
Abstract
Influenza A viruses cause a mild-to-severe respiratory disease that affects millions of people each year. One of the many determinants of disease outcome is the innate immune response to the viral infection. While antiviral responses are essential for viral clearance, excessive innate immune activation promotes lung damage and disease. The influenza A virus RNA polymerase is one of viral proteins that affect innate immune activation during infection, but the mechanisms behind this activity are not well understood. In this review, we discuss how the viral RNA polymerase can both activate and suppress innate immune responses by either producing immunostimulatory RNA species or directly targeting the components of the innate immune signalling pathway, respectively. Furthermore, we provide a comprehensive overview of the polymerase residues, and their mutations, associated with changes in innate immune activation, and discuss their putative effects on polymerase function based on recent advances in our understanding of the influenza A virus RNA polymerase structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A 100 years after the devastating influenza pandemic of 1918, influenza A viruses continue to pose a serious threat to human health. They infect up to twenty percent of the global population each year, resulting in several hundred thousand deaths [1]. In addition, influenza A viruses have a well-established zoonotic potential and ability to cause pandemics in naïve populations. In humans, influenza disease typically manifests itself as a mild-to-severe respiratory disease, but influenza A virus infections can occasionally spread beyond the respiratory tract. In humans and animal models, viral antigens have also been detected in the nervous system and heart [2,3,4].
Many factors contribute to influenza disease severity, including underlying risk factors, bacterial co-infections, and the innate immune response to infection [5, 6]. During an influenza virus infection, the innate immune response is typically activated when viral RNA (vRNA) is detected by the cytoplasmic or nuclear retinoic acid-inducible gene I (RIG-I) [7,8,9]. Subsequent signalling events lead to the production of interferons and other cytokines, which together mount a robust antiviral defence, and attract leukocytes and lymphocytes to clear the IAV infection. Counterintuitively, the virus can benefit from the pro-inflammatory responses in the lung, if it can use recruited leukocytes as additional targets for replication, as was shown for a low-dose influenza A virus infection [10]. Another outcome of infection is a disproportional and prolonged innate immune activation, commonly referred to as ‘cytokine storm’, that can cause tissue damage, acute lung injury, or severe acute respiratory distress syndrome [11, 12]. Prolonged exposure to IFNs, which are one group of cytokines overproduced during the ‘cytokine storm’, has also been linked to impaired lung repair and an increased susceptibility to bacterial infection after influenza virus infection in mice [13]. The cytokine storm is particularly common in infections with highly virulent influenza A virus strains, such as highly pathogenic avian H5N1 and H7N9 viruses, and the 1918 pandemic H1N1 strain, which trigger an overproduction of interferons and pro-inflammatory cytokines in the lower respiratory tract [14,15,16,17]. Several viral and host determinants of the immune dysregulation have been proposed, but the molecular mechanisms that allow these factors to start a cytokine storm are still poorly understood (reviewed in [18]).
Activation of the innate immune response is triggered by the replication of the viral genome [19]. The influenza A virus genome consists of eight segments of negative-sense single-stranded vRNA that vary in length from 890 to 2341 nucleotides and code for ten major proteins, such as nucleoprotein (NP), non-structural protein 1 (NS1), and polymerase subunits [polymerase acidic (PA), polymerase basic 1 and 2 (PB1 and PB2)], as well as various accessory proteins, such as PA-X and PB1-F2 [20]. Each vRNA segment is encapsidated by a double-helical filament of NP and capped by a copy of the viral RNA polymerase, forming a viral ribonucleoprotein (vRNP) (Fig. 1A). It is in the context of these RNPs that the viral RNA polymerase copies and transcribes the genome segments [21]. In addition to making full-length copies of the viral genes, the viral RNA polymerase produces various aberrant RNA products that contain deletions in the genome segments, such as defective viral genomes (DVGs), mini viral RNAs (mvRNAs) and small viral RNAs (svRNAs) [22]. Various lines of research suggest that DVGs and mvRNAs play a role in activating the innate immune response through RIG-I [23, 24].

