
Abstract
Rheumatoid arthritis (RA) is an autoimmune disease that mainly affects the joints but also leads to systemic inflammation. Auto-reactivity and dysregulation of self-tolerance are thought to play a vital role in disease onset. In the pathogenesis of autoimmune diseases, disturbed immunosuppressive properties of regulatory T cells contribute to the dysregulation of immune homeostasis. In RA patients, the functions of Treg cells and their frequency are reduced. Therefore, focusing on the re-establishment of self-tolerance by increasing Treg cell frequencies and preventing a loss of function is a promising strategy for the treatment of RA. This approach could be especially beneficial for those patients who do not respond well to current therapies. In this review, we summarize and discuss the current knowledge about the function, differentiation and regulation of Treg cells in RA patients and in animal models of autoimmune arthritis. In addition, we highlight the therapeutic potential as well as the challenges of Treg cell targeting treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disorder characterized by chronic inflammation in multiple joints, inducing synovitis, cartilage damage and bone erosion. Joint destruction can lead to disability, reduced life quality, reduced life expectancy, and a high burden on the healthcare systems [1, 2]. RA affects about 1% of the population worldwide, occurs at any age and affects women two to three times more often than men [3,4,5]. Currently, conventional synthetic disease-modifying antirheumatic drugs (DMARDs), targeted synthetic DMARDs, and biological DMARDs are used in clinical practice and can induce remission in many patients. However, in approximately 30% of the patients remission cannot be achieved and RA remains an incurable disease due to the complexity of its pathogenesis [6,7,8,9]. Thus, it is necessary to identify new therapeutic targets for the treatment of RA patients, especially for those who do not respond to current therapies.
In the past years, reports about the frequency of Treg cells in the peripheral blood and in the synovial fluid of RA patients have shown contradictory results [10,11,12]. The differences between the reported results might be due to different approaches used to identify Treg cells. Treg cells seem to be significantly decreased in the peripheral blood at the early stage of RA [13]. Moreover, Treg cells accumulate in the synovial fluid and synovial membrane of inflamed joints of RA patients [14]. The frequency of Treg cells in synovium, peripheral blood and synovial fluid might affect the cell contact-mediated suppressive function of Treg cells in RA [15]. Therefore, both the lack of Treg cells and impaired Treg cell functions contribute to the imbalance between effector T cells and regulatory T cells in RA. Importantly, in humans and mice with Foxp3 deficiency can lead to a proliferative autoimmune disorder, whereas the transfer of Foxp3+ Treg cells helps to prevent the development of a fatal lymphoproliferative syndrome with inflammation in many organs caused by the deficiency of foxp3 gene in mice [16]. Lentiviral overexpression of FoxP3 in CD4+ T cells from RA patients induces increased levels of CD25 and CTLA-4 and decreased levels of CD127 and TNF-alpha [17]. Considering the important role of Treg cells in immune homeostasis, reestablishment of self-tolerance by Treg cell therapy seems to be a promising approach to reduce autoimmunity in RA patients. In this review, we will discuss the current knowledge about the function, differentiation and regulation of Treg cells in RA and murine arthritis models In addition, we will highlight the therapeutic potential and challenges of Treg cells and the progress in this field.
Suppressive properties of regulatory T cells
Regulatory T cells are a CD4+ T cell subset that is characterized by expression of its master transcription factor Foxp3 (Forkhead box protein 3), high expression of IL-2 receptor (CD25) and low or negative expression of CD127 [18, 19]. In addition, a small subset of Foxp3 negative CD4+ T cells which is characterized by TGF-beta1 and IL-10 secretion has similar suppressive abilities as Foxp3+ Treg cells [20,21,22]. This subset is called type 1 regulatory T cells (Tr1 cells). However, the surface markers and transcription factors of this subset have not been fully identified [23]. To date, CD4+CD25highCD127lowFoxp3+ cells remain the most studied Treg group. Our review will, therefore, mainly focus on this cell type, which is crucial for the prevention of autoimmunity despite its low frequency in the peripheral blood [24,25,26,27].
Treg cell transcription factors
Foxp3 promotes the differentiation of naïve CD4+ T cells into Treg cells and is the most important transcription factor for the development and function of Treg cells [28]. Scurfy mice which are deficient for Foxp3 die early due to highly activated CD4+ T cells and overwhelming proinflammatory cytokine production [29]. Humans with a mutation in the Foxp3 gene can develop the autoimmune inflammatory syndrome IPEX which is characterized by immune dysregulation, polyendocrinopathy and enteropathy [30]. Furthermore, the overexpression of Foxp3 helps to increase the absolute number of Treg cells and CD4+CD25−Foxp3− T cells transfected with Foxp3 show immune suppressive properties and prevent autoimmunity in a mouse model This evidence indicates that the transcriptional factor Foxp3 is crucial for maintaining the suppressive activity of Treg cells, both in human and mice. However, it seems that Foxp3 is not the only gene required for the maintenance of Treg cell development and function. It has been shown that Helios enhances Treg cell function in cooperation with Foxp3. Helios increases the suppressive function of induced Treg cells and upregulates various Treg cell-related molecules [31]. Moreover, Zheng et al. reported an important role of conserved non-coding DNA sequence (CNS) elements at the Foxp3 locus in the determination of Treg cell frequency, stability and characteristics in mice [32]. Furthermore, CTLA4-Ig and vasoactive intestinal peptide (VIP) are also reported to play a role in the development of Treg cells [33, 34]. However, it remains elusive how these proteins work together to determine Treg cell development and suppressive activity.
Suppressive mechanisms
The suppressive activity of Treg cells is mediated by different mechanisms, including cytokine production, direct cell–cell contact suppression and the regulation of antigen-presenting cells (APCs), which induce effector T cells apoptosis and immunosuppression [35,36,37,38,39].
