Immunol 8:153. https://doi.org/10.3389/fimmu.2017.00153
Li Q, Lan P (2023) Activation of immune signals during organ transplantation. Signal Transduct Target Ther 8:110. https://doi.org/10.1038/s41392-023-01377-9
Sacreas A, Yang JYC, Vanaudenaerde BM, Sigdel TK, Liberto JM, Damm I et al (2018) The common rejection module in chronic rejection post lung transplantation. PLoS One 13:e0205107. https://doi.org/10.1371/journal.pone.0205107
Gieseck RL, Wilson MS, Wynn TA (2017) Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18:62–76. https://doi.org/10.1038/nri.2017.90
Siu JHY, Surendrakumar V, Richards JA, Pettigrew GJ (2018) T cell allorecognition pathways in solid organ transplantation. Front Immunol 9:2548. https://doi.org/10.3389/fimmu.2018.02548
Dai H, Lan P, Zhao D, Abou-Daya K, Liu W, Chen W et al (2020) PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science 368:1122–1127. https://doi.org/10.1126/science.aax4040
Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD et al (2014) Non-self recognition by monocytes initiates allograft rejection. J Clin Invest 124:3579–3589. https://doi.org/10.1172/JCI74370
Liu W, Xiao X, Demirci G, Madsen J, Li XC (2012) Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol 188:2703–2711. https://doi.org/10.4049/jimmunol.1102997
Dai H, Friday AJ, Abou-Daya KI, Williams AL, Mortin-Toth S, Nicotra ML et al (2017) Donor SIRPalpha polymorphism modulates the innate immune response to allogeneic grafts. Sci Immunol. https://doi.org/10.1126/sciimmunol.aam6202
Ochando J, Ordikhani F, Boros P, Jordan S (2019) The innate immune response to allotransplants: mechanisms and therapeutic potentials. Cell Mol Immunol 16:350–356. https://doi.org/10.1038/s41423-019-0216-2
Domínguez-Andrés J, Netea MG (2020) The specifics of innate immune memory. Science 368:1052–1053. https://doi.org/10.1126/science.abc2660
Fu J, Lehmann CHK, Wang X, Wahlbuhl M, Allabauer I, Wilde B, Amon L, Dolff S, Cesnjevar R, Kribben A, Woelfle J, Rascher W, Hoyer PF, Dudziak D, Witzke O, Hoerning A (2021) CXCR4 blockade reduces the severity of murine heart allograft rejection by plasmacytoid dendritic cell-mediated immune regulation. Sci Rep 11(1):23815. https://doi.org/10.1038/s41598-021-03115-z
Braza MS, Conde P, Garcia M, Cortegano I, Brahmachary M, Pothula V et al (2018) Neutrophil derived CSF1 induces macrophage polarization and promotes transplantation tolerance. Am J Transplant 18:1247–1255. https://doi.org/10.1111/ajt.14645
Wu C, Zhao Y, Xiao X, Fan Y, Kloc M, Liu W et al (2016) Graft-infiltrating macrophages adopt an M2 phenotype and are inhibited by purinergic receptor P2X7 antagonist in chronic rejection. Am J Transplant 16:2563–2573. https://doi.org/10.1111/ajt.13808
Zhu J, Wang Y, Li J, Das P, Zhang H, Passang T et al (2022) Donor plasmacytoid dendritic cells limit graft-versus-host disease through vasoactive intestinal polypeptide expression. Blood 140:1431–1447. https://doi.org/10.1182/blood.2021012561
Mortaz E, Amani S, Mumby S, Adcock IM, Movassaghi M, Folkerts J et al (2018) Role of mast cells and type 2 innate lymphoid (ILC2) cells in lung transplantation. J Immunol Res 2018:2785971. https://doi.org/10.1155/2018/2785971
Keren A, Shemer A, Ginzburg A, Ullmann Y, Schrum AG, Paus R et al (2018) Innate lymphoid cells 3 induce psoriasis in xenotransplanted healthy human skin. J Allergy Clin Immunol 142:305–8 e6. https://doi.org/10.1016/j.jaci.2018.02.015
Kang J, Loh K, Belyayev L, Cha P, Sadat M, Khan K et al (2021) Type 3 innate lymphoid cells are associated with a successful intestinal transplant. Am J Transplant 21:787–797. https://doi.org/10.1111/ajt.16163