Structure and function of the influenza A virus RNA polymerase. A A schematic representation of an RNP, in which both termini of the vRNA are bound by the viral polymerase and the rest is associated with NP. B A surface representation of the human H3N2 influenza A virus RNA polymerase (PDB 6RR7) bound to the 3′ and 5′ promoter and the capped RNA (black; labelled as product). PA C-terminal (PA-C) and endonuclease (PA-endo) domains and PB2 cap-binding (PB2-cap), Mid-link and 627 domains are indicated. C A model of influenza A virus polymerase showing the active site cavity within the PB1 subunit. The location of the four channels that lead to the active site and the binding pockets of the 3′ and 5′ RNA termini are indicated. D Schematic representation of influenza virus genome transcription and replication. During transcription PA-C interacts with the Ser5-phosphorylated CTD of a transcribing RNA Pol II. PB2-cap binds the cap structure of the nascent cellular RNA, which is subsequently cleaved by PA and used to prime viral mRNA synthesis. Capped and polyadenylated viral mRNAs are translated in the cytoplasm by the host ribosomes. The newly made components of the viral RNP are transported into the nucleus by importins where they promote viral replication. During cRNA synthesis from a vRNA template, a newly made encapsidating polymerase forms a dimer with a replicating polymerase, while ANP32A acts as a bridge between the polymerases. During vRNA synthesis from a cRNA template a newly made trans-activating polymerase forms a dimer with the replicating polymerase, stimulating replication. An encapsidating-replicating polymerase dimer is also likely to form during vRNA synthesis with the aid of ANP32A to promote vRNP assembly. Examples of other cellular factors that are involved in either transcription or replication are shown at the bottom of the diagram
Several viral proteins, such as NS1, PA-X and PB1-F2, have well-established functions as innate immune modulators [25, 26]. However, these proteins do not explain the full complexity of the activation and inhibition of the innate immune response during infection. A factor, whose role in triggering and antagonising the innate immune response has been less comprehensively analysed, is the influenza A virus RNA polymerase. The role of the influenza virus RNA polymerase in innate immune responses to infection became evident in genome segment reassortment studies, in which polymerase encoding segments were exchanged between different virus strains or isolates. Together those studies demonstrated that the polymerase genes not only significantly affect virulence and innate immune signalling, but also likely contribute to cytokine dysregulation during infection with highly pathogenic virus strains [27,28,29,30,31] (full list in Supplementary Table 1). Similarly, innate immune activation can be induced by single mutations in the polymerase genes or combinations of mutations in one or more genes [24, 32, 33] (full list in Supplementary Table 2). Additional studies have shown that elimination of the interferon pressure during infection leads to substitution of conserved polymerase amino acids, indicating that the RNA polymerase amino acid composition is under a selection pressure from the immune response [34, 35].
In the last decade, significant advances have been made in understanding influenza A virus genetics, vRNA and vRNP structure, and the catalytic activity of the influenza A virus RNA polymerase [21]. However, the mechanisms underlying the immuno-stimulatory and -inhibitory effects of the viral polymerase are complex and diverse, and they have, to our best knowledge, not been comprehensively reviewed in light of these recent advances. Here, we aim to bring together our current understanding of the RNA polymerase, the role of the RNA polymerase in the innate immune response during influenza virus infection, and the polymerase mutations that affect innate immune signalling and host adaptation. By framing these concepts within our understanding of the RNA polymerase structure, we hope to extend our knowledge of the innate immune activation during influenza virus infection and the outcome of influenza disease.
Structure of the viral RNA polymerase
The viral RNA polymerase transcribes and replicates the vRNA segments in the context of vRNPs (Fig. 1A). Within an RNP, the RNA polymerase binds to the viral promoter, which is formed by partially complementary 5′ and 3′ termini of the bound vRNA segment [36,37,38]. This organisation ensures that the vRNA termini as well as the RNA polymerase reside at one end of the RNP.
The influenza virus RNA polymerase is comprised of three subunits: PB1, PB2 and PA (Fig. 1B). The PB1 subunit contributes most to the RNA polymerase core and contains a typical RNA-dependent RNA polymerase (RdRp) domain fold, with fingers, thumb and palm subdomains. The three subdomains give rise to the structure of the active site and provide the residues that coordinate nucleotide incorporation [39,40,41]. The carboxy-terminal domain of PA and the amino-terminal third of PB2 also contribute to the RNA polymerase core, and in particular the thumb subdomain of the active site. The active site can be accessed by four channels that direct the movement of the RNA template in and out of the active site, the RNA product out of the active site, and nucleotides towards the active site (Fig. 1C). The RNA polymerase core is surrounded by several additional domains that are attached to linkers and support the transcriptional activity of the RNA polymerase [40,41,42]. These additional domains include an endonuclease domain that resides in the amino-terminal third of PA and a cap-binding domain that resides in the C-terminal third of PB2. Other important PB2 domains include the N-terminal domain, Mid-link, 627 domain, and nuclear localisation signal (NLS) domain [40].
