
Abstract
Due to activation of fibroblast into cancer-associated fibroblasts, there is often an increased deposition of extracellular matrix and fibrillar collagens, e.g. type III collagen, in the tumor microenvironment (TME) that leads to tumor fibrosis (desmoplasia). Tumor fibrosis is closely associated with treatment response and poor prognosis for patients with solid tumors. To assure that the best possible treatment option is provided for patients, there is medical need for identifying patients with high (or low) fibrotic activity in the TME. Measuring unique collagen fragments such as the pro-peptides released into the bloodstream during fibrillar collagen deposition in the TME can provide a non-invasive measure of the fibrotic activity. Based on data from 8 previously published cohorts, this review provides insight into the prognostic value of quantifying tumor fibrosis by measuring the pro-peptide of type III collagen in serum of a total of 1692 patients with different solid tumor types and discusses the importance of tumor fibrosis for understanding prognosis and for potentially guiding future drug development efforts that aim at overcoming the poor outcome associated with a fibrotic TME.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to the extracellular matrix (ECM) and collagens in the tumor microenvironment (TME)
The tumor microenvironment (TME) is important for tumor progression and patient survival. The extracellular matrix (ECM) comprises an important component of the TME in addition to the tumor cells, stromal cells and immune infiltrate [1]. The ECM is the non-cellular component of tissues and organs that provides crucial physical, bio-mechanical and bio-chemical properties that is required for tissue morphogenesis, differentiation and homeostasis [2]. The major components of the ECM are the collagens, of which 28 different types have been described, each with a unique role in supporting the tissue microarchitecture [3]. Under normal conditions, a homeostatic state of collagen turnover is maintained by a refined balance between synthesis, degradation and post-translational modifications that maintains tissue integrity. In contrast to a normal healthy stroma, this collagen homeostasis is disrupted in the TME as the composition and quality of the tumor tissue becomes altered [4]. Changes in the composition of the ECM/collagens have been shown to modulate the hallmarks of cancer and are thought to play a vital role in tumor progression and metastasis as well as in defining the likelihood of responding to anti-cancer therapies [5,6,7,8].
Overall, the ECM can be divided into the basement membrane and the interstitial matrix [2, 3]. The basement membrane underlies the epithelial and endothelial cells and supports glandular structures and blood vessels [9]. It is a relatively loose ECM with so-called network forming collagens, where type IV collagen is the most abundant protein together with laminins. The basement membrane allows nutrients and oxygen to diffuse through. In the context of cancer, loss of basement membrane structures has been associated with tumor cell invasion and angiogenesis [10, 11]. It has been well investigated and documented since the early discoveries of Mina Bissel and colleagues that the basement membrane is important for cell function and can even revert a malignant cell phenotype [12, 13]. Recent findings support that the basement membrane is key for determining the metastatic potential of cancer [14]. Cellular invasion through the basement membrane is a key factor in tumorigenesis and is driven primarily by the increased matrix metalloprotease (MMP) activity in the TME that degrade e.g. type IV collagen and alters cellular adhesion and integrin-signaling and hereby affects cell behavior [4, 15,16,17,18,19,20,21].
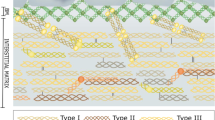
Below the basement membrane appears the interstitial matrix [2, 3]. The interstitial matrix consists of a fibrillar collagen network of type I, III, V, and XI collagens that form a 3D lattice to support tissue structure and cell function. The two major fibrillar collagens in the interstitial matrix are type I collagen and type III collagen. Type I collagen is the most abundant protein in the body and can be found in bone and connective tissues [3]. Type III collagen is the second most abundant collagen, found primarily in connective tissues.
In the TME, there is often an increased interstitial matrix deposition and remodeling of fibrillar collagens due to activation of quiescent fibroblasts into cancer-associated fibroblasts (CAFs) that not only synthesize excess amount of ECM and collagen but also contribute to MMP mediated fibrillar collagen degradation [22]. This chronic-active scarring process is also known as tumor fibrosis, or desmoplasia. As described below, tumor fibrosis has been shown to be closely associated with tumor aggressiveness, treatment response and prognosis for patients. However, we are only beginning to understand the potential impact of a fibrotic TME, the ECM and associated collagens.
Major drivers and impact of fibrosis in the TME
The major pathological signature of tumor fibrosis is a fibrous connective tissue of interstitial matrix formed by proliferation and activation of fibroblasts which takes place inside, adjacent to, and around a solid tumor [23]. All the fibrillar collagens associated with tumor fibrosis are produced by CAFs resulting in increased deposition of a cross-linked dense and stiff collagen matrix that is impermeable for treatment, nutrients, and oxygen and therefore associated with poor outcome [24,25,26]. The CAF and tumor fibrosis biology builds on lessons learned from fibrotic disorders such as idiopathic pulmonary fibrosis, non-alcoholic steatohepatitis, primary sclerosing cholangitis, systemic sclerosis as well as liver, heart, lung and kidney fibrosis [4, 5, 27,28,29,30,31,32,33,34,35] and it has been shown that ECM turnover is generally higher in liver cancer versus cirrhosis, lung cancer versus idiopathic pulmonary fibrosis and pancreatic cancer versus chronic pancreatitis [36,37,38]. The CAFs promote tumorigenesis by contributing to ECM remodeling as well as secreting e.g. cytokines and growth factors to crosstalk with the immune cells and cancer cells. Among the growth factors and cytokines, transforming growth factor-β (TGF-β) is considered as the major pro-fibrotic cytokine and inducer of fibrogenesis because it promotes CAF development and increased collagen synthesis [39, 40]. Other cytokines such as interleukin (IL)-4, IL-13, and platelet-derived growth factor (PDGF) are pro-fibrotic as well and affect collagen expression [41, 42]. MMPs can also activate and release latent TGF-β stored in the ECM and hence can drive tumor fibrosis indirectly [43].