Cytokines produced by Treg cells
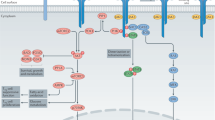
Treg cells produce a series of cytokines that contribute to their suppressive function. These cytokines include IL-10, TGF-beta and IL-35 (as shown in Fig. 1) [40,41,42,43,44]. In the DBA/1 mouse line, heterozygous (IL-10±) and homozygous (IL-10−/−) mice develop worse arthritis compared to wildtype (WT) mice following induction of collagen type II [45]. Similar results have been observed in C57BL/10 mice and IL-10 deficient mice [46]. Anti-IL-10 antibodies inhibit the expansion of Treg cells in mice with collagen-induced arthritis (CIA), whereas human IL-10 gene transduction can ameliorate the symptoms of established experimental autoimmune arthritis [47, 48]. In addition, it has been reported that Tr1 cells characterized by IL-10 production have the suppressive capacity in mice and humans [49]. Moreover, TGF-beta produced by Treg cells promotes their suppressive function, the expression of Foxp3 and immune homeostasis in vivo [50]. Treg cells also consistently express a high level of TGF-beta on their cell surface after stimulation, which contributes to the inhibition of effector T cells activation and proliferation in both, human and mice [51, 52]. TGF-beta1 produced also mediates immunosuppression by restricting the production of immunoglobulins (Ig) by B cell. These inhibitory effects can be disrupted by the treatment with anti-TGF-beta antibodies [52]. In addition, IL-35 is constitutively expressed in murine Treg cells and contributes to the inhibitory function of Treg cells [43]. Finally, Nakano et al. reported that IL-35 induces suppression of peripheral T cells in RA patients [53]. Taken together, various cytokines help Treg cells to exert maximal suppression of activated immune cells.

The mechanisms of immunosuppressive function mediated by Treg cells. Treg cells secret the cytokines TGF-beta, IL-10 and IL-35 to directly inhibit the activation and proliferation of effector T cells. CTLA-4, LAG-3 and PD1 on Treg cells mediate the downregulation of APC cell functions, which prevents the activation of naïve T cells and effector T cells. CD25 expressed on Treg cells outcompetes IL-2 which is necessary for T cell in the peripheral to prevent its activation and proliferation. Adenosine produced by the hydrolyzation of CD39 and CD73 from ATP or ADP binds to A2A receptor on effector T cells, thereby inhibiting the proliferation and production of inflammatory cytokines. Treg cells also mediate the cytolysis of effector T cells by granzymes and perforin
Cell–cell contact-mediated suppression
A two-step mode of cell contact-mediated suppression by Treg cells was proposed by Onishi et al.: First, leukocyte function-associated antigen-1 (LFA-1) dependent formation of Treg cells facilitates the interaction with immature dendritic cells, followed by subsequent upregulation of LFA-1 and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) on activated Treg cells. These molecules can interact with antigen-presenting dendritic cells (DC) and lead to reduced expression levels of CD80 and CD86 on DCs, thus preventing the activation and proliferation of antigen-reactive naïve T cells by decreasing the ability of APC both in vivo and in vitro [54]. Like CTLA-4, the transmembrane protein lymphocyte activation gene-3 (LAG-3) binds MHC II on antigen-presenting cells and inhibits DC activation through an ITAM-mediated inhibitory signaling pathway [55]. Furthermore, the CD28 superfamily member PD-1 binds to its ligands, thereby preventing proliferation and IFN-γ production in T cells [56]. Importantly, granzyme B expressing Treg cells have cytotoxic effects on effector T cells [57] and the apoptosis of effector T cells can be induced by Treg cells in a cell–cell contact-dependent manner [58]. In summary, these suppressive pathways work together to induce cell contact-dependent suppressive function of Treg cells (as shown in Fig. 1). Recently, it was reported that murine Treg cells also mediate suppressive functions through CD40L and P-selectin-dependent pathways during cell–cell contact-dependent inhibition [59]. So far, it is not fully understood how this cell contact-dependent suppression is regulated.
Regulation of metabolism in target cells
Another mechanism of Treg cell suppression is the metabolic disruption of the targeted cells. Treg cells are characterized by high expression levels of IL-2R (CD25) [60]. This helps Treg cells to bind IL-2 and to prevent IL-2 consumption by effector T cells. IL-2 is crucial for the maintenance of effector T cells activation and proliferation. Lack of IL-2 induces apoptosis of effector T cells [39]. Moreover, Treg cells also mediate immunosuppression by the ectonucleotidases CD39 and CD73. CD39 is constitutively expressed on Treg cells and can hydrolyze ATP and ADP [61]. It also acts together with CD73 to produce adenosine (Fig. 1) [62]. Once binding to adenosine receptor A2A on activated T cells [63], adenosine can induce metabolic disruption through the following mechanisms: cyclic adenosine monophosphate (cAMP) in Treg cells drives the inhibition of T cell proliferation and promotes the synthesis of IL-2 by gap junction formation, which prevents the production of proinflammatory cytokines in effector T cells [64].
Development of Treg cells
To date, three different phenotypes of Treg cells have been identified based on their origins: natural Treg (nTreg) cells derived from the thymus, peripheral Treg cells from peripheral lymphoid organs and induced Treg (iTreg) cells differentiated in vitro from naïve T cells under Treg skewing conditions [65]. All of them are characterized by the expression of Foxp3 [30].
Natural Treg cells
In the thymus, nTreg cells are derived from hematopoietic progenitor cells through a two-step mechanism [66] and migrate into the peripheral blood to maintain immune tolerance and to prevent autoimmunity. Early T cell development occurs in the cortex whereas the later phases of maturation occur in the medulla [67]. Immature CD4+ single positive T cells get depleted by strong TCR activation, whereas the cells receiving intermediate TCR activation escape from negative depletion and are able to differentiate into Treg cells [68]. First, TCR stimulation and cytokines drive the CD4+ single positive T cells to upregulation of IL-2R and TNF receptor superfamily members (GITR, OX40, and TNFR2). Next, the expression of FoxP3 is upregulated by recognition of self-antigen-MHC II complexes presented by thymic antigen-presenting cells, which results in the maturation of Treg cells [69,70,71,72]. Natural Treg cells are fairly stable and Foxp3 is stabilized by demethylation of the CNS2 region of the Foxp3 locus that leads to recruitment of various transcription factors including Foxp3 itself [73, 74].
Peripheral and induced Treg cells
During the development of peripheral Treg cells, naïve CD4+ T cells first migrate into the peripheral blood without any TCR activation. Once stimulated by antigens in the peripheral blood, naïve CD4+ T cells can differentiate into Foxp3+ Treg cells in the presence of both, TGF-beta and IL-2 [75]. Despite their low frequency, peripheral Treg cells can prevent inflammation in barrier tissues [72, 76, 77]. When Treg cells are generated in vitro, they are termed iTreg cells [78]. These cells are also suppressive and can maintain immune homeostasis [79]. Moreover, retinoic acid in the gut is also reported to promote peripheral Treg cells differentiation [78]. Mucosal dendritic cells induce Foxp3+ Treg cells by producing TGF-beta and retinoic acid [80, 81]. The microbial metabolites are short-chain fatty acids (SCFAs) and were reported to facilitate peripheral Treg cell development. However, TGF-beta is required for regulatory effects [82]. It has to be emphasized that iTreg cells are not so stable compared to nTreg cells.