Di Campli M, Azouz A, Assabban A, Scaillet J, Splittgerber M, Van Keymeulen A et al (2021) The mononuclear phagocyte system contributes to fibrosis in post-transplant obliterans bronchiolitis. Eur Respir J. https://doi.org/10.1183/13993003.00344-2020
Wang Y-Y, Jiang H, Pan J, Huang X-R, Wang Y-C, Huang H-F et al (2017) Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol 28:2053–2067. https://doi.org/10.1681/asn.2016050573
Pascual-Gil S, Epelman S (2018) Monocyte-derived macrophages: the missing link in organ transplantation. Immunity 49:783–785. https://doi.org/10.1016/j.immuni.2018.11.005
Mildner A, Yona S, Jung S (2013) A close encounter of the third kind: monocyte-derived cells. Adv Immunol 120:69–103. https://doi.org/10.1016/b978-0-12-417028-5.00003-x
Goudot C, Coillard A, Villani A, Gueguen P, Cros A, Sarkizova S et al (2017) Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity 47:582–96.e6. https://doi.org/10.1016/j.immuni.2017.08.016
Bakri Y, Sarrazin S, Mayer UP, Tillmanns S, Nerlov C, Boned A et al (2005) Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood 105:2707–2716. https://doi.org/10.1182/blood-2004-04-1448
Lakkis FG, Li XC (2018) Innate allorecognition by monocytic cells and its role in graft rejection. Am J Transplant 18:289–292. https://doi.org/10.1111/ajt.14436
Menon MC, Heeger PS (2017) Donor SIRP-alpha polymorphisms: widening the innate-to-adaptive continuum in allograft rejection. Kidney Int 92:1305–1308. https://doi.org/10.1016/j.kint.2017.10.006
Takizawa H, Manz M (2007) Macrophage tolerance: CD47-SIRP-alpha-mediated signals matter. Nat Immunol 8:1287–1289. https://doi.org/10.1038/ni1207-1287
Pengam S, Durand J, Usal C, Gauttier V, Dilek N, Martinet B et al (2019) SIRPalpha/CD47 axis controls the maintenance of transplant tolerance sustained by myeloid-derived suppressor cells. Am J Transplant 19:3263–3275. https://doi.org/10.1111/ajt.15497
Christen T, Nahrendorf M, Wildgruber M, Swirski FK, Aikawa E, Waterman P et al (2009) Molecular imaging of innate immune cell function in transplant rejection. Circulation 119:1925–1932. https://doi.org/10.1161/CIRCULATIONAHA.108.796888
Poulter L, Bradley N, Turk J (1971) The role of macrophages in skin allograft rejection. I. Histochemical studies during first-set rejection. Transplantation 12:40–44. https://doi.org/10.1097/00007890-197107000-00006
Eardley KS, Zehnder D, Quinkler M, Lepenies J, Bates RL, Savage CO et al (2006) The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int 69:1189–1197. https://doi.org/10.1038/sj.ki.5000212
Croker B, Clapp W, Abu Shamat A, Kone B, Peterson J (1996) Macrophages and chronic renal allograft nephropathy. Kidney Int Suppl 57:S42–S49
Bleriot C, Chakarov S, Ginhoux F (2020) Determinants of resident tissue macrophage identity and function. Immunity 52:957–970. https://doi.org/10.1016/j.immuni.2020.05.014
Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M (2015) Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 42:145–158. https://doi.org/10.1016/j.immuni.2014.12.020
Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y et al (2019) Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity 51:655–70 e8. https://doi.org/10.1016/j.immuni.2019.09.002
Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A et al (2017) Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol 322:74–83. https://doi.org/10.1016/j.cellimm.2017.10.006
Liu Y, Kloc M, Li XC (2016) Macrophages as effectors of acute and chronic allograft injury. Curr Transplant Rep 3:303–312. https://doi.org/10.1007/s40472-016-0130-9
Tang PM, Nikolic-Paterson DJ, Lan HY (2019) Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol 15:144–158. https://doi.org/10.1038/s41581-019-0110-2
Pakshir P, Noskovicova N, Lodyga M, Son DO, Schuster R, Goodwin A et al (2020) The myofibroblast at a glance. J Cell Sci. https://doi.org/10.1242/jcs.227900