Function of the viral RNA polymerase
Upon viral entry and release of the vRNPs from the virion into the cytoplasm, the vRNPs are transported by host cell importins into the nucleus where primary transcription takes place (Fig. 1D) [43]. The process of viral transcription is initiated when the PA C-terminus of the incoming vRNP associates with the C-terminal domain (CTD) of an initiating, serine-5 phosphorylated cellular RNA polymerase II (Pol II). Following this interaction, the cap-binding domain of PB2 binds the cap-structures of nascent Pol II RNAs, ensuring that they can be cleaved by the endonuclease domain of PA. The resulting capped primers are 10–14 nucleotides long and used by PB1 as primers for viral transcription [41, 44, 45]. Viral transcription terminates when the viral RNA polymerase stutters on a poly-uridine track at the 5′ terminus of the vRNA template, producing a polyA tail [41, 46]. The resulting viral mRNAs are translated by host ribosomes into new viral proteins required for viral replication (Fig. 1D). These new viral proteins are transported from the cytoplasm to the nucleus by host cell importins [47].
In contrast to viral transcription, viral replication is a two-step process that starts with the generation of a complementary RNA (cRNA) intermediate using a primer-independent initiation mechanism (Fig. 1D). As the nascent cRNA leaves the active site through the product exit channel, it must be encapsidated by a newly synthesized RNA polymerase and nucleoproteins [21, 22]. To start the encapsidation process, the replicating polymerase and new polymerase must form a dimer that is stabilised by host protein Acidic Nuclear Phosphoprotein 32 Family Member A (ANP32A) [48,49,50,51,52,53,54]. Next, the new RNA polymerase in the resulting cRNP uses the cRNA as template for vRNA synthesis [55]. However, the initiation of vRNA synthesis requires a trans-activating or regulatory polymerase, as well as an encapsidating RNA polymerase and ANP32A. The trans-activating polymerase and the replicating polymerase form a dimer that is distinct from the encapsidating-replicating polymerase dimer (Fig. 1D). This dimer is likely formed to ensure that enough newly made viral proteins are available to assemble the nascent vRNA into a vRNP, and to trigger an essential realignment step during the initiation of vRNA synthesis when this condition is met [48, 56, 57].
Besides Pol II and ANP32A, viral transcription and replication are assisted by a number of other cellular proteins, many of which also directly interact with the viral polymerase (reviewed in [58]). These host factors participate in different stages of RNA synthesis, such as cap snatching (rRNA processing 1 homolog B or RRP1B, RNA exosome), general transcription (chromodomain helicase DNA-binding 1 or CHD1, hCLE and NXP2) polyadenylation (splicing factor proline-glutamine rich or SFPQ), cRNA synthesis (minichromosome maintenance or MCM) nuclear import and assembly of the polymerase (importins, long non-coding RNA-PAAN or lncRNA-PAAN, heat shock protein 90 or Hsp90), and vRNP assembly and transport (LYAR, FMRP and CLUH) (Fig. 1D) [58].
RIG-I signalling pathway activation by IAV
Pathogen receptors, or pattern recognition receptors (PRRs), start the innate immune response during an infection. PRRs function by binding to conserved structures, or pathogen-associated molecular patterns (PAMPs), and activating signalling cascades that trigger expression of innate immune genes, such as type I and type III interferons. There are at least three PRR protein families involved in the recognition of an influenza virus infection, including toll-like receptors (TLRs), the nucleotide oligomerization domain (NOD)-like receptors (NLRs) and RIG-I-like receptors (RLRs) [59]. In addition, influenza A virus RNA is bound by Z-DNA binding protein 1, an activator of necroptosis [60].