Other suppressive CD4+ T cell populations
There is an additional CD4+ T cell subset that mediates immunosuppression in vitro and which attracted a lot of attention in the past years. The cells are characterized by secretion of TGF-beta1 and IL-10 without expressing the transcription factor of Foxp3 [20]. This subset is termed type 1 regulatory T cells (Tr1 cells). However, no specific surface biomarker or transcription factors for this subset have been identified so far, although some promising candidates have been reported [23]. The suppressive mechanisms used by Tr1 cells are similar to those of nTreg cells and include the production of immunosuppressive cytokines, cell–cell contact-mediated suppression, cytotoxicity, and metabolic disruption [23]. IL-10 and other cytokines, including IFN-α, IL-6 and IL-27, are required for the generation of Tr1 cells [83,84,85,86]. Importantly, Tr1 Treg cells are found to be less suppressive compared to nTreg cells in the early stage of life, because they fail to rescue IPEX patients with a complete lack of Foxp3 [21]. In addition, it has been also reported that Foxp3+ CD8+ T cells, CD4−CD8− cells and gamma/delta T cells also share some suppressive properties, but no evidence has shown that they play an important role in self-tolerance [30, 87].
Regulation of Treg cells in rheumatoid arthritis
Compared to healthy individuals, significant lower frequencies of Treg cells are found in the peripheral blood of patients with RA at an early stage of disease [88]. Moreover, a negative correlation between CD4+CD25highCD127low Treg cell numbers and disease activity as assessed by disease activity score 28 (DAS-28) has been reported [89]. Treg cells are also found in the synovial fluid of RA patients, but their suppressive function is impaired [90]. In a study comparing Treg cell numbers in the synovial fluid of RA patients and osteoarthritis patients, the number of Treg cells were increased in the synovium of RA patients but failed to suppress dendritic cells activation and maturation [91]. In addition, Treg cells in the synovial fluid express higher levels of CTLA-4, GITR, OX40, and Foxp3 despite their impaired ability to suppress the proliferative response of effector T cells [12]. Treg cell plasticity has been reported to allow Treg cells to develop into highly inflammatory Th17 cells under physiological and pathogenic conditions. Interestingly, some Treg cells in the peripheral blood of RA patients produce IL-17 while they maintain their suppressive function [92]. However, it remains unknown if IL-17-producing Treg cells contribute to the pathogenesis of RA. Ex-Foxp3 T cells represent another T cell subset that is found in RA patients, Their phenotype is less stable in inflamed joints with high IL-6 concentration and they do not express Foxp3. These ex-FoxP3 T cells can convert into more osteoclastogenic Th17 cells as compared to Th17 cells which have been differentiated from naïve T cells [93]. Furthermore, deficiency of genes encoding for IL-2, IL-2 receptor (CD25) or CTLA-4 are associated with autoimmune diseases, indicating that these Treg associated cytokines and molecules are important for the prevention of autoimmune disorders [94,95,96,97]. In the murine model of collagen-induced arthritis (CIA), the suppressive capacity of Treg cells from mice immunized with complete Freud’s adjuvant (CFA) and collagen type II is impaired. Disruption of Treg cells by anti-CD25 antibodies in these mice has been shown to accelerate the onset of joint inflammation and to increase disease severity. Moreover, iTreg cells from CIA mice upregulate FoxP3 following cell contact with dendritic cells (DC). DC-stimulated Treg cells can prevent effector T cell proliferation and Th17 cell differentiation more efficiently than iTreg cells without DC stimulation, leading to less severe arthritis [98]. In summary, various studies indicate that dysregulation and deficiency of Treg cells play an important role in the pathogenesis of RA.
Therapeutic potential of Treg cells
Treg-based therapies are a promising approach to overcome impaired Treg cell functions and reduced frequencies of Treg cells in RA. The therapeutic potential of Treg cells has been shown in other autoimmune disorders, including experimental autoimmune diabetes and encephalomyelitis [99, 100]. Preclinical research indicates that Treg cell therapies can delay the onset of autoimmune inflammation and reduce graft-versus-host reactions in organ transplantation [101]. Application of type 1 regulatory T cells to symptomatic patients with refractory Crohn’s disease has been proved to be safe and to achieve dose-dependent effects [102]. Moreover, ex vivo expansion of polyclonal Treg cells from patients with early type 1 diabetes resulted in a significant increase in Treg cells numbers, C-peptide levels, prolonged survival of beta-cells, and lower requirement for insulin [103]. In addition, regulatory T-cell therapy proved to be safe in patients who received kidney transplantation, leading to less infectious complications despite an equivalent rejection rate in the first year [104]. Currently, Treg cell therapies are evaluated in several phase I/II clinical trials involving patients with cutaneous lupus, type 1 diabetes, Crohn’s disease, autoimmune hepatitis, and organ transplantation [105]. The results of these trials will help to improve the design of future Treg-based therapies for RA patients. The current strategies to increase Treg cell numbers and their function in vivo and in vitro are shown in Table 1 and in Fig. 2.

Strategies to increase Treg cells frequencies and the suppressive function in vivo and in vitro. In vivo and in vitro strategies can be used to increase Treg cells number or function with immunomodulatory drugs. Low dose IL-2, engineered IL-2 muteins, the complex of IL-2/IL-2 receptor, IL-4, IL-5, IL-7, IL-12, IL-15, and IFN-γ are shown to have the potential to increase the activity of natural Treg cells or peripheral Treg cells against effector T cells in vivo. In addition, selective depletion of effector T cells by anti-CD3 antibodies can restore Treg cell predominance over effector T cells. Treg cells or naïve T cells isolated from the peripheral blood or the thymus of pediatric cardiac patients can be expanded and genetically modified in vitro for adaptive transfer to increase Treg cell numbers or improve specificity
In vivo strategies
Immunomodulatory drugs targeting Treg cells in vivo represent a possible strategy to improve Treg cell functions and to maintain immune tolerance in RA. This approach includes the application of cytokines as well as the treatment with specific antibodies.
Preclinical studies using animal models
A series of preclinical studies revealed that it is possible to increase absolute Treg cell numbers with cytokines or antibodies and to improve Treg cell functions. For example, oral 1,25-dihydroxyvitamin D3 was reported to be able to stabilize Treg cells by enhancing TGF-beta and Foxp3 gene expression, thereby increasing absolute Treg cell numbers and reducing IL-6 expression in mice with lupus-like disease [106]. In addition, short-chain fatty acids (SCFAs) contribute to Treg cell expansion in vitro and in vivo [107]. Moreover, progesterone promotes the suppressive activity of Treg cells and increases the stability of Treg cells in inflammatory tissues through the mTOR pathway [108]. Furthermore, vitamin C was reported to be necessary for Foxp3 expression [109]. All these drugs prove that the suppressive capacity of Treg cells can be modulated in vivo.