Ikezumi Y, Suzuki T, Yamada T, Hasegawa H, Kaneko U, Hara M et al (2015) Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatr Nephrol 30:1007–1017. https://doi.org/10.1007/s00467-014-3023-0
LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C et al (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19:1047–1053. https://doi.org/10.1038/nm.3218
van Amerongen MJ, Bou-Gharios G, Popa E, van Ark J, Petersen AH, van Dam GM et al (2008) Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol 214:377–386. https://doi.org/10.1002/path.2281
Suga H, Rennert RC, Rodrigues M, Sorkin M, Glotzbach JP, Januszyk M et al (2014) Tracking the elusive fibrocyte: identification and characterization of collagen-producing hematopoietic lineage cells during murine wound healing. Stem Cells 32:1347–1360. https://doi.org/10.1002/stem.1648
Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W et al (2016) Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest 126:3063–3079. https://doi.org/10.1172/jci85328
Yang F, Chang Y, Zhang C, Xiong Y, Wang X, Ma X et al (2021) UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int Immunopharmacol. https://doi.org/10.1016/j.intimp.2021.107396
Haider N, Bosca L, Zandbergen HR, Kovacic JC, Narula N, Gonzalez-Ramos S et al (2019) Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J Am Coll Cardiol 74:3124–3135. https://doi.org/10.1016/j.jacc.2019.10.036
Wei J, Xu Z, Yan X (2022) The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front Immunol. https://doi.org/10.3389/fimmu.2022.934377
Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, et al. (2016) TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 7:8809–8822. https://doi.org/10.18632/oncotarget.6604
Panzer SE (2022) Macrophages in transplantation: a matter of plasticity, polarization, and diversity. Transplantation 106:257–267. https://doi.org/10.1097/TP.0000000000003804
Wang H, Xi Z, Deng L, Pan Y, He K, Xia Q (2021) Macrophage polarization and liver ischemia-reperfusion injury. Int J Med Sci 18:1104–1113. https://doi.org/10.7150/ijms.52691
Ordikhani F, Pothula V, Sanchez-Tarjuelo R, Jordan S, Ochando J (2020) Macrophages in organ transplantation. Front Immunol. https://doi.org/10.3389/fimmu.2020.582939
Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11:750–761. https://doi.org/10.1038/nri3088
Chen S, Lakkis FG, Li XC (2020) The many shades of macrophages in regulating transplant outcome. Cell Immunol. https://doi.org/10.1016/j.cellimm.2020.104064
Gkika E, Vach W, Adebahr S, Schimek-Jasch T, Brenner A, Brunner T et al (2017) Is serum level of CC chemokine ligand 18 a biomarker for the prediction of radiation induced lung toxicity (RILT)? PloS one 12:e0185350. https://doi.org/10.1371/journal.pone.0185350
Wynn TA, Barron L (2010) Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30:245–257. https://doi.org/10.1055/s-0030-1255354
Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseno CG et al (2016) Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat Commun 7:12597. https://doi.org/10.1038/ncomms12597
MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC et al (2010) An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 116:3955–3963. https://doi.org/10.1182/blood-2010-02-266296
Hume DA, MacDonald KP (2012) Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119:1810–1820. https://doi.org/10.1182/blood-2011-09-379214
Jose M, Le Meur Y, Atkins R, Chadban S (2003) Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg 3:294–300. https://doi.org/10.1034/j.1600-6143.2003.00068.x