Tumor fibrosis may result in reduced treatment effect by forming a barrier for treatment that hinders drug penetration [26]. The interaction between tumor fibrosis, CAFs and immune cells infiltrating the tumor microenvironment directly and indirectly inhibit antitumor immunity with the activation of fibroblasts and excessive collagen deposition linked to the lack of T-cell infiltration and activity in the tumor that is a prerequisite for efficient response to immunotherapies [44,45,46,47,48,49,50]. This fibrosis associated T-cell exclusion from the tumor core may be due to entrapment in the collagen-rich peritumoral stroma and/or due to leukocyte-specific collagen receptor 1 (LAIR1) dependent T-cell exhaustion [48, 51, 52]. Tumor fibrosis may also limit the anti-tumor activity of effector T-cells by mediating the recruitment, and the activation of secretory programs of immunosuppressive cells such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) [47, 53, 54]. Higher collagen density within the tumor ECM promotes the polarization of TAMs to a more tumor-promoting functional phenotype characterized by enhanced expression of immunosuppressive genes and secreted proteins [55]. Furthermore, single cell sequencing studies of tumors reveal that similar to CAFs, TAMs are also capable of upregulating the expression of ECM genes, suggesting that they may themselves influence the fibrotic composition of the tumor stroma [56]. Figure 1 illustrates this major pathological tissue signature and associated clinical impact of tumor fibrosis.

Overview of the major pathological signature of tumor fibrosis and the associated clinical impact. Tumor fibrosis is driven by pro-fibrotic signaling such as transforming growth factor beta (TGF-β) that activates quiescent fibroblasts into activated cancer associated fibroblasts (CAFs) that synthesize excess amounts of collagens resulting in a fibrotic extracellular matrix (ECM). Tumor fibrosis can be observed in many solid tumor types and forms a barrier for treatment, hindering drug penetration and T-cell recruitment to the tumor cells as well as directly impacts and regulates anti-tumor immunity due to increased recruitment of myeloid-derived suppressor cells (MDSCs) and changes in tumor associated macrophages (TAMs) composition from a pro-inflammatory (M1-TAM) to an anti-inflammatory (M2-TAM) phenotype. Fibrotic tumors are generally much less responsive to anti-cancer therapy than non-fibrotic tumors are
Quantifying tumor fibrosis in a liquid biopsy: potential prognostic value of measuring type III collagen pro-peptides non-invasively in patients with cancer
The common standard for assessing tumor fibrosis in patients diagnosed with cancer is by use of Sirius red or trichrome staining of total collagen content in tissue biopsies, or by staining for type I collagen and III collagen with antibodies for more detailed immunohistochemical assessments. The measurement of fibroblast activation markers, e.g. alpha smooth muscle actin (α-SMA) and fibroblast activation protein (FAP), and stromal gene signatures in the immuno-oncology setting, recently have been added to this portfolio [48, 51, 52, 56,57,58]
To describe the dynamics of tumor fibrosis, a range of novel technologies are emerging which quantify specific collagen fragments in blood [59,60,61]. By targeting unique fibrillar collagen degradation fragments, or pro-peptides, one may provide a dynamic measure of tumor fibrosis with the ability to quantify the collagen turnover or synthesis rate (fibrotic activity). As collagens are degraded or built into fibers, there is a release of unique epitopes that may provide information about the ongoing pathological processes of damage and repair with some epitopes being released during collagen formation (e.g. pro-peptides) and other epitopes being released during collagen degradation (e.g. MMP-generated peptide fragments) [62]. Such epitopes/peptides can be identified by mass spectrometry, then targeted by antibodies, and ultimately quantified by an immunoassay or alike. As the bone consist primarily of type I collagen, the pro-peptide from type I collagen is often used as a surrogate for bone formation whereas the degradation fragment CTX-I, is often used as a surrogate for bone degradation. As type III collagen is almost exclusively found in soft tissue and not in bone, and is derived from activated fibroblasts, it may be a superior fibrosis marker than the pro-peptide from type I collagen. An illustration of the biology and dynamics supporting this non-invasive biomarker approach to quantify tumor fibrosis is shown in Fig. 2. The rationale for investigating the prognostic value of quantifying tumor fibrosis through measurements of type III collagen fragments emerged through a hypothesis-driven approach supported by observations that type III collagen pro-peptides have been found significantly increased (> fivefold) in conditioned media from the ‘scar-in-a-jar’ in vitro culture of highly fibrotic CAFs as compared to normal fibroblast [63], and highly elevated in fibrotic disorders [4]. The monoclonal antibody used to quantify the pro-peptide of type III collagen in all the studies presented here was originally described and developed by Nielsen et al., to reflect true formation of type III collagen [64]. As this particular monoclonal antibody was raised specifically against the N-protease cleavage site of the pro-collagen (i.e. only targeting released pro-peptides) it differs from other available similar assays that either employs polyclonal antibodies or monoclonal antibodies targeting internal sequences of the pro-peptide and therefore cannot differentiate between type III collagen formation and degradation as the removal of the pro-peptide is sometimes incomplete resulting in abnormal fibrils that are prone to rapid metabolic turnover [65,66,67,68].