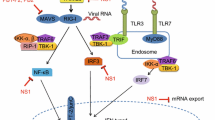
In most cell types, the RIG-I signalling pathway (Fig. 2) plays a key role in detecting influenza A virus RNA [8, 9, 61]. Only in plasmocytoid dendritic cells are the influenza virus RNA molecules mainly detected by the endosomal TLR7 [62, 63]. RIG-I is activated when its C-terminal domain (CTD) binds the partially double-stranded 3′ and 5′ termini of the vRNA or cRNA promoter, so-called ‘panhandle’ [7, 64, 65] (Fig. 2). This binding leads to an ATP-dependent conformational change in RIG-I that exposes the N-terminal caspase activation and recruitment domains (CARDs) [66]. At the same time, the RNA-binding domains of RIG-I (CTD and helicase) translocate along the RNA ligand, bringing several RIG-I molecules and their CARDs into proximity [67,68,69]. The exposed CARDs of RIG-I are next polyubiquitinated by the tripartite motif-containing protein 25 (TRIM25), which promotes formation of CARD tetramers [70, 71]. CARD tetramers of RIG-I subsequently bind to the CARDs of mitochondrial antiviral-signalling protein (MAVS), nucleating MAVS filament formation—a step necessary for subsequent signal transduction [72]. MAVS oligomerisation leads to recruitment of downstream signalling molecules, such as TNF receptor-associated factor (TRAF) family E3 ubiquitin ligases and inhibitor of NF-κB kinase (IKK) family members. IKKs subsequently activate interferon-regulatory factors 3 and 7 (IRF3, IRF7) and NF-κB, which translocate into the nucleus to promote the transcription of interferon and pro-inflammatory cytokine genes [73].

RIG-I signalling pathway and its interaction with the influenza A virus polymerase subunits. Centre left to centre bottom: the RIG-I signalling pathway is activated upon binding of an RNA ligand, such as the 5′ppp-dsRNA region of the influenza virus promoter, by the CTD of RIG-I. A subsequent ATP-dependent conformational change exposes two N-terminal CARDs of RIG-I and promotes RIG-I translocation along the RNA and formation of a RIG-I oligomer. One of the host proteins that potentiates RIG-I signalling is PKR activating protein (PACT), which binds to the CTD domain of RIG-I and enhances ATPase activity. RIG-I filament formation brings RIG-I CARDs into close proximity, facilitating formation of a CARD tetramer. The tetramer is stabilised by ubiquitin chains that are added by TRIM25. The RIG-I complex then migrates towards the mitochondrial outer membrane, where it associates with MAVS. The interaction between the CARD domains of RIG-I and MAVS nucleates MAVS filament formation. MAVS aggregation induces binding of TRAF family E3 ubiquitin ligases to MAVS, which potentiate recruitment of IKKs. Activated IKKε and TBK1 phosphorylate transcriptional factors IRF3 and IRF7, while transcriptional factor NF-κB is activated by the IKKα/β/γ complex. The activated IRF3, IRF7 and NF-κB translocate into the nucleus where they induce transcriptional activation of IFN and pro-inflammatory cytokine genes. Top left: the influenza virus RNA polymerase shields viral promoter from RIG-I recognition. Polymerase subunits also bind RIG-I in an ‘ESIE’-motif-dependent manner and interact with PACT. Middle: PB2 binding to MAVS is associated with inhibition of IFN expression and is attributed either to its N-terminal domain or to PB2 amino acids 588 and 292. PB1 induces degradation of MAVS by forming a complex with MAVS, RNF5 and NBR1. PB1 induces Lys27-linked ubiquitination of MAVS by RNF5, which is then recognised by NBR1 that targets ubiquitinated MAVS for autophagic degradation. Bottom right: PB2 (residues 490–759) binds to TRAF3 and prevents its Lys63-linked polyubiquitination by TRIM35, disrupting the formation of TRAF3-MAVS complex and preventing activation of TBK1 and IKKε kinases. TRIM35, in turn, induces Lys48-linked polyubiquitination of PB2, targeting it for proteasomal degradation. PB2 is also targeted to mitochondria via residues L7, L10 and N9. Bottom left: PA inhibits IFN production by binding to IRF3 and precluding its phosphorylation and nuclear translocation
Our understanding of the mechanisms behind the interactions between RIG-I and the viral promoter are complicated by the fact that the viral RNA polymerase shields the partially double-stranded promoter from RIG-I recognition in the context of an RNP. The RNA polymerase binds the first 10 residues of the 5′ terminus in a hook structure within a binding pocket consisting of PA and PB1 residues. The 3′ terminus, on the other hand, exists in at least three positions: either bound above the active site (A-site), on the outside of the PB2-N1 and PB1 thumb subdomain (B-site), or in the active site (Fig. 3) [36, 41, 56, 74]. Even during viral genome replication, nascent RNA is likely directly encapsidated by a new polymerase [48, 55]. Nevertheless, base-pairing of the terminal promoter region, which occurs in the absence of polymerase, is important for RIG-I activation [7]. It has been proposed that influenza viruses evolved a promoter that is not completely double-stranded, such as observed in other negative-strand RNA viruses, and that interruptions in the duplex reduce RIG-I activation relative to a fully base-paired promoter [7, 75]. It, therefore, remains unknown at which stage of the viral life cycle RIG-I is able to gain access to the viral promoter and initiate signalling.