In vivo strategies targeting Treg cells
IL-2 receptor alpha chain (CD25) is highly expressed on Foxp3 Treg cells compared to effecter CD4+ T cells. Enhancing IL-2 signaling in Treg cells improves the immunosuppression mediated by Treg cells. However, IL-2 signaling is also necessary for the activation and proliferation of proinflammatory T cells. Therefore, some research groups developed immunomodulatory drugs which can prevent the activation of effector T cells by IL-2. Low-dose IL-2 leads to expansion of Treg cell populations and recovery of Treg cell suppression in vivo [110,111,112,113]. Engineered IL-2 has the ability to selectively activate and expand regulatory T cells [114, 115]. Furthermore, IL-2/IL-2 mAb complex also showed potent selective immune regulation on T cells in vivo probably by conformational changes, although the mechanism of this effect remains unclear [116, 117]. In addition, other cytokine-based strategies have the potential to improve Treg cell functions. These cytokines include IL-4, IL-5, IL-7, IL-12, IL-15, and IFN-γ [118,119,120,121,122,123]. Although these strategies have been tested in mouse models of organ transplantation, type 1 diabetes and other autoimmune disorders, these cytokines have not been used to ameliorate experimental autoimmune arthritis or RA, so far. However, promising results observed in other autoimmune diseases justify further research on the effect of immunosuppressive cytokines on arthritis. Interestingly, selective depletion of effector T cells by anti-CD3 antibodies helps to improve immune tolerance in autoimmune patients and mouse models by preferentially depleting pathogenic cells while preserving Treg cells [124, 125].
In vitro strategies
Preclinical studies involving mouse models
Asnagli et al. reported that collagen-induced Treg cells was effective in the treatment of experimental autoimmune arthritis [126]. The efficiency of Treg cell transfer in rats with CIA varies dependent on the stage of disease. In the early stages, Treg cell transfer reduces the symptoms while it shows no effect in later stages because the ability of Treg cells to migrate to lymph nodes is impaired [127]. A recent report has shown that transfusion of exogenous Treg cells in CIA mice can significantly improve the severity of arthritis by increasing the proportion of Treg cells in the spleen and in the peripheral blood [128]. Several phase I/II clinical trials with Treg cells have been performed, so far [101, 129, 130]. Th17 cells are a highly proinflammatory T cell subset in autoimmune arthritis [24, 131,132,133], Haque et al. have shown that the transfer of functional antigen-specific Treg cells, which were obtained by reprogramming induced pluripotent stem cells, ameliorated the development of CIA by suppressing pro-inflammatory Th17 cells [134]. Ex vivo generated iTreg cells from purified mouse CD4 + T splenocytes showed equivalent suppressive activity as freshly isolated nTreg cells in a mouse model of chronic colitis [135]. Moreover, adoptive cell therapy (ACT) with Treg cells from the peripheral blood, which are expanded in vitro by stimulation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, has shown promising results. ACT can prevent the development of CIA [136] by inhibiting T cells, B cells and osteoclast-mediated bone destruction [137, 138].
Clinical trials with Treg cells in RA patients
Several preclinical studies in arthritis mouse models have shown the effectiveness and safety of adoptive Treg cells and their potential to ameliorate clinical symptoms. Clinical trials have shown that adoptive Treg cell transfer was safe and achievable in kidney transplant recipients (NCT02091232) [104] and was associated with a reduced rate of infectious complications in type 1 diabetes (NCT01210664) [139]. There are some ongoing clinical trials involving patients with other types of diseases, including acute graft-versus-host disease (NCT01795573), steroid-refractory chronic graft-versus-host-disease (NCT03683498), and pemphigus (NCT03239470). These clinical trials may provide useful information for the development of Treg cells-based clinical trials in RA. So far, adoptive Treg cell transfer has not been used in clinical trials involving RA patients.
Limitations, challenges and opportunities for Treg-based therapies
Despite recent advances, Treg cell-based therapies remain challenging. First, the number of nTreg cells is low in the peripheral blood and the required number of cells can hardly be obtained without ex vivo expansion of the Treg cell population. Second, iTreg cells are not as stable as nTreg cells. This means that iTreg cells will lose their suppressive function by reducing Foxp3 expression in the absence of stimulating cytokines [140]. However, several pieces of evidence show that it is possible to expand Treg cells which have effective suppressive functions in vitro based on good manufacturing practice-compliant (GMP-compliant) protocols [141,142,143,144,145,146,147,148]. These GMP-compliant protocols can be used for adoptive Treg cell transfer. In addition, Treg cells should be specific enough to target cells. Chimeric antigen receptors can help to increase the specificity, thereby re-directing Treg cells against immunogenic antigens and restore immune tolerance.
Limited number of available Treg cells in the peripheral blood
Lanni et al. reported that freshly isolated Treg cells can be used for clinical applications [149], but the low frequency of Treg cells in the peripheral blood restricts their use in clinical trials. It has been shown that conventional T cells can convert into Treg cells in mice by forced expression of Foxp3. In humans, the situation seems to be by far more complicated as it is not possible to generate potent suppressive human Treg cells in vitro by retroviral gene transfer mediated overexpression of Foxp3 [150,151,152,153]. However, Allan et al. reported the successful use of lentivirus for the transduction of FoxP3, thereby efficiently converting effector T cells into Treg cells [154]. However, further confirmation is required and safety issues regarding viral gene transfer into human T cells have to be solved. Interestingly, Dijke et al. reported that discarded human thymuses from pediatric cardiac surgery can serve as a new efficient source of Treg cells. These Treg cells are characterized by enhanced survival, stable expression of FoxP3 and immunosuppressive functions [101] (See Table 1).
Instability of Treg cells
Transduction of Foxp3 into iTreg cells seems to be a promising approach to induce stable FoxP3 expression, to stabilize the Treg cell phenotype and to maintain the full suppressive ability of Treg cells [154]. Recently, Honaker et al. used homology-directed repair (HDR)-based gene editing to enforce stable and robust expression of endogenous Foxp3, showing suppressive activity of iTreg cells both in human and mice with inflammatory diseases [155]. The stability of iTreg cells might be important to develop the full potential of adoptive Treg transfer in future clinical trials. However, it remains unclear whether the efficiency of genetically engineered iTreg cells is comparable to freshly isolated ex vivo nTreg cells or peripheral Treg cells [156]. It has also been reported that the transcription factors Helios, Eos, IRF4, Satb1, Lef1, and GATA-1 are necessary to maintain the suppressive property of Treg cells [157,158,159,160]. These findings demonstrate the complexity of Treg cell functions and the challenges which have to be faced to obtain suppressive iTreg cells. It has been reported that epigenetic modifications are involved in the regulation of Treg cell stability [161], but the underlying mechanisms remain unclear.