Biology and dynamics supporting the biomarker approach to quantify tumor fibrosis non-invasively. Cancer associated fibroblasts (CAFs) synthesize excess amount of fibrillar collagens such as type III collagen upon activation by for example transforming growth factor beta (TGF-β). These fibrillar collagens contain pro-peptides that are released into circulation when the collagens are deposited as collagen fibrils in the tissue. It is the excess accumulation of bundles of collagen fibrils that make up excess of collagens fibers that ultimately result in tumor fibrosis. In the blood, the pro-peptides are quantifiable biomarkers of tumor fibrosis
Data from clinical studies encompassing 1692 patients suffering from breast cancer, pancreatic cancer, colorectal cancer, liver cancer and malignant melanoma are summarized in the forest plot in Fig. 3. In all studies, the patients with high net fibrotic activity (type III collagen pro-peptides) at baseline had poor overall survival (OS). Approximately, two to threefold increased risk of death was observed in patients with high levels of type III collagen pro-peptides. An overview of the different patient cohorts are depicted in Table 1.

Forest plot summarizing the prognostic value of high vs low fibrotic activity. Type III collagen pro-peptides were measured in pre-treatment serum or plasma and were associated with overall survival (OS) outcomes in patients with different cancer types. All studies, except Chen et al., applied cutoffs that were based on dichotomizing patients in to ´high’ and ‘low’ levels of type III collagen pro-peptides and the exact cutoff value varied from study to study. In the study by Chen et al., the HR calculations were based on a continuous scale (*). See additional study details in Table 1
All studies, except Chen et al., applied cutoffs that were based on dichotomizing patients in to ´high’ and ‘low’ levels of type III collagen pro-peptides and the exact cutoff value varied from study to study. In the study by Chen et al., the hazard ratio (HR) calculations were based on a continuous scale and may therefore partly explain the relatively lower HR compared to the other study cohorts. Importantly, full clinical utility of type III collagen pro-peptides as a prognostic tumor fibrosis biomarker needs additional exploration of a specific cut-off per indication and treatment modality which warrants additional prospective studies. Altogether, measuring fibroblast derived type III collagen pro-peptides in serum seems not only to be a tumor agnostic, prognostic, tumor fibrosis biomarker (liquid biopsy) but also points to the need of focusing particularly on the fibroblast-derived interstitial matrix in the context of cancer [61, 63, 69, 70].
It is worth emphasizing that most of the studies listed here included patients with advanced/metastatic disease which mostly carries a very poor prognosis. Nonetheless, based on the data published from the study by Chen et al., including approximately 800 patients with pancreatic cancer this tumor fibrosis prognostic signature seems independent of stage of disease and tumor burden [38]. In addition, preliminary results of type III collagen pro-peptides measured in the early colorectal cancer setting showed associations with disease-free survival (DFS) as defined by the time interval between surgery and recurrence and is aligned with the fact that ECM composition and quality impacts and modulates the metastatic potential and hence risk of relapse (prognosis) [7]. This suggest that fibrotic activity in patients with cancer should be considered alongside more commonly assessed risk factors when attempting to provide the best possible prognosis for patients. Of interest, measuring type III collagen pro-peptides in serum was recently reported to be stable under conditions conforming with hospital sample-handling requirements and with levels not associated with sex, age, body mass index (BMI), or ethnicity [71]. In addition to the solid tumor types addressed here, elevated serum levels of collagen fragments have been found in patients with head and neck cancer, non-small cell lung cancer, gastric cancer and ovarian cancer supporting the tumor agnostic nature of altered collagen turnover and tumor fibrosis [37, 72,73,74,75,76].
There is a major medical need for defining this ‘fibrotic’ group of cancer patients. The first step is to differentiate those with ongoing tumor fibrosis from those without. A liquid biopsy approach as presented here for evaluating collagen peptides associated with tumor fibrosis may provide a novel and clinically applicable tool for patient stratification according to their fibrotic activity. As with any liquid biopsy, given its systemic nature there is a potential need of a concurrent (or upfront) tissue-based assessment for full histological diagnosis. However, a liquid biopsy-based approach is less invasive, quicker, and generally more frequently accessible than the gold standard tumor biopsy-based approach (which is further limited by tumor heterogeneity and is challenging, or impossible, to obtain) [77].
Future perspectives
As highlighted above, the prognostic value of quantifying tumor fibrosis non-invasively can be obtained by measuring the pro-peptide of type III collagen in serum/plasma. The prognostic value was demonstrated across various solid tumor types including notoriously hard to treat cancers such as pancreatic cancer, and prevalent cancer types such as breast cancer, colorectal cancer, liver cancer and malignant melanoma, and for multiple treatment modalities. This supports the importance of fibrosis as a tumor agnostic process and points toward a broadly applicable biomarker approach for future clinical cancer research. While type III collagen pro-peptides is reflective of tumor fibrosis and CAF activity, type III collagen has also been shown to maintain tumor dormancy depending on context and composition [69]. Similarly, type VI collagen, another fibroblast derived collagen, can be both pro- and anti-tumorigenic, depending on context [69, 78]. In fact, there are emerging subtypes of CAFs, fibrosis types, and collagen profiles, which may have a unique function in either supporting or inhibiting cancer growth depending on context. Fibroblasts heterogeneity and the existence of different fibroblast subsets, their transcriptional profiles, and lineages are being extensively studied and where in particular iCAFs and myCAFs has been introduced as two subtypes of CAFs that play an inflammatory and myofibroblast like role, respectively, and differ in their functionality and localization within the TME [79,80,81,82,83,84,85,86,87,88,89,90,91]. Moreover, in several mouse models, in particular PDAC models, it has been shown that attenuating collagen synthesis in cancer associated fibroblasts increases tumor growth and spread, but at the same time may also leave tumors more prone to therapeutic intervention [92,93,94,95]. Altogether indication that there are not only good and bad fibroblast subtypes but also good and bad collagens [4].