The conformations of the viral promoter when bound to the polymerase heterotrimer. Left: bat H17N10 influenza A polymerase with bound vRNA promoter (PDB 4WSB). Middle: bat H17N10 influenza A polymerase pre-termination complex with 3′ terminus threaded through the template exit channel and bound at the polymerase surface (top view) (PDB 6SZU). Right: human H3N2 influenza A virus polymerase bound to a vRNA promoter and a capped primer with 3′ terminus entering the active site (PDB 6RR7). Viral RNA polymerases are shown in surface representation and the viral promoter strands as a cartoon
Interference of the viral RNA polymerase with the components of RIG-I signalling pathway
Besides its major role in genome replication and transcription, the influenza A virus polymerase and its individual subunits specifically interact with and inhibit several components of the RIG-I signalling pathway (Fig. 2).
Viral polymerase binds RIG-I and its adaptor protein PACT
Several studies have demonstrated a direct interaction between the RNA polymerase or the vRNPs and RIG-I [32, 76, 77]. Weber et al. [76] showed that incoming vRNPs with an avian-adapted 627E residue in PB2 were more efficiently bound by RIG-I than those with the mammalian-adapted 627K signature. However, this difference in binding did not affect innate immune signalling. Instead, the authors proposed that the interaction allows RIG-I to block replication of 627E-containing viruses in mammalian cells [76]. Similarly, Li et al. [77] showed that all three polymerase subunits of a H9N2 virus strain bind RIG-I, and that this interaction does not result in innate immune activation. By contrast, Liedmann et al. [32, 78] identified an ‘ESIE’ motif consisting of PB1 (398E/524S/563I) and PA (351E) residues, which not only enhances the binding of the viral RNA polymerase to RIG-I, but also inhibits innate immune activation when compared to the ‘GGRK’ variant of the same motif (Fig. 2). Except PB1 398E, the motif’s residues are conserved and located on the thumb subdomain side of the RNA polymerase. By contrast, PB1 398E is localised at the opposite side of the RNA polymerase, and it is therefore unknown how these residues collectively contribute to RIG-I binding and whether separate residues might exhibit different immunomodulatory effects.
All three subunits of the viral RNA polymerase also interact with the PKR activating protein (PACT), an activator of PKR and RIG-I [79, 80]. In the case of RIG-I, PACT binds to the RIG-I CTD and triggers ATPase activity [80]. Chan et al. [81] showed that overexpression of the polymerase subunits diminishes IFN-β promoter activation during overexpression of RIG-I and PACT in the absence of vRNA. Additional experiments showed that the overexpressed polymerase subunits can co-precipitate with PACT, in the absence of vRNA, and that knockdown of endogenous PACT stimulates influenza A virus polymerase activity. The interaction between the RNA polymerase and PACT can be interpreted as a viral strategy to interfere with host innate immune signalling as well as an antiviral strategy of the host cell [81]. At present, it is unclear whether the observed immunomodulatory effects are directly linked to the interaction of the viral polymerase with PACT or that they derive from a reduced activation of RIG-I, or both.
Polymerase subunits target MAVS, TRAF3 and IRF3
The PB1 subunit of the RNA polymerase was recently found to inhibit RIG-I signalling by inducing autophagic degradation of MAVS [82]. Specifically, PB1 forms a complex with MAVS and E3 ligase RNF5. This complex allows RNF5 to add Lys27-linked ubiquitin to MAVS (Fig. 2), which is recognised by an autophagic receptor, neighbour BRCA1 (NBR1). NBR1 recognition subsequently targets MAVS for autophagic degradation, inhibiting MAVS-mediated signalling [82].