Opportunities for Treg cells expansion
It has been reported that freshly isolated Treg cells can be expanded ex vivo under Treg skewing conditions [162], and expanded polyclonal suppressive human Treg cells can prevent graft rejection [163]. During the past several years, there are some pieces of evidence showing that it is feasible to expand a satisfying amount of Treg cells that keep their suppressive ability in vitro using GMP-compliant protocols (see Table 2). This approach seems promising for future clinical trials in this field. Some reports suggest different approaches, including retroviral gene transfer mediated overexpression of FoxP3 or engineered iTreg cells to increase the stability of their suppressive function. In most reports, Treg cells were expanded from natural Treg cells obtained from peripheral blood mononuclear cells whereas in one of these reports cryopreserved umbilical cord blood was used to obtain naïve Treg cells. Anti-CD3/CD28 beads, IL-2 and rapamycin or everolimus were used in these expansion conditions. It is worth highlighting that Landwehr et al. reported that they used an allogeneic B cell bank for the expansion of allospecific natural Treg cells, which showed a superior suppressive ability compared to polyclonal natural Treg cells [143]. Even though the expansion protocols vary among these reports, they all show that a satisfying amount of Treg cells can be obtained and that Treg cells keep their suppressive function. However, further investigation is required to determine how these Treg cells could contribute to the restoration of immune tolerance in clinical trials.
Antigen-specificity of Treg cells and use of chimeric antigen receptors
Treg cells express a spectrum of molecules that helps them to transmigrate into inflammatory sites, including CCR6, CXCR4 and CXCR5 [164]. Evidence obtained from animal studies shows that antigen-specific Treg cells are more effective than polyclonal Tregs [155, 165,166,167,168,169,170,171]. A study about Treg cell transfer in a mouse model of myocardial infarction-induced ventricular remodeling has demonstrated that transferred Treg cells predominantly accumulate in the spleen rather than in inflammatory tissues [172]. Therefore, antigen-specific Treg cells are believed to be more suitable for clinical applications. Recently, we reported the role of post-translational modification of VASP for reduced migration of Treg cells in RA [24]. VASP might serve as a new potential specific target for engineered Treg cells. High VASP expression could increase the migration of Treg cells into inflamed synovial tissues in RA patients. To date, several strategies have been developed to obtain antigen-specific Treg cells: T cell receptor overexpression, antigen-stimulated expansion and chimeric antigen receptors (CAR). However, the first two methods are limited by low cell numbers, complex manufacture procedures and patient-specific TCRs. In contrast, CARs seem to be a promising tool for the generation of antigen-specific Treg cells. Various CARs have been used for the experimental treatment of autoimmune diseases. For example, CD19-CAR transfer was reported to be effective to suppress pathogenic B cells in autoantibody-mediated autoimmune disease. CD19-CAR Treg cells derived from naïve CD4+CD25highCD127lowCD45RA+ cells maintained their suppressive functions, including high expression levels of TGF-beta and Foxp3 as compared to CD19-CAR Treg cells derived from CD4+CD25highCD127lowCD45RO+ memory cells [173].
Recently, CAR-T cells targeting specific citrullinated peptide epitopes, including citrullinated vimentin (cVIM), citrullinated type II collagen (cCOII), citrullinated fibrinogen (cFib), citrullinated tenascin-C (cTNC-5), and cyclocitrulline peptide (CCP-1) were tested in vitro using T cells from CIA mice or RA patients. These CAR-T cells not only specifically recognized and killed anti-COII antibody-secreting B cells from CIA mice but also diminished specific autoreactive B cells in RA patients [174]. Noteworthy, there are individual autoantigen expression profiles in RA patients, making it difficult to develop specific CAR-T cells for a larger cohort of patients. However, it is possible to develop customized CAR-T cells for patients with severe diseases and organ manifestations. Moreover, Whittington et al. reported recently that DR1-CII CAR-T cells can specifically and effectively recognize and kill CD4+ T cells which are specific for the CII autoantigen in vivo in CIA mice. This leads to reduced autoantibody production, decreased collagen II-specific CD4+ T cell response and diminished severity of autoimmune arthritis in mice [175]. Furthermore, HLA-A*02 CAR-Treg cells were generated and have shown to be highly effective in preventing immune responses mediated by HLA-A*02 in both, human and mice [176]. Currently, there is no clear evidence showing that CAR-Treg cells could be used to cure RA, even though the aforementioned specific CAR-T cells seem to be very promising. Therefore, further investigations are need, especially to test the efficacy of CAR-Treg cells in vivo in RA patients and in arthritis mouse models.
In addition, co-stimulation with CD28/CTLA-4/B7 domains plays an important role in the homeostasis and function of Treg cells [177, 178]. The intracellular domain of CD28 is often integrated in CARs along with the intracellular domain of CD3. Dawson et al. reported that co-receptor signaling domains improve the efficacy of CAR-Treg cells [179]. The authors compared ten different co-receptor signaling domain-CARs and analyzed the gene expression profiles and the functions of human Treg cells. They demonstrated that a CAR encoding CD28 is superior to CARs with other intracellular signaling domains, including ICOS, CTLA-4, PD-1, OX40, GITR, 4-1BB, and TNFR2 [179]. Interestingly, 4-1BB and TNFR2 CARs are less effective regulator of the methylation status of TSDR within Foxp3, thereby inducing Treg cells with reduced stability. Furthermore, Boroughs et al. have reported that CD28-based CAR-Treg cells maintain their suppressive function in contrast to 4-1BB-CARs [180]. Due to the enhanced specificity of antigen-specific Treg cells, lower numbers of engineered iTreg cells are needed for adaptive Treg cell transfer as compared to nTreg cells.
The half-life time of CAR-T cells was reported to be relatively short after passive transfer into CIA mice and it was not possible to detect CAR-T cells in mice later than day 10 after adoptive cell transfer [175]. However, CAR-CD8 + Treg cells are still functional and can activate T cells in vivo for at least 80 days [181]. Moreover, CAR-CD4 + Treg cells can still be recruited at least 40 days after transfer into mice with skin grafts [170]. The mechanisms regulating the survival time of CAR-T cells and CAR-Treg cells is not clear yet, but it seems that the half-life time of CAR-Treg cells is superior to the half-life time of CAR-T cells. Further efforts will help to improve antigen-specific Treg transfer and to use CAR Treg cells for clinical application.