The impact that tumor fibrosis may have on clinical outcome and in shaping the future of clinical cancer research needs to be considered. A significant percentage of patients with cancer that are included in clinical trials do not benefit from treatment, and consequently, there is a need for predictive biomarkers to treat the right patients with the right drugs at the right time [50, 96]. Intriguingly, type III collagen turnover, measured retrospectively at baseline in plasma from a discovery and validation cohort of patients with metastatic pancreatic cancer has been shown to have the capacity to also predict treatment benefit of a stromal modifier (PEGPH20) when used in combination with chemotherapy hereby providing evidence for potential predictive value [97]. In detail, both the objective response rate and survival outcomes improved significantly with PEGPH20 as an add-on to chemotherapy compared to chemotherapy alone in the patients with a high ratio of type III collagen degradation to formation, whereas in the remaining patients with a low ratio, there was no effect of adding PEGPH20 to chemotherapy. Hence, a tumor fibrosis liquid biopsy may not only be used prognostically but may also predict response to anti-fibrotic treatments. Another clinical utility may be to identify high risk patients in earlier stages of disease that may need more aggressive treatments and frequent monitoring.
In recent years, 85% of US Food and Drug Administration (FDA) approved cancer treatments have been related to the cancer-immunity cycle [98]. And while immune checkpoint inhibitors have been proven to be efficacious, underpinning the importance of the immune cells in the TME, significant differences in how inflammation is present in tumors, from complete absence to active inflammation that overlaps with the fibrotic component, have been associated with large differences in efficacy of intervention [45, 47, 56, 99,100,101,102]. Consequently, there has been much work in the immuno-oncology field to identify predictive biomarkers including measurement of programmed death-ligand 1 (PD-L1) expression, tumor mutational burden (TMB) and inflammatory gene expression profiles in the TME [99, 101,102,103,104,105,106]. While there is a common thread that such measurements are in many cases predictive of cancer immunotherapeutic efficacy, the immune responses within these microenvironments are likely to be highly complex and there is sufficient reason to believe that fibrotic activity is playing a role in establishing an immune excluded and immune suppressed TME. This may provide an opportunity for approaches to patient management that could complement current patient selection methods for improved outcomes. As measurement methods for fibrosis could potentially be deployed using conventional immunoassays and serum/plasma specimens as exemplified here, there would be significant cost and logistical advantages over current patient selection methods.
Importantly, future research is warranted to determine if altering the fibrotic status of the TME could lead to therapeutics with either monotherapy effect, enabling the host’s immune system, or in combination to enhance efficacy of anti-cancer- and immuno-therapeutics. Numerous therapeutic strategies that target aspects of tumor fibrosis to unleash the immune system against the tumor are currently under investigation [107]. For example, Jiang et al. have shown that modulating the fibroblast-derived collagen expression and deposition in the TME renders pancreatic cancers responsive to checkpoint inhibitor immunotherapy [108]. In other approaches, collagen-binding domains (CBDs) are being used as drug conjugates for more efficient drug delivery and reduced toxicity [109,110,111]. The anti-TGF-β compounds currently in clinical testing may also prove to be anti-fibrotic as part of the mode-of-action and are often tested in combination with immunotherapies [112]. As another example, losartan (an angiotensin II receptor antagonist) has been shown to inhibit type I collagen formation and reduce the desmoplastic reaction in mice with breast, pancreatic, and skin cancers and thereby enhance the efficacy of different compounds [113]. Losartan has also shown promising results in the clinic in combination with chemotherapy [114]. Likewise, metformin-induced depletion of collagen has been shown to enhance penetration of gemcitabine-loaded nanoparticles in pancreatic cancer [115]. Thus, modulating collagens and tumor fibrosis may also affect conventional treatment approaches such as chemotherapies. As the depletion of specific collagens or fibroblasts/CAFs may influence other TME components and lead to immune suppression, tumor progression and other inadvertent effects, a homeostatic restoration of the fibrotic stroma rather than its ablation may be the best approach for eliminating tumor progression [92, 95, 116,117,118]. Perhaps lessons learned from the organ fibrosis field can be leveraged to overcome challenges in drug development associated with tumor fibrosis.
Conclusion
High baseline levels of type III collagen pro-peptides in serum/plasma from patients with solid tumors treated with chemotherapy, targeted therapy, or immunotherapy is a promising prognostic tumor fibrosis biomarker. These results underline the impact that tumor fibrosis may have on clinical outcome and for shaping the future of clinical cancer research towards anti-fibrotic modalities in where the type III collagen pro-peptides measured in serum/plasma could provide a new potential strategy for stratifying patients.
Availability of data and materials
Not applicable.