The PB2 subunit of the RNA polymerase also inhibits innate immune signalling by targeting MAVS [83, 84]. The MAVS-interacting region of PB2 was mapped to the last 37 residues of its N-terminus, but mutations outside of the N-terminal region also affect PB2-MAVS binding [85,86,87]. One of those mutations is PB2 T588I, which was identified in a swine isolate of the 2009 pandemic H1N1 virus strain (pdm09) that was highly pathogenic in mice [85]. Upon closer examination, the T588I mutation was found to improve polymerase activity and increase viral replication in cell culture and murine lungs. Interestingly, the mutation also led to a decrease in IFN-β expression [85]. This decrease correlated with the stronger binding of the T588I mutant to MAVS [85]. Another mutation implicated in MAVS binding, and gaining prevalence among circulating avian H9N2 viruses in recent years, is I292V in the Mid-link domain of PB2 [86]. I292V improved PB2-MAVS binding and decreased IFN-β expression, resulting in a more severe disease in mice [86]. Although it is unknown how PB2 inhibits innate immune activation upon MAVS binding, it is possible that PB2 prevents a correct subcellular localisation of MAVS, limits MAVS oligomerization, or induces MAVS degradation [88, 89].
Another mechanism through which PB2 modulates MAVS-mediated signalling is by targeting TNF receptor-associated factor 3 (TRAF3), the adaptor protein of MAVS that is required for optimal signal transduction [90]. TRAF3 interacts with MAVS (Fig. 2), catalysing recruitment of the TBK1 and IKKε kinases, which in turn phosphorylate IRF3 promoting expression of IFN genes [90, 91]. To achieve this, TRAF3 needs to be activated by TRIM35 through the addition of Lys63-linked polyubiquitin. PB2 prevents polyubiquitination of TRAF3 by binding to TRAF3 with PB2 residues 490–759. The binding between PB2 and TRAF3 disrupts formation of the TRAF3-MAVS complex, and inhibits downstream IFN-β promoter activation. TRIM35, on the other hand, counters this immunomodulatory activity of PB2 by adding Lys48-linked polyubiquitin to Lys736 of PB2, which targets PB2 for proteasomal degradation [90].
The PA subunit of the viral RNA polymerase also inhibits innate immune signalling by binding to IRF3. This interaction prevents phosphorylation and nuclear translocation of IRF3, both of which are central for IFN expression [92]. The same study also showed that a D108A mutation in PA inhibits PA-IRF3 interaction. However, it is unclear how D108 could be involved in this interaction as it is a catalytic residue of the endonuclease and not located on the surface of the domain.
PB2 is targeted to mitochondria
Mitochondria serve as platform for the interaction between RIG-I and MAVS and thus play a prominent role in innate immune signalling [88, 93, 94]. Influenza A viruses encode an accessory protein, called PB1-F2, that specifically localises to mitochondria and interferes with their function in innate immune signalling [26]. However, there is also a small pool of PB2 that localises to the mitochondrial matrix. The purpose of this localisation remains controversial [84, 95,96,97].
The mitochondrial targeting signal of PB2 has been mapped to PB2 residues L7 and L10, or N9 [84, 95]. It has also been shown that the PB2 proteins of different influenza A virus strains have different mitochondrial localisation tendencies, with seasonal human strains localising to mitochondria and avian strains showing reduced localisation, suggesting that mitochondrial localisation might play a role in host adaptation [84]. The strain-specific localisation of PB2 dependents on the PB2 residue nine, which is typically an aspartate (D) in the PB2 proteins of avian strains and an asparagine (N) in the PB2 proteins of human-adapted strains. In vitro, introduction of an avian-like N9D mutation in the PB2 of the A/WSN/33 virus strain increases IFN-β expression in lung epithelial cells. In mice, however, the N9D mutation decreased IFN-β expression, likely because of the impaired growth of the N9D mutant in vivo [84]. Presently, the mechanisms behind innate immune modulation by the mitochondrial PB2 remain unclear, but they might involve changes in mitochondrial dynamics or mitochondrial membrane potential [95, 96].
Aberrant replication products as innate immune activators
Aberrant replication products and their synthesis
Besides full-length replication products, the influenza virus RNA polymerase also generates aberrant replication products, including DVGs, mvRNAs, and svRNAs (Fig. 4A) [24, 98,99,100]. svRNA are 21–27 nucleotides long and only contain the 5′ terminus of the vRNA template. By contrast, DVGs and mvRNAs both contain the conserved 5′ and 3′ terminal ends that are present in each vRNA segment and bound by the viral RNA polymerase. However, they lack internal sequences and can be distinguished from full-length vRNAs by their size, with DVGs being typically 178 to several hundred nucleotides long, and mvRNAs being 56–125 nucleotides long [24, 101, 102].