Conclusion
Treg cell transfer and maintenance of Treg cell functions is a promising strategy to improve the treatment of RA. Preclinical and clinical trials have demonstrated the efficiency of Treg cell transfer. CAR-Treg cells may be a helpful tool to treat or even cure autoimmune diseases. However, a better understanding of Treg cells and their regulatory mechanisms is still required to improve the clinical application of Treg therapy in RA. The best time point for Treg cell therapies and the optimal dosage of Treg cells or Treg cell promoting drugs need to be determined. Taken together, Treg cell therapies have the potential to revolutionize RA therapy but intensive research is still needed to evaluate and improve this therapeutic approach.
Data availability
Not applicable.
References
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis. Lancet 388(10055):2023–2038
Chang MH, Nigrovic PA (2019) Antibody-dependent and -independent mechanisms of inflammatory arthritis. JCI Insight 4(5):1–15
Cross M et al (2014) The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis 73(7):1316–1322
Almutairi K et al (2021) The global prevalence of rheumatoid arthritis: a meta-analysis based on a systematic review. Rheumatol Int 41(5):863–877
Helmick CG et al (2008) Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I Arthritis Rheum 58(1):15–25
Feldmann M, Maini RN (2003) Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med 9(10):1245–50
Smolen JS et al (2010) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 69(6):964–975
Meyer A, Kofler DM (2019) Failure of a T cell regulator: CD6 contributes to the aggravation of autoimmune inflammation. Cell Mol Immunol 16(9):733–734
Lewis MJ et al (2019) Molecular portraits of early rheumatoid arthritis identify clinical and treatment response phenotypes. Cell Rep 28(9):2455-2470.e5
van Amelsfort JM et al (2004) CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum 50(9):2775–2785
Jiao Z et al (2007) Accumulation of FoxP3-expressing CD4+CD25+ T cells with distinct chemokine receptors in synovial fluid of patients with active rheumatoid arthritis. Scand J Rheumatol 36(6):428–433
Möttönen M et al (2005) CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol 140(2):360–367
Lawson CA et al (2006) Early rheumatoid arthritis is associated with a deficit in the CD4+CD25high regulatory T cell population in peripheral blood. Rheumatology (Oxford) 45(10):1210–1217
Moradi B et al (2014) CD4+CD25+/highCD127low/– regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints–analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res Ther 16(2):R97
Behrens F et al (2007) Imbalance in distribution of functional autologous regulatory T cells in rheumatoid arthritis. Ann Rheum Dis 66(9):1151–1156
Huter EN et al (2008) TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol 38(7):1814–1821
Beavis PA et al (2011) Resistance to regulatory T cell-mediated suppression in rheumatoid arthritis can be bypassed by ectopic foxp3 expression in pathogenic synovial T cells. Proc Natl Acad Sci USA 108(40):16717–16722
Sakaguchi S et al (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155(3):1151–64
Khattri R et al (2003) An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 4(4):337–342
Vieira PL et al (2004) IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol 172(10):5986–5993
Gregori S, Goudy KS, Roncarolo MG (2012) The cellular and molecular mechanisms of immuno-suppression by human type 1 regulatory T cells. Front Immunol 3:30
Chen Y et al (1994) Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265(5176):1237–1240
Song Y et al (2021) Tr1 cells as a key regulator for maintaining immune homeostasis in transplantation. Front Immunol 12:671579
Yan S et al (2021) Membrane-bound IL-6R is upregulated on Th17 cells and inhibits Treg cell migration by regulating post-translational modification of VASP in autoimmune arthritis. Cell Mol Life Sci 79(1):3
Meyer A et al (2021) Kinase activity profiling reveals contribution of G-protein signaling modulator 2 deficiency to impaired regulatory T cell migration in rheumatoid arthritis. J Autoimmun 124:102726
Scheinecker C, Göschl L, Bonelli M (2020) Treg cells in health and autoimmune diseases: new insights from single cell analysis. J Autoimmun 110:102376
Ko HJ et al (2010) CTLA4-Ig modifies dendritic cells from mice with collagen-induced arthritis to increase the CD4+CD25+Foxp3+ regulatory T cell population. J Autoimmun 34(2):111–120
Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299(5609):1057–1061
Brunkow ME et al (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27(1):68–73
Sakaguchi S et al (2008) Regulatory T cells and immune tolerance. Cell 133(5):775–787
Takatori H et al (2015) Helios enhances treg cell function in cooperation with FoxP3. Arthritis Rheumatol 67(6):1491–1502
Zheng Y et al (2010) Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463(7282):808–812
Razmara M et al (2008) CTLA-4 x Ig converts naive CD4+CD25- T cells into CD4+CD25+ regulatory T cells. Int Immunol 20(4):471–483
Gonzalez-Rey E, Delgado M (2007) Vasoactive intestinal peptide and regulatory T-cell induction: a new mechanism and therapeutic potential for immune homeostasis. Trends Mol Med 13(6):241–251
Wing JB, Sakaguchi S (2012) Multiple treg suppressive modules and their adaptability. Front Immunol 3:178
Shevach EM (2009) Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30(5):636–645
Putnam AL et al (2009) Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 58(3):652–662
Okeke EB, Uzonna JE (2019) The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol 10:680
Thornton AM, Shevach EM (1998) CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 188(2):287–296
Fahlén L et al (2005) T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med 201(5):737–746
Hara M et al (2001) IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol 166(6):3789–3796
Asseman C et al (1999) An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med 190(7):995–1004
Collison LW et al (2007) The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450(7169):566–569
Anderson AC et al (2004) IL-10 plays an important role in the homeostatic regulation of the autoreactive repertoire in naive mice. J Immunol 173(2):828–834
Finnegan A et al (2003) Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res Ther 5(1):R18-24
Johansson AC et al (2001) IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J Immunol 167(6):3505–12
Fellowes R et al (2000) Amelioration of established collagen induced arthritis by systemic IL-10 gene delivery. Gene Ther 7(11):967–977
Park MJ et al (2018) Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep 8(1):3753
Roncarolo MG et al (2006) Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 212:28–50
Marie JC et al (2005) TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 201(7):1061–1067
Bennett CL et al (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27(1):20–21
Nakamura K, Kitani A, Strober W (2001) Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med 194(5):629–644
Nakano S et al (2015) Immunoregulatory role of IL-35 in T cells of patients with rheumatoid arthritis. Rheumatology (Oxford) 54(8):1498–1506
Onishi Y et al (2008) Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA 105(29):10113–10118
Liang B et al (2008) Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol 180(9):5916–5926
Fife BT, Pauken KE (2011) The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci 1217:45–59
Grossman WJ et al (2004) Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 104(9):2840–2848