Abbreviations
- α-SMA:
-
Alpha smooth muscle actin
- BMI:
-
Body mass index
- CAF:
-
Cancer-associated fibroblast
- CBD:
-
Collagen-binding domain
- DFS:
-
Disease free survival
- ECM:
-
Extracellular matrix
- FAP:
-
Fibroblast activation protein
- FDA:
-
Food and Drug Administration
- HR:
-
Hazard ratio
- IHC:
-
Immunohistochemistry
- IL:
-
Interleukin
- LAIR1:
-
Leukocyte-specific collagen receptor
- MDSC:
-
Myeloid-derived suppressor cell
- OS:
-
Overall survival
- PDGF:
-
Platelet derived growth factor
- PD-L1:
-
Programmed death-ligand 1
- TAM:
-
Tumor-associated macrophages
- TGF-β:
-
Transforming growth factor-β
- TMB:
-
Tumor mutational burden
- TME:
-
Tumor microenvironment
References
Balkwill FR, Capasso M, Hagemann T (2012) The tumor microenvironment at a glance. J Cell Sci 125:5591–5596
Frantz C, Stewart KM, Weaver VM (2010) The extracellular matrix at a glance. J Cell Sci 123:4195–4200
Karsdal M (2016) Biochemistry of collagens: structure, function and biomarkers. Academic Press, Cambridge
Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T et al (2017) The good and the bad collagens of fibrosis—their role in signaling and organ function. Adv Drug Deliv Rev 121:43–56
Pickup MW, Mouw JK, Weaver VM (2014) The extracellular matrix modulates the hallmarks of cancer. EMBO 15:1243–1253
Brassart-Pasco S, Brézillon S, Brassart B, Ramont L, Oudart JB, Monboisse JC (2020) Tumor microenvironment: extracellular matrix alterations influence tumor progression. Front Oncol 10:397
Kai FB, Drain AP, Weaver VM (2019) The extracellular matrix modulates the metastatic journey. Dev Cell 49:332–346
Henke E, Nandigama R, Ergün S (2020) Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci 6:1–24
Sekiguchi R, Yamada KM (2018) Basement membranes in development and disease. Curr Top Dev Biol 130:143–191
Rowe RG, Weiss SJ (2008) Breaching the basement membrane: who, when and how? Trends Cell Biol 18:560–574
Kalluri R (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 3:422–433
Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C et al (1997) Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol 137:231–245
Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ (1987) Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc Natl Acad Sci USA 84:136–140
Reuten R, Zendehroud S, Nicolau M, Fleischhauer L, Laitala A, Kiderlen S et al (2021) Basement membrane stiffness determines metastases formation. Nat Mater 20:892–903
Mercurio AM, Bachelder RE, Chung J, O’Connor KL, Rabinovitz I, Shaw LM et al (2001) Integrin laminin receptors and breast carcinoma progression. J Mammary Gland Biol Neoplasia 6:299–309
Hood JD, Cheresh DA (2002) Role of integrins in cell invasion and migration. Nat Rev Cancer 2:91–100
Craig SEL, Brady-Kalnay SM (2011) Cancer cells cut homophilic cell adhesion molecules and run. Cancer Res 71:303–309
Poincloux R, Lizarraga F, Chavrier P (2009) Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci 1:3015–3024
Nascimento CF, Gama-De-Souza LN, Freitas VM, Jaeger RG (2010) Role of MMP9 on invadopodia formation in cells from adenoid cystic carcinoma. Study by laser scanning confocal microscopy. Microsc Res Tech 73:99–108
Jacob A, Jing J, Lee J, Schedin P, Gilbert SM, Peden AA et al (2013) Rab40b regulates MMP2 and MMP9 trafficking during invadopodia formation and breast cancer cell invasion. J Cell Sci 126(20):4647–4658
Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K (2009) Lipid rafts and caveolin-1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res 69(22):8594–8602
Thorseth M-L, Carretta M, Jensen C, Mølgaard K, Jürgensen HJ, Engelholm LH et al (2022) Uncovering mediators of collagen degradation in the tumor microenvironment. Matrix Biol Plus 13:100101
Lin S-C, Kaufmann WK, Brandt BH, Hsieh S-L, Lin W-W, End C et al (2011) Desmoplasia. Encyclopedia of Cancer [Internet]. Springer, Berlin, pp 1093–1095
Longo DL, Rockey DC, Bell PD, Hill JA (2015) Fibrosis—a common pathway to organ injury and failure. N Engl J Med 372:1138–1149
Pankova D, Chen Y, Terajima M, Schliekelman MJ, Baird BN, Fahrenholtz M et al (2016) Cancer-associated fibroblasts induce a collagen cross-link switch in tumor stroma. Mol Cancer Res 14:287–295
Yamauchi M, Barker TH, Gibbons DL, Kurie JM (2018) The fibrotic tumor stroma. J Clin Investig 128:16–25
Hirschfield GM, Chazouillères O, Drenth JP, Thorburn D, Harrison SA, Landis CS et al (2019) Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: a multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol 70:483–493
Bonnans C, Chou J, Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15:786–801
Karsdal MA, Nielsen MJ, Sand JM, Henriksen K, Genovese F, Bay-Jensen AC et al (2013) Extracellular matrix remodeling: the common denominator in connective tissue diseases possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol 11:70–92
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM et al (2020) A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20:174–186
Villesen IF, Daniels SJ, Leeming DJ, Karsdal MA, Nielsen MJ (2020) Review article: the signalling and functional role of the extracellular matrix in the development of liver fibrosis. Aliment Pharmacol Ther 52:85–97
Reese-Petersen AL, Olesen MS, Karsdal MA, Svendsen JH, Genovese F (2020) Atrial fibrillation and cardiac fibrosis: a review on the potential of extracellular matrix proteins as biomarkers. Matrix Biol 91–92:188–203
Karsdal MA, Daniels SJ, Holm Nielsen S, Bager C, Rasmussen DGK, Loomba R et al (2020) Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int 40:736–750
Karsdal MA, Kraus VB, Shevell D, Bay-Jensen AC, Schattenberg J, Rambabu Surabattula R et al (2021) Profiling and targeting connective tissue remodeling in autoimmunity—a novel paradigm for diagnosing and treating chronic diseases. Autoimmun Rev 20:102706