Boersema M, van den Born JC, van Ark J, Harms G, Seelen MA, van Dijk MC et al (2015) CD16(+) monocytes with smooth muscle cell characteristics are reduced in human renal chronic transplant dysfunction. Immunobiology 220:673–683. https://doi.org/10.1016/j.imbio.2014.11.011
Ishida Y, Kimura A, Nosaka M, Kuninaka Y, Hemmi H, Sasaki I et al (2017) Essential involvement of the CX3CL1-CX3CR1 axis in bleomycin-induced pulmonary fibrosis via regulation of fibrocyte and M2 macrophage migration. Sci Rep 7:16833. https://doi.org/10.1038/s41598-017-17007-8
Liang H, Liu B, Gao Y, Nie J, Feng S, Yu W et al (2022) Jmjd3/IRF4 axis aggravates myeloid fibroblast activation and m2 macrophage to myofibroblast transition in renal fibrosis. Front Immunol. https://doi.org/10.3389/fimmu.2022.978262
Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y et al (2016) Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis 7:e2495. https://doi.org/10.1038/cddis.2016.402
Doloff JC, Veiseh O, Vegas AJ, Tam HH, Farah S, Ma M et al (2017) Colony stimulating factor-1 receptor is a central component of the foreign body response to biomaterial implants in rodents and non-human primates. Nat Mater 16:671–680. https://doi.org/10.1038/nmat4866
Moore C, Gao B, Roskin K, Vasilescu E, Addonizio L, Givertz M et al (2020) B cell clonal expansion within immune infiltrates in human cardiac allograft vasculopathy. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg 20:1431–1438. https://doi.org/10.1111/ajt.15737
Li W, Gauthier JM, Higashikubo R, Hsiao HM, Tanaka S, Vuong L et al (2019) Bronchus-associated lymphoid tissue-resident Foxp3+ T lymphocytes prevent antibody-mediated lung rejection. J Clin Invest 129:556–568. https://doi.org/10.1172/JCI122083
Lodhi SA, Lamb KE, Meier-Kriesche HU (2011) Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant 11:1226–1235. https://doi.org/10.1111/j.1600-6143.2011.03539.x
Loupy A, Hill GS, Jordan SC (2012) The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol 8:348–357. https://doi.org/10.1038/nrneph.2012.81
Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG et al (2009) Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant 9:2520–2531. https://doi.org/10.1111/j.1600-6143.2009.02799.x
Bergler T, Jung B, Bourier F, Kuhne L, Banas MC, Rummele P et al (2016) Infiltration of macrophages correlates with severity of allograft rejection and outcome in human kidney transplantation. PLoS One 11:e0156900. https://doi.org/10.1371/journal.pone.0156900
Xu L, Collins J, Drachenberg C, Kukuruga D, Burke A (2014) Increased macrophage density of cardiac allograft biopsies is associated with antibody-mediated rejection and alloantibodies to HLA antigens. Clin Transplant 28:554–560. https://doi.org/10.1111/ctr.12348
von der Thusen JH, Vandermeulen E, Vos R, Weynand B, Verbeken EK, Verleden SE (2018) The histomorphological spectrum of restrictive chronic lung allograft dysfunction and implications for prognosis. Mod Pathol 31:780–790. https://doi.org/10.1038/modpathol.2017.180
Vandermeulen E, Lammertyn E, Verleden SE, Ruttens D, Bellon H, Ricciardi M et al (2017) Immunological diversity in phenotypes of chronic lung allograft dysfunction: a comprehensive immunohistochemical analysis. Transpl Int 30:134–143. https://doi.org/10.1111/tri.12882
Morrell MR, Pilewski JM, Gries CJ, Pipeling MR, Crespo MM, Ensor CR et al (2014) De novo donor-specific HLA antibodies are associated with early and high-grade bronchiolitis obliterans syndrome and death after lung transplantation. J Heart Lung Transplant 33:1288–1294. https://doi.org/10.1016/j.healun.2014.07.018
Misumi K, Wheeler DS, Aoki Y, Combs MP, Braeuer RR, Higashikubo R et al (2020) Humoral immune responses mediate the development of a restrictive phenotype of chronic lung allograft dysfunction. JCI Insight. https://doi.org/10.1172/jci.insight.136533