Gondek DC et al (2005) Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol 174(4):1783–1786
Rupp MC et al (2021) The posttraumatic response of CD4+ regulatory T cells is modulated by direct cell-cell contact via CD40L- and P-selectin-dependent pathways. Cent Eur J Immunol 46(3):283–294
de la Rosa M et al (2004) Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol 34(9):2480–2488
Bours MJ et al (2006) Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112(2):358–404
Borsellino G et al (2007) Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 110(4):1225–1232
Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414(6866):916–920
Bopp T et al (2007) Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med 204(6):1303–1310
Campbell DJ, Koch MA (2011) Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol 11(2):119–130
Owen DL, Sjaastad LE, Farrar MA (2019) Regulatory T cell development in the thymus. J Immunol 203(8):2031–2041
Savage PA, Klawon DEJ, Miller CH (2020) Regulatory T cell development. Annu Rev Immunol 38:421–453
Famili F, Wiekmeijer AS, Staal FJ (2017) The development of T cells from stem cells in mice and humans. Future Sci OA 3(3):Fso186
Lio CW, Hsieh CS (2008) A two-step process for thymic regulatory T cell development. Immunity 28(1):100–111
Burchill MA et al (2008) Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 28(1):112–121
Jordan MS et al (2001) Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2(4):301–306
Apostolou I et al (2002) Origin of regulatory T cells with known specificity for antigen. Nat Immunol 3(8):756–763
Feng Y et al (2014) Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 158(4):749–763
Sekiya T et al (2016) Roles of transcription factors and epigenetic modifications in differentiation and maintenance of regulatory T cells. Microbes Infect 18(6):378–386
Chen W et al (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198(12):1875–1886
Abbas AK et al (2013) Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol 14(4):307–308
Hadis U et al (2011) Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34(2):237–246
Kanamori M et al (2016) Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol 37(11):803–811
de Curotto Lafaille MA, Lafaille JJ (2009) Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30(5):626–35
Mucida D et al (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835):256–260
Coombes JL et al (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764
Smith PM et al (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341(6145):569–573
Groux H et al (1997) A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389(6652):737–742
Jin JO, Han X, Yu Q (2013) Interleukin-6 induces the generation of IL-10-producing Tr1 cells and suppresses autoimmune tissue inflammation. J Autoimmun 40:28–44
Awasthi A et al (2007) A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol 8(12):1380–1389
Vasanthakumar A, Kallies A (2013) IL-27 paves different roads to Tr1. Eur J Immunol 43(4):882–885
Shevach EM et al (2006) The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev 212:60–73
Morita T et al (2016) The proportion of regulatory T cells in patients with rheumatoid arthritis: a meta-analysis. PLoS ONE 11(9):e0162306
Khattab SS et al (2016) CD4+ CD25+ CD127low regulatory T cells as indicator of rheumatoid arthritis disease activity. Egypt J Immunol 23(2):87–95
Flores-Borja F et al (2008) Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA 105(49):19396–19401
E XQ et al (2012) Distribution of regulatory T cells and interaction with dendritic cells in the synovium of rheumatoid arthritis. Scand J Rheumatol 41(6):413–20
Wang T et al (2015) Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann Rheum Dis 74(6):1293–1301
Komatsu N et al (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20(1):62–68
Romo-Tena J, Gómez-Martín D, Alcocer-Varela J (2013) CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev 12(12):1171–1176
Goudy K et al (2013) Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol 146(3):248–261
Matesanz F et al (2007) IL2RA/CD25 polymorphisms contribute to multiple sclerosis susceptibility. J Neurol 254(5):682–684
Vella A et al (2005) Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet 76(5):773–779
Yang J et al (2017) Adoptive cell therapy of induced regulatory T cells expanded by tolerogenic dendritic cells on murine autoimmune arthritis. J Immunol Res 2017:7573154
Kohm AP et al (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169(9):4712–4716
Mukherjee R et al (2003) CD4+CD25+ regulatory T cells generated in response to insulin B:9–23 peptide prevent adoptive transfer of diabetes by diabetogenic T cells. J Autoimmun 21(3):221–237
Dijke IE et al (2016) Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant 16(1):58–71
Desreumaux P et al (2012) Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 143(5):1207-1217.e2
Marek-Trzonkowska N et al (2014) Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets—results of one year follow-up. Clin Immunol 153(1):23–30
Sawitzki B et al (2020) Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 395(10237):1627–1639
Romano M et al (2019) Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol 10:43
Lavi Arab F et al (2015) Assessment of 1,25-dihydroxyvitamin D3 effects on Treg cells in a mouse model of systemic lupus erythematosus. Immunopharmacol Immunotoxicol 37(1):12–18
Furusawa Y et al (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–450
Lee JH, Lydon JP, Kim CH (2012) Progesterone suppresses the mTOR pathway and promotes generation of induced regulatory T cells with increased stability. Eur J Immunol 42(10):2683–2696
Sasidharan Nair V, Song MH, Oh KI (2016) Vitamin C facilitates demethylation of the Foxp3 enhancer in a tet-dependent manner. J Immunol 196(5):2119–2131
Saadoun D et al (2011) Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 365(22):2067–2077
Hartemann A et al (2013) Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 1(4):295–305
Castela E et al (2014) Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol 150(7):748–751
von Spee-Mayer C et al (2016) Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 75(7):1407–1415
Peterson LB et al (2018) A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun 95:1–14
Spangler JB et al (2018) Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol 201(7):2094–2106
Boyman O et al (2006) Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311(5769):1924–1927
Trotta E et al (2018) A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med 24(7):1005–1014
Yang WC et al (2017) Interleukin-4 supports the suppressive immune responses elicited by regulatory T cells. Front Immunol 8:1508
Tran GT et al (2012) IL-5 promotes induction of antigen-specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood 119(19):4441–4450
Gratz IK et al (2013) Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol 190(9):4483–4487
Verma ND et al (2014) Interleukin-12 (IL-12p70) promotes induction of highly potent Th1-like CD4(+)CD25(+) T regulatory cells that inhibit allograft rejection in unmodified recipients. Front Immunol 5:190
Tosiek MJ et al (2016) IL-15-dependent balance between Foxp3 and RORγt expression impacts inflammatory bowel disease. Nat Commun 7:10888
Wang Z et al (2006) Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25- T cells to CD4+ Tregs. J Clin Invest 116(9):2434–2441
Kuhn C, Weiner HL (2016) Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy 8(8):889–906
Penaranda C, Tang Q, Bluestone JA (2011) Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol 187(4):2015–2022