Kristensen JH, Karsdal MA, Genovese F, Johnson S, Svensson B, Jacobsen S et al (2014) The role of extracellular matrix quality in pulmonary fibrosis. Respiration 88:487–499
Jensen C, Holm Nielsen S, Eslam M, Genovese F, Nielsen MJ, Vongsuvanh R et al (2020) Cross-linked multimeric pro-peptides of type III collagen (PC3X) in hepatocellular carcinoma—a biomarker that provides additional prognostic value in AFP positive patients. J Hepatocell Carcinoma 7:301–313
Willumsen N, Bager CL, Leeming DJ, Smith V, Christiansen C, Karsdal MA et al (2014) Serum biomarkers reflecting specific tumor tissue remodeling processes are valuable diagnostic tools for lung cancer. Cancer Med 3:1136–1145
Chen IM, Willumsen N, Dehlendorff C, Johansen AZ, Jensen BV, Hansen CP et al (2019) Clinical value of serum hyaluronan and propeptide of type III collagen in patients with pancreatic cancer. Int J cancer 146:2913–2922
Meng X, Nikolic-Paterson DJ, Lan HY (2016) TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338
Bourgot I, Primac I, Louis T, Noël A, Maquoi E (2020) Reciprocal interplay between fibrillar collagens and collagen-binding integrins: implications in cancer progression and metastasis. Front Oncol 10:1–28
Borthwick LA, Wynn TA, Fisher AJ (2013) Cytokine mediated tissue fibrosis. Biochim Biophys Acta Mol Basis Dis 1832:1049–1060
Ricard-Blum S, Baffet G, Théret N (2018) Molecular and tissue alterations of collagens in fibrosis. Matrix Biol 68–69:122–149
Krstic J, Santibanez JF (2014) Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. Sci World J 2014:521754
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J et al (2021) Crosstalk between cancer—associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer 131:1–30
Joyce JA, Fearon DT (2015) T cell exclusion, immune privilege, and the tumor microenvironment. Science 348:74–80
Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT et al (2008) Collagen density promotes mammary tumor initiation and progression. BMC Med 6:11
Jiang H, Hegde S, DeNardo DG (2017) Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother 66:1037–1048
Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA et al (2020) Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8+ T cell exhaustion. Nat Commun 11:4520
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA et al (2016) Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22:851–860
Mushtaq MU, Papadas A, Pagenkopf A, Flietner E, Morrow Z (2018) Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. JITC 6:1–14
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y et al (2018) TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554:544–548
Wang L, Saci A, Szabo PM, Chasalow SD, Castillo-Martin M, Domingo-Domenech J et al (2018) EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun 9:1–12
Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES (2020) Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol 9:1512
DeNardo DG, Ruffell B (2019) Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol [Internet] 19:369–382
Larsen AMH, Kuczek DE, Kalvisa A, Siersbæk MS, Thorseth M-L, Johansen AZ et al (2020) Collagen density modulates the immunosuppressive functions of macrophages. J Immunol 205:1461–1472
Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD (2018) TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 9:4692
Okrah K, Tarighat S, Liu B, Koeppen H, Wagle MC, Cheng G et al (2018) Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol 15:25
Han C, Liu T, Yin R (2020) Biomarkers for cancer-associated fibroblasts. Biomark Res 8:64
Cavaco ACM, Dâmaso S, Casimiro S, Costa L (2020) Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev 39:603–623
Giussani M, Triulzi T, Sozzi G, Tagliabue E, Giussani M, Triulzi T et al (2019) Tumor extracellular matrix remodeling: new perspectives as a circulating tool in the diagnosis and prognosis of solid tumors. Cells 8:81
Nissen NI, Karsdal M, Willumsen N (2019) Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res 38:115
Karsdal M, Genovese F, Rasmussen D, Bay-Jensen A, Mortensen J, Holm Nielsen S et al (2021) Considerations for understanding protein measurements: Identification of formation, degradation and more pathological relevant epitopes. Clin Biochem 97:11–24
Nissen NI, Johansen AZ, Chen I, Johansen JS, Pedersen RS, Hansen CP et al (2022) Collagen biomarkers quantify fibroblast activity in vitro and predict survival in patients with pancreatic ductal adenocarcinoma. Cancers 14:819
Nielsen MJ, Nedergaard AF, Sun S, Veidal SS, Larsen L, Zheng Q et al (2013) The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am J Transl Res 5:303–315
Rohde H, Vargas L, Eckharthahn KH, Bruguera M, Timpl R (1979) Radioimmunoassay for type III procollagen peptide and its application to human liver disease. Eur J Clin Investig 9:451–459
Brocks DG, Steinert C, Gerl M, Knolle J, Neubauer HP, Günzler V (1993) A radioimmunoassay for the N-terminal propeptide of rat procollagen type III. Application to the study of the uptake of the N-terminal propeptide of procollagen type III in isolated perfused rat liver. Matrix 13:381–387
Niemelä O, Risteli L, Parkkinen J, Risteli J (1985) Purification and characterization of the N-terminal propeptide of human type III procollagen. Biochem J 232:145–150
Wang W-M, Ge G, Lim NH, Nagase H, Greenspan DS (2006) TIMP-3 inhibits the procollagen N-proteinase ADAMTS-2. Biochem J 398:515–519
Di Martino JS, Nobre AR, Mondal C, Taha I, Farias EF, Fertig EJ et al (2022) A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat Cancer 3:90–107
Belhabib I, Zaghdoudi S, Lac C, Bousquet C, Jean C (2021) Extracellular matrices and cancer-associated fibroblasts: targets for cancer diagnosis and therapy? Cancers 13:3466