Smirnova NF, Conlon TM, Morrone C, Dorfmuller P, Humbert M, Stathopoulos GT et al (2019) Inhibition of B cell-dependent lymphoid follicle formation prevents lymphocytic bronchiolitis after lung transplantation. JCI Insight. https://doi.org/10.1172/jci.insight.123971
Peng X, Yong Z, Xiaoyan W, Yuanshan C, Guangzhu W, Xuehuan L (2021) Mechanism of graft damage caused by NTPDase1-activated macrophages in acute antibody-mediated rejection. Transplant Proc 53:436–442. https://doi.org/10.1016/j.transproceed.2020.06.033
Wei X, Valenzuela NM, Rossetti M, Sosa RA, Nevarez-Mejia J, Fishbein GA et al (2020) Antibody-induced vascular inflammation skews infiltrating macrophages to a novel remodeling phenotype in a model of transplant rejection. Am J Transplant 20:2686–2702. https://doi.org/10.1111/ajt.15934
Wheeler D, Misumi K, Walker N, Vittal R, Combs M, Aoki Y et al (2021) Interleukin 6 trans-signaling is a critical driver of lung allograft fibrosis. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg 21:2360–2371. https://doi.org/10.1111/ajt.16417
Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483. https://doi.org/10.1146/annurev.immunol.021908.132532
Chu Z, Sun C, Sun L, Feng C, Yang F, Xu Y et al (2020) Primed macrophages directly and specifically reject allografts. Cell Mol Immunol 17:237–246. https://doi.org/10.1038/s41423-019-0226-0
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T et al (2002) Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415:536–541. https://doi.org/10.1038/415536a
Wynn TA (2004) Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol 4:583–594. https://doi.org/10.1038/nri1412
Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040. https://doi.org/10.1038/nm.2807
Ulloa L, Doody J, Massagué J (1999) Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397:710–713. https://doi.org/10.1038/17826
Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A (2003) Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Investig 112:1486–1494. https://doi.org/10.1172/jci200319270
Chen X, Chen Y, Wang Z, Dong Z, Yao Y, Li Y et al (2022) Adipose-derived stem cells regulate CD4+ T-cell-mediated macrophage polarization and fibrosis in fat grafting in a mouse model. Heliyon 8:e11538. https://doi.org/10.1016/j.heliyon.2022.e11538
Gomez-Massa E, Talayero P, Utrero-Rico A, Laguna-Goya R, Andres A, Mancebo E et al (2020) Number and function of circulatory helper innate lymphoid cells are unaffected by immunosuppressive drugs used in solid organ recipients - a single centre cohort study. Transpl Int 33:402–413. https://doi.org/10.1111/tri.13567
Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M et al (2011) Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med 208:1069–1082. https://doi.org/10.1084/jem.20101709
Chu Z, Feng C, Sun C, Xu Y, Zhao Y (2021) Primed macrophages gain long-term specific memory to reject allogeneic tissues in mice. Cell Mol Immunol 18:1079–1081. https://doi.org/10.1038/s41423-020-00521-7
Jorch SK, Kubes P (2017) An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23:279–287. https://doi.org/10.1038/nm.4294
Sayah D, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A et al (2015) Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 191:455–463. https://doi.org/10.1164/rccm.201406-1086OC
Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L et al (2017) Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol 28:1753–1768. https://doi.org/10.1681/ASN.2016080925
Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J et al (2010) In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A 107:18073–18078. https://doi.org/10.1073/pnas.1008737107