Asnagli H et al (2014) Type 1 regulatory T cells specific for collagen type II as an efficient cell-based therapy in arthritis. Arthritis Res Ther 16(3):R115
Felcenloben I et al (2015) Adoptively transferred Tregs accumulate in a site-specific manner and ameliorate signs of less advanced collagen-induced arthritis progress in rats. Immunotherapy 7(3):215–228
Li S et al (2020) Therapeutic effect of exogenous regulatory T cells on collagen-induced arthritis and rheumatoid arthritis. Cell Transplant 29:963689720954134
Romano M et al (2017) Treg therapy in transplantation: a general overview. Transpl Int 30(8):745–753
Vaikunthanathan T et al (2017) Regulatory T cells: tolerance induction in solid organ transplantation. Clin Exp Immunol 189(2):197–210
Kotschenreuther K et al (2021) Cannabinoids drive Th17 cell differentiation in patients with rheumatic autoimmune diseases. Cell Mol Immunol 18(3):764–766
Klasen C et al (2019) Prostaglandin receptor EP4 expression by Th17 cells is associated with high disease activity in ankylosing spondylitis. Arthritis Res Ther 21(1):159
Meyer A et al (2021) Regulatory T cell frequencies in patients with rheumatoid arthritis are increased by conventional and biological DMARDs but not by JAK inhibitors. Ann Rheum Dis 80(12):e196
Haque M et al (2016) Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci Rep 6:20588
Karlsson F et al (2011) Ex vivo generation of regulatory T cells: characterization and therapeutic evaluation in a model of chronic colitis. Methods Mol Biol 677:47–61
Brunstein CG et al (2011) Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117(3):1061–1070
Morgan ME et al (2005) Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum 52(7):2212–2221
Zaiss MM et al (2007) Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum 56(12):4104–4112
Bluestone JA et al (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7(315):315ra189
Schmidl C et al (2018) Epigenetic mechanisms regulating T-cell responses. J Allergy Clin Immunol 142(3):728–743
Seay HR et al (2017) Expansion of human Tregs from cryopreserved umbilical cord blood for GMP-compliant autologous adoptive cell transfer therapy. Mol Ther Methods Clin Dev 4:178–191
Fraser H et al (2018) A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev 8:198–209
Landwehr-Kenzel S et al (2014) Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am J Transplant 14(3):594–606
Wiesinger M et al (2017) Good manufacturing practice-compliant production and lot-release of ex vivo expanded regulatory T cells as basis for treatment of patients with autoimmune and inflammatory disorders. Front Immunol 8:1371
Janssens I et al (2022) Engineering of regulatory T cells by means of mRNA electroporation in a GMP-compliant manner. Cytotherapy 24(6):659–672
Gedaly R et al (2019) mTOR inhibitor everolimus in regulatory T cell expansion for clinical application in transplantation. Transplantation 103(4):705–715
Alsuliman A et al (2016) A robust, good manufacturing practice-compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy 18(10):1312–1324
Lavazza C et al (2022) Process development and validation of expanded regulatory T cells for prospective applications: an example of manufacturing a personalized advanced therapy medicinal product. J Transl Med 20(1):14
Di Ianni M et al (2011) Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117(14):3921–3928
Karim M et al (2004) Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol 172(2):923–928
Allan SE et al (2005) The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest 115(11):3276–3284
Ocklenburg F et al (2006) UBD, a downstream element of FOXP3, allows the identification of LGALS3, a new marker of human regulatory T cells. Lab Invest 86(7):724–737
Yagi H et al (2004) Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol 16(11):1643–1656
Allan SE et al (2008) Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther 16(1):194–202
Honaker Y et al (2020) Gene editing to induce FOXP3 expression in human CD4(+) T cells leads to a stable regulatory phenotype and function. Sci Transl Med 12(546):1–18
Rosado-Sánchez I, Levings MK (2020) Building a CAR-Treg: going from the basic to the luxury model. Cell Immunol 358:104220
Nakagawa H et al (2016) Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci U S A 113(22):6248–6253
Seng A et al (2020) Coexpression of FOXP3 and a Helios isoform enhances the effectiveness of human engineered regulatory T cells. Blood Adv 4(7):1325–1339
Fu W et al (2012) A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol 13(10):972–980
Kim HJ et al (2015) Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 350(6258):334–339
Polansky JK et al (2008) DNA methylation controls Foxp3 gene expression. Eur J Immunol 38(6):1654–1663
Trenado A et al (2006) Ex vivo-expanded CD4+CD25+ immunoregulatory T cells prevent graft-versus-host-disease by inhibiting activation/differentiation of pathogenic T cells. J Immunol 176(2):1266–1273
Kingsley CI et al (2002) CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol 168(3):1080–1086
Huehn J, Hamann A (2005) Homing to suppress: address codes for Treg migration. Trends Immunol 26(12):632–636
Dawson NAJ, Levings MK (2017) Antigen-specific regulatory T cells: are police CARs the answer? Transl Res 187:53–58
Fujio K et al (2006) Gene therapy of arthritis with TCR isolated from the inflamed paw. J Immunol 177(11):8140–8147
Fransson M et al (2012) CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation 9:112
Elinav E, Waks T, Eshhar Z (2008) Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology 134(7):2014–2024
Noyan F et al (2017) Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant 17(4):917–930
MacDonald KG et al (2016) Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest 126(4):1413–1424
Sun G et al (2018) Adoptive induced antigen-specific Treg cells reverse inflammation in collagen-induced arthritis mouse model. Inflammation 41(2):485–495
Matsumoto K et al (2011) Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int Heart J 52(6):382–387
Imura Y et al (2020) CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight 5(14):1–16
Zhang B et al (2021) In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann Rheum Dis 80(2):176–184
Whittington KB et al (2022) CD8(+) T cells expressing an HLA-DR1 chimeric antigen receptor target autoimmune CD4(+) T cells in an antigen-specific manner and inhibit the development of autoimmune arthritis. J Immunol 208(1):16–26
Vimond N et al (2021) Genetic engineering of human and mouse CD4(+) and CD8(+) Tregs using lentiviral vectors encoding chimeric antigen receptors. Mol Ther Methods Clin Dev 20:69–85
Bour-Jordan H, Bluestone JA (2009) Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev 229(1):41–66
Azuma M (2019) Co-signal molecules in T-cell activation : historical overview and perspective. Adv Exp Med Biol 1189:3–23
Dawson NAJ et al (2020) Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med 12(557):1–16
Boroughs AC et al (2019) Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight 5(8):1–19
Boardman DA et al (2017) Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant 17(4):931–943
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
SY, KK, SD, and DMK performed a literature search. SY and DMK wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, S., Kotschenreuther, K., Deng, S. et al. Regulatory T cells in rheumatoid arthritis: functions, development, regulation, and therapeutic potential. Cell. Mol. Life Sci. 79, 533 (2022). https://doi.org/10.1007/s00018-022-04563-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04563-0