Erhardtsen E, Rasmussen DGK, Frederiksen P, Leeming DJ, Shevell D, Gluud LL et al (2021) Determining a healthy reference range and factors potentially influencing PRO-C3—a biomarker of liver fibrosis. JHEP Rep 3:100317
Nurmenniemi S, Koivula M-KK, Nyberg P, Tervahartiala T, Sorsa T, Mattila PS et al (2012) Type I and III collagen degradation products in serum predict patient survival in head and neck squamous cell carcinoma. Oral Oncol 48:136–140
Nielsen SH, Willumsen N, Brix S, Sun S, Manon-Jensen T, Karsdal M et al (2018) Tumstatin, a matrikine derived from collagen type IVα3, is elevated in serum from patients with non-small cell lung cancer. Transl Oncol 11:528–534
Thorlacius-Ussing J, Manon-Jensen T, Sun S, Leeming DJ, Sand JM, Karsdal M et al (2020) Serum type xix collagen is significantly elevated in non-small cell lung cancer: a preliminary study on biomarker potential. Cancers 12:1–14
Brachmann CB, Zhang Y, Zavodovskaya M, Hu J, Maltzman JD, Smith V et al (2017) Evaluating collagen neoepitopes as pharmacodynamic biomarkers of GS-5745, an MMP9 inhibitor, in advanced gastric cancer. J Clin Oncol 35:58–58
Bager CL, Willumsen N, Leeming DJ, Smith V, Karsdal MA, Dornan D et al (2015) Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: a preliminary study. Cancer Biomark 15:783–788
Wang J, Chang S, Li G, Sun Y (2017) Application of liquid biopsy in precision medicine: opportunities and challenges. Front Med 11:522–527
Piao X-M, Hwang B, Jeong P, Byun YJ, Kang HW, Seo SP et al (2021) Collagen type VI-α1 and 2 repress the proliferation, migration and invasion of bladder cancer cells. Int J Oncol 59:37
Öhlund D, Elyada E, Tuveson D (2014) Fibroblast heterogeneity in the cancer wound. J Exp Med 211:1503–1523
Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M et al (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214(3):579–596
Biffi G, Tuveson DA (2021) Diversity and biology of cancer-associated fibroblasts. Physiol Rev 101:147–176
Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW et al (2021) Cross-tissue organization of the fibroblast lineage. Nature 593:575–579
Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S et al (2020) Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov 10:232–253
Yamauchi M, Gibbons DL, Zong C, Fradette JJ, Bota-Rabassedas N, Kurie JM (2020) Fibroblast heterogeneity and its impact on extracellular matrix and immune landscape remodeling in cancer. Matrix Biol 91–92:8–18
Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16:582–598
Gascard P, Tlsty TD (2016) Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev 30:1002–1019
Hanley CJ, Noble F, Ward M, Bullock M, Drifka C, Mellone M et al (2016) A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 7:6159–6174
Wagner EF (2016) Cancer: fibroblasts for all seasons. Nature 530:42–43
Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K (2018) Fibroblasts in the tumor microenvironment: shield or spear? Int J Mol Sci 19:1–21
Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan A-C et al (2017) Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res 23:1710–1721
Kawase T, Yasui Y, Nishina S, Hara Y, Yanatori I, Tomiyama Y et al (2015) Fibroblast activation protein-α-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol BioMed Cent 15:109
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR et al (2014) Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25:719–734
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA et al (2014) Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25:735–747
Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S et al (2021) Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Investig 131:e146987
Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H et al (2021) Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39:548–565
Willumsen N, Thomsen LB, Bager CL, Jensen C, Karsdal MA (2018) Quantification of altered tissue turnover in a liquid biopsy: a proposed precision medicine tool to assess chronic inflammation and desmoplasia associated with a pro-cancerous niche and response to immuno-therapeutic anti-tumor modalities. Cancer Immunol Immunother 67:1–12
Wang S, Bager CL, Karsdal MA, Chondros D, Taverna D, Willumsen N (2021) Blood-based extracellular matrix biomarkers as predictors of survival in patients with metastatic pancreatic ductal adenocarcinoma receiving pegvorhyaluronidase alfa. J Transl Med 19:39
de Miguel M, Calvo E (2020) Clinical challenges of immune checkpoint inhibitors. Cancer Cell 38:326–333
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH et al (2018) The immune landscape of cancer. Immunity 48:812–830
Waldman AD, Fritz JM, Lenardo MJ (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20:651–668
Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O et al (2021) Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39:845–865
Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R et al (2021) Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184:596–614
Osipov A, Lim SJ, Popovic A, Azad NS, Laheru DA, Zheng L et al (2020) Tumor mutational burden, toxicity, and response of immune checkpoint inhibitors targeting PD(L)1, CTLA-4, and combination: a meta-regression analysis. Clin Cancer Res 26:4842–4851
Wu Y, Xu J, Du C, Wu Y, Xia D, Lv W et al (2019) The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: a systematic review and meta-analysis. Front Oncol 9:1161
Zhu J, Zhang T, Li J, Lin J, Liang W, Huang W et al (2019) Association between tumor mutation burden (TMB) and outcomes of cancer patients treated with PD-1/ PD-L1 inhibitions: a meta-analysis. Front Pharmacol 10:673
Davis AA, Patel VG (2019) The role of PD-L1 expression as a predictive biomarker: an analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer 7:1–8
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L et al (2019) Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol 12:86