Nakamura K, Kageyama S, Kupiec-Weglinski JW (2019) The evolving role of neutrophils in liver transplant ischemia-reperfusion injury. Curr Transplant Rep 6:78–89. https://doi.org/10.1007/s40472-019-0230-4
Scozzi D, Ibrahim M, Menna C, Krupnick AS, Kreisel D, Gelman AE (2017) The role of neutrophils in transplanted organs. Am J Transplant 17:328–335. https://doi.org/10.1111/ajt.13940
Tiriveedhi V, Banan B, Deepti S, Nataraju A, Hachem R, Trulock E et al (2014) Role of defensins in the pathogenesis of chronic lung allograft rejection. Hum Immunol 75:370–377. https://doi.org/10.1016/j.humimm.2013.12.014
Christoffersson G, Vagesjo E, Vandooren J, Liden M, Massena S, Reinert RB et al (2012) VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 120:4653–4662. https://doi.org/10.1182/blood-2012-04-421040
Sablik KA, Jordanova ES, Pocorni N, Clahsen-van Groningen MC, Betjes MGH (2019) Immune cell infiltrate in chronic-active antibody-mediated rejection. Front Immunol 10:3106. https://doi.org/10.3389/fimmu.2019.03106
Yang C, Lei L, Collins JWM, Briones M, Ma L, Sturdevant GL et al (2021) Chlamydia evasion of neutrophil host defense results in NLRP3 dependent myeloid-mediated sterile inflammation through the purinergic P2X7 receptor. Nat Commun 12:5454. https://doi.org/10.1038/s41467-021-25749-3
Pan D, Acevedo-Cintron JA, Sayanagi J, Snyder-Warwick AK, Mackinnon SE, Wood MD (2020) The CCL2/CCR2 axis is critical to recruiting macrophages into acellular nerve allograft bridging a nerve gap to promote angiogenesis and regeneration. Exp Neurol. https://doi.org/10.1016/j.expneurol.2020.113363
Eberl G, Colonna M, Di Santo JP, McKenzie AN (2015) Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. https://doi.org/10.1126/science.aaa6566
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G et al (2013) Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149. https://doi.org/10.1038/nri3365
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12:21–27. https://doi.org/10.1038/ni.1962
Cheng H, Jin C, Wu J, Zhu S, Liu YJ, Chen J (2017) Guards at the gate: physiological and pathological roles of tissue-resident innate lymphoid cells in the lung. Protein Cell 8:878–895. https://doi.org/10.1007/s13238-017-0379-5
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY (2015) Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350:981–985. https://doi.org/10.1126/science.aac9593
Eberl G, Di Santo JP, Vivier E (2015) The brave new world of innate lymphoid cells. Nat Immunol 16:1–5. https://doi.org/10.1038/ni.3059
Panda SK, Colonna M (2019) Innate lymphoid cells in mucosal immunity. Front Immunol 10:861. https://doi.org/10.3389/fimmu.2019.00861
Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A et al (2019) Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 570:528–532. https://doi.org/10.1038/s41586-019-1276-2
Hsu AT, Gottschalk TA, Tsantikos E, Hibbs ML (2021) The role of innate lymphoid cells in chronic respiratory diseases. Front Immunol. https://doi.org/10.3389/fimmu.2021.733324
Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H (2017) Innate lymphoid cells in autoimmunity: emerging regulators in rheumatic diseases. Nat Rev Rheumatol 13:164–173. https://doi.org/10.1038/nrrheum.2016.218
Bruchard M, Ghiringhelli F (2019) Deciphering the roles of innate lymphoid cells in cancer. Front Immunol 10:656. https://doi.org/10.3389/fimmu.2019.00656
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K et al (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14:221–229. https://doi.org/10.1038/ni.2534
Quatrini L, Tumino N, Moretta F, Besi F, Vacca P, Moretta L (2020) Helper innate lymphoid cells in allogenic hematopoietic stem cell transplantation and graft versus host disease. Front Immunol. https://doi.org/10.3389/fimmu.2020.582098