Aberrant viral RNA formation and RNA polymerase residues putatively implicated in this process. A A schematic representation of different types of aberrant RNAs produced by the influenza A virus polymerase. B A possible mechanism for the DVG and mvRNA synthesis during replication. C A surface representation of the bat H17N10 influenza A polymerase (PDB 6T0V) during transcription elongation. The 3′ terminus of the template (orange) can be seen exiting through the template exit channel. PB2 residues 9 and 81 are highlighted in red and PB2 80–90 loop is shown in purple. D A cartoon representation of the template exit channel of the bat H17N10 influenza A polymerase at the stage of transcription pre-initiation (PDB 6T0N) and during transcription elongation (PDB 6T0V). The 80–90 loop (purple) undergoes an outward movement, allowing opening of the template exit channel during elongation. Direction of the exiting template is indicated with an arrow. E A surface representation of the replicating-encapsidating polymerase dimer of the influenza C virus (ICV) polymerase in the complex with chicken ANP32A (PDB 6XZR). Residue numbering is as in ICV RNA polymerase. The corresponding IAV RNA polymerase residues are shown in parenthesis. PB2 residue F228 of ICV (A221 IAV) is located in an RNA path (yellow arrow) between the product exit channel of the replicating polymerase and the 5′ binding site of the encapsidated polymerase. Hydrogen bonds between the PA E513 of ICV (D529 in IAV) and two lysines of ANP32A (K99 and K101) are shown. F A surface representation of the bat H17N10 influenza A virus bound to the CTD of Pol II (PDB 5M3H). Two binding sites for Pol II CTD (orange) are shown. Residue numbering as in bat IAV, and corresponding human and avian residues are shown in parenthesis. PA residues, K630 and R633 of the bat IAV polymerase (corresponding to K635 and R638 of the human or avian IAV polymerase, respectively) are shown to form hydrogen bonds with phosphorylated Ser5 of the Pol II CTD. PA residue C448 in the bat IAV polymerase (C453 in the human or avian IAV) is indicated in purple
vRNAs, DVGs and mvRNAs all contain the conserved RNA promoter structure with 5′ triphosphate that forms a ‘panhandle’ in solution [103]. In vitro and in vivo, this RNA structure is recognised by the cellular RIG-I sensor and able to activate the MAVS signalling cascade [7, 19, 24, 66, 104]. RIG-I binds different influenza A virus RNA species with different efficiencies. Aberrant RNAs of 56–125 nucleotides long are bound by RIG-I more efficiently than longer aberrant RNAs, and shorter vRNA segments are bound more efficiently than longer vRNA segments [24, 104]. Interestingly, aberrant RNAs shorter than 56 nucleotides are not bound by RIG-I at all, even though short artificial hairpins are potent RIG-I agonists [24, 105]. Although, both mvRNAs and DVGs are potent inducers of IFN expression, they are thought to have opposite effects on disease, with mvRNAs having been linked to virulence and the cytokine storm, both common for the highly pathogenic influenza virus strains, and DVGs to protective IFN responses and a reduction of viral virulence [24, 106, 107].
The molecular mechanisms underlying DVG and mvRNA formation are currently poorly defined. In one model, the internal deletions are generated when the viral RNA polymerase pauses during elongation, back-tracks to separate template and nascent strand, and finally translocates to a downstream template sequence to realign the nascent strand and continue nascent RNA extension (Fig. 4B) [22, 98]. Such polymerase translocations might be affected or directed by A/U-rich sequences, which have been observed near DVG breakpoints [108, 109]. It is possible that such A/U-rich sequences facilitate separation of the template and nascent strand prior to translocation. Other models for DVG and mvRNA synthesis, which involve, for instance, endonucleolytic cleavage and ligation of the product RNA, are not supported by experimental data [98].
Several viral factors have been associated with the formation of aberrant RNAs. Recent studies have shown that elongation defects can be induced experimentally by limiting the availability of NP, an important elongation factor and key component of RNPs, suggesting that impaired elongation or aberrant encapsidation play a role in DVG or mvRNA formation [24, 110, 111]. In addition, mutations in several viral proteins, such as nuclear export protein, matrix protein 1 and 2, and the RNA polymerase subunits, also promote the formation of DVGs [34, 112,113,114].