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA et al (2016) Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22:1–13
Ishihara J, Fukunaga K, Ishihara A, Larsson HM, Potin L, Hosseinchi P et al (2017) Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci Transl Med 9:eaan0401
Ishihara J, Ishihara A, Sasaki K, Lee SSY, Williford JM, Yasui M et al (2019) Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med 11:eaau3259
Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A et al (2020) Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat Biomed Eng 4:531–543
Ciardiello D, Elez E, Tabernero J, Seoane J (2020) Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol 31:1336–1349
Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK (2011) Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci 108:2909–2914
Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY et al (2019) Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol 5:1020–1027
Han H, Hou Y, Chen X, Zhang P, Kang M, Jin Q et al (2020) Metformin-induced stromal depletion to enhance the penetration of gemcitabine-loaded magnetic nanoparticles for pancreatic cancer targeted therapy. J Am Chem Soc 142:4944–4954
Froeling FEM, Kocher HM (2015) Homeostatic restoration of desmoplastic stroma rather than its ablation slows pancreatic cancer progression. Gastroenterology 148:849–850
Hauge A, Rofstad EK (2020) Antifibrotic therapy to normalize the tumor microenvironment. J Transl Med 18:207
Jiang H, Torphy RJ, Steiger K, Hongo H, Ritchie AJ, Kriegsmann M et al (2020) Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J Clin Investig 130:4704–4709
Lipton A, Leitzel K, Ali SM, Polimera HV, Nagabhairu V, Marks E et al (2018) High turnover of extracellular matrix reflected by specific protein fragments measured in serum is associated with poor outcomes in two metastatic breast cancer cohorts. Int J Cancer 143:3027–3034
Willumsen N, Ali SM, Leitzel K, Drabick JJ, Yee N, Polimera HV et al (2019) Collagen fragments quantified in serum as measures of desmoplasia associate with survival outcome in patients with advanced pancreatic cancer. Sci Rep 9:19761
Jensen C, Madsen DH, Hansen M, Schmidt H, Svane IM, Karsdal MA et al (2018) Non-invasive biomarkers derived from the extracellular matrix associate with response to immune checkpoint blockade (anti-CTLA-4) in metastatic melanoma patients. J Immunother Cancer 6:1–10
Hurkmans DP, Jensen C, Koolen SLW, Aerts J, Karsdal MA, Mathijssen RHJ et al (2020) Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J Immunother Cancer 8:e001193
Nissen NI, Kehlet S, Boisen MK, Liljefors M, Jensen C, Johansen AZ et al (2021) Prognostic value of blood-based fibrosis biomarkers in patients with metastatic colorectal cancer receiving chemotherapy and bevacizumab. Sci Rep 11:865
Funding
No funding to be declared.
Author information
Authors and Affiliations
Contributions
NW collected the related studies and drafted the manuscript. NW, CJ, GG, NIN, JN, DMN, RSP, PF, CC, MBL and MK participated in the design of the review. NW, CJ, GG, NIN, JN, DMN, PF, IMC, DHM, IMS, AL, KL, SMA, JTE, DPH, RHJM, JG, CC, MBL, MK interpreted the summarized patient data. CJ, GG, NIN, JN, DMN, RSP, IMC, MKB, AZJ, DHM, IMS, AL, KL, SMA, DPH, RHJM, JA, ME, JG, CC, MBL, MK substantively revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
NW is employed and have stockownership and patents in Nordic Bioscience. CJ is employed at Nordic Bioscience. GG is employed and have stockownership and patents or other related intellectual property in Bristol Myers Squibb. NIN is employed in Nordic Bioscience. JN is employed and have stockownership in Bristol Myers Squibb. DMN is employed and have stockownership and patents or other related intellectual property in Bristol Myers Squibb. PF is employed in Nordic Bioscience. RSP is employed in Nordic Bioscience. IMC declares a consultancy/advisory role at Amgen, to have received research funding from Bristol Myers Squibb, Roche, Genis TILT, Lytix and travels expenses paid by Bristol Myers Squibb, Roche, Bayer. MKB declares no competing interests. AZJ declares no competing interests. DHM declares no competing interests. IMS declares paid honoraria from IO Biotech, MSD, a consultancy/advisory role at Pierre Fabre and Novartis, to have received research funding from Bristol Myers Squibb, Roche, TILT, Lytix and travels expenses paid by MSD. AL declares no competing interests. KL declares no competing interests. SMA declares no competing interests. JTE is a founder and CEO of LOXiGen ApS and was CEO of LOXiPharm IVS. DPH declares no competing interests. RHJM declares a consultancy/advisory role at Servier, to have received research funding from Astellas, Bayer, BI, Cristal tx, Pamgene, Pfizer, Novastis, Roche, Servier, Sanofi and to have patents or other related intellectual property pending at Pamgene. JA declares to have patents or other related intellectual property at Amphera and travels expenses paid by MSD. ME declares paid honoraria from Sanofi and a consultancy/advisory role at Pfizer. JG declares paid honoraria and a consultancy/advisory role at Roche, MSD, Pfizer, Abbvie, AstraZeneca, Novartis, Pharmaxis, Cincera, Novo, Gilead, Norgine, Eisai. CC have stockownership and patents in Nordic Bioscience. MBL declares no competing interests. MK is employed and have stockownership and patents in Nordic Bioscience.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Willumsen, N., Jensen, C., Green, G. et al. Fibrotic activity quantified in serum by measurements of type III collagen pro-peptides can be used for prognosis across different solid tumor types. Cell. Mol. Life Sci. 79, 204 (2022). https://doi.org/10.1007/s00018-022-04226-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04226-0