Monticelli LA, Diamond JM, Saenz SA, Tait Wojno ED, Porteous MK, Cantu E et al (2020) Lung innate lymphoid cell composition is altered in primary graft dysfunction. Am J Respir Crit Care Med 201:63–72. https://doi.org/10.1164/rccm.201906-1113OC
Kim J, Chang Y, Bae B, Sohn KH, Cho SH, Chung DH et al (2019) Innate immune crosstalk in asthmatic airways: Innate lymphoid cells coordinate polarization of lung macrophages. J Allergy Clin Immunol 143:1769–82 e11. https://doi.org/10.1016/j.jaci.2018.10.040
Calabrese DR, Lanier LL, Greenland JR (2019) Natural killer cells in lung transplantation. Thorax 74:397–404. https://doi.org/10.1136/thoraxjnl-2018-212345
Pontrelli P, Rascio F, Castellano G, Grandaliano G, Gesualdo L, Stallone G (2020) The role of natural killer cells in the immune response in kidney transplantation. Front Immunol 11:1454. https://doi.org/10.3389/fimmu.2020.01454
Zecher D, Li Q, Oberbarnscheidt MH, Demetris AJ, Shlomchik WD, Rothstein DM et al (2010) NK cells delay allograft rejection in lymphopenic hosts by downregulating the homeostatic proliferation of CD8+ T cells. J Immunol 184:6649–6657. https://doi.org/10.4049/jimmunol.0903729
Kondo T, Morita K, Watarai Y, Auerbach M, Taub D, Novick A et al (2000) Early increased chemokine expression and production in murine allogeneic skin grafts is mediated by natural killer cells. Transplantation 69:969–977. https://doi.org/10.1097/00007890-200003150-00051
Prosser AC, Kallies A, Lucas M (2018) Tissue-resident lymphocytes in solid organ transplantation: innocent passengers or the key to organ transplant survival? Transplantation 102:378–386. https://doi.org/10.1097/TP.0000000000002001
Huang Q, Ma X, Wang Y, Niu Z, Wang R, Yang F, et al (2020) IL-10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol Med 12:e12305. https://doi.org/10.15252/emmm.202012305
Chen WY, Wu YH, Tsai TH, Li RF, Lai AC, Li LC et al (2021) Group 2 innate lymphoid cells contribute to IL-33-mediated alleviation of cardiac fibrosis. Theranostics 11:2594–2611. https://doi.org/10.7150/thno.51648
Wen Q, Kong Y, Zhao HY, Zhang YY, Han TT, Wang Y et al (2019) G-CSF-induced macrophage polarization and mobilization may prevent acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 54:1419–1433. https://doi.org/10.1038/s41409-019-0449-9
Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y et al (2013) A T-bet gradient controls the fate and function of CCR6−RORγt+ innate lymphoid cells. Nature 494:261–265. https://doi.org/10.1038/nature11813
Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F et al (2008) Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29:958–970. https://doi.org/10.1016/j.immuni.2008.11.001
Sonnenberg GF, Fouser LA, Artis D (2011) Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12:383–390. https://doi.org/10.1038/ni.2025
Kruse PH, Matta J, Ugolini S, Vivier E (2014) Natural cytotoxicity receptors and their ligands. Immunol Cell Biol 92:221–229. https://doi.org/10.1038/icb.2013.98
Mjosberg J, Spits H (2016) Human innate lymphoid cells. J Allergy Clin Immunol 138:1265–1276. https://doi.org/10.1016/j.jaci.2016.09.009
Sonnenberg GF, Artis D (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21:698–708. https://doi.org/10.1038/nm.3892
Mathur R, Alam MM, Zhao XF, Liao Y, Shen J, Morgan S et al (2019) Induction of autophagy in Cx3cr1(+) mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol 12:612–623. https://doi.org/10.1038/s41385-019-0146-4
Rutz S, Eidenschenk C, Ouyang W (2013) IL-22, not simply a Th17 cytokine. Immunol Rev 252:116–32. https://doi.org/10.1111/imr.12027
Brembilla NC, Dufour AM, Alvarez M, Hugues S, Montanari E, Truchetet ME et al (2016) IL-22 capacitates dermal fibroblast responses to TNF in scleroderma. Ann Rheum Dis 75:1697–1705. https://doi.org/10.1136/annrheumdis-2015-207477
Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP et al (2018) Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol. https://doi.org/10.1126/sciimmunol.aar7754
Tanaka S, Gauthier JM, Fuchs A, Li W, Tong AY, Harrison MS et al (2019) IL-22 is required for the induction of bronchus-associated lymphoid tissue in tolerant lung allografts. Am J Transplant 20:1251–1261. https://doi.org/10.1111/ajt.15701
Talayero P, Mancebo E, Calvo-Pulido J, Rodriguez-Munoz S, Bernardo I, Laguna-Goya R et al (2016) Innate lymphoid cells groups 1 and 3 in the epithelial compartment of functional human intestinal allografts. Am J Transplant 16:72–82. https://doi.org/10.1111/ajt.13435
Weiner J, Zuber J, Shonts B, Yang S, Fu J, Martinez M et al (2017) Long-term persistence of innate lymphoid cells in the gut after intestinal transplantation. Transplantation 101:2449–2454. https://doi.org/10.1097/TP.0000000000001593
Bauche D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM et al (2018) LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity 49:342–52 e5. https://doi.org/10.1016/j.immuni.2018.07.007
Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER et al (2014) CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med 211:1571–1583. https://doi.org/10.1084/jem.20140678
Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU et al (2013) CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol 6:177–188. https://doi.org/10.1038/mi.2012.61
Pallett LJ, Burton AR, Amin OE, Rodriguez-Tajes S, Patel AA, Zakeri N et al (2020) Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J Exp Med. https://doi.org/10.1084/jem.20200050
de Leur K, Dieterich M, Hesselink DA, Corneth OBJ, Dor F, de Graav GN et al (2019) Characterization of donor and recipient CD8+ tissue-resident memory T cells in transplant nephrectomies. Sci Rep 9:5984. https://doi.org/10.1038/s41598-019-42401-9
Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E et al (2019) Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol. https://doi.org/10.1126/sciimmunol.aav5581
Deng X, Yang Q, Wang Y, Zhou C, Guo Y, Hu Z et al (2020) CSF-1R inhibition attenuates ischemia-induced renal injury and fibrosis by reducing Ly6C(+) M2-like macrophage infiltration. Int Immunopharmacol. https://doi.org/10.1016/j.intimp.2020.106854
Denny WA, Flanagan JU (2021) Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin Ther Pat 31:107–117. https://doi.org/10.1080/13543776.2021.1839414
Hume DA, Irvine KM, Pridans C (2019) The mononuclear phagocyte system: the relationship between monocytes and macrophages. Trends Immunol 40:98–112. https://doi.org/10.1016/j.it.2018.11.007
Funding
This work was supported by the National Key Research and Development Program (Grant number 2021YFF0704800), the National Natural Science Foundation of China (Grant number 82273186), the Scientific and Technological Developing Plan of Jilin Province (Grant number 20210402009GH and YDZJ202201ZYTS098), the Norman Bethune Program of Jilin University (Grant number 2022B34), and the Jilin Provincial Department of Education Scientific Research Project (JJKH20231213KJ).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. JC had the idea for the article and XL performed the literature search and data analysis. JC, JW, and SZ critically revised the work. The first draft of the manuscript was written by XL and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Wu, J., Zhu, S. et al. Intragraft immune cells: accomplices or antagonists of recipient-derived macrophages in allograft fibrosis?. Cell. Mol. Life Sci. 80, 195 (2023). https://doi.org/10.1007/s00018-023-04846-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04846-0