RNA polymerase mutations that affect aberrant RNA synthesis
We can learn more about the potential mechanisms underlying aberrant RNA generation and their role in innate immune activation by studying the RNA polymerase mutations that affect their formation. High levels of mvRNA production by the polymerases of highly pathogenic avian H5N1 and 1918 pandemic viruses are in part determined by avian-adaptive mutations in the PB2 polymerase subunit of those strains [24]. Introducing such avian-specific PB2 mutations, e.g., N9D and M81T, into the lab-adapted A/WSN/33 (H1N1) strain significantly increased mvRNA production and IFN-β promoter activation [24]. Both these residues are located at the top of the RNA polymerase core, near the interaction interface of the PB1 C-terminus and the PB2 N-terminus (Fig. 4C). Of the two residues, PB2 residue 81 stands out as it is located within the PB2 80–90 loop that undergoes an outward conformational change to allow template egress during elongation (Fig. 4D) [41]. Because of the role of the 80–90 loop in elongation, it is tempting to speculate that a mutation of residue 81 could trigger elongation defects, which may contribute to mvRNA production.
Synthesis of aberrant RNAs is also affected by the fidelity of the viral polymerase. A V43I mutation in the PB1 subunit, which reduces the mutation rate of RNA synthesis by approximately twofold in some genetic backgrounds, was also shown to lower levels of mvRNA synthesis by the polymerases of the 1918 pandemic and H5N1 strains [24, 115]. Interestingly, V43I change in H5N1 strain also decreased neurotropism and lowered lethality in mice [116]. V43I is located near the NTP entry channel of the polymerase and may increase polymerase fidelity by improving nucleoside selectivity [116]. However, it remains unclear whether the same mechanisms could also contribute to the production of aberrant RNA species.
Similar to mvRNA synthesis, DVG formation is affected by mutations in the RNA polymerase. Two of these mutations, PB2 A221T and PA D529N, were identified in a virus isolated from a fatal case of pdm09 (H1N1) influenza [114]. Interestingly, the two mutations demonstrated opposite effects on DVG generation and immune activation when studied in more detail. PB2 A221T increased DVG formation and enhanced protective antiviral responses, while PA D529N counteracted both effects [114]. Analysis of the localisation of PA A221 in various influenza virus RNA polymerase structures shows that this residue can be involved in the interaction between the N-terminal and the 627 domains of PB2 (PDB: 6T0V) or it can be residing in the path that the nascent RNA takes when it emerges from the replicating polymerase to bind the encapsidating polymerase (PDB: 6XZR) (Fig. 4E). While it is not clear if those localisations are directly involved in DVG production, it is tempting to speculate that the A221T mutation could affect encapsidation of the nascent RNA, thereby reducing processivity. Alternatively, the mutation may lead to aberrant RNA formation by affecting NP recruitment to the nascent strand, which was proposed to occur near the RNA transition path [48]. By contrast, PA D529 resides above the Pol II binding interface of the transcriptionally active polymerase (PDB: 6T0V), while in the ANP32A-supported dimer, PA D529, is located at the ANP32A interaction interface (Fig. 4E) [48]. Thus, in the dimer, PA D529N is ideally positioned to compensate for defects in polymerase processivity or encapsidation by stabilizing ANP32A binding and RNA polymerase dimer formation.
Aberrant polymerase activity can also be the result of defects in viral transcription. Influenza virus transcription is dependent on cap-snatching and the binding of the C-terminal domain of PA to a Ser5 phosphorylated CTD of Pol II. Mutations in key PA residues involved in this interaction, K635A and R638A (Fig. 4F), not only decrease the activity of the A/WSN/33 (H1N1) polymerase, but also promote DVG formation [108, 113, 117]. On the other hand, a PA C453R mutation at same site (Fig. 4F) reduces DVG formation, because it may restore the binding of PA to the Pol II CTD, as suggested by the structural analysis [113, 117]. What might be the mechanism behind DVG formation in this case? As mentioned above, elongation defects can be induced experimentally by limiting the availability of NP during viral replication [24, 110, 111]. Although not experimentally confirmed, the transcriptional defects induced by reduced Pol II binding likely result in lower NP levels, which subsequently promote aberrant polymerase activity [24, 110, 111].
Host-adaptive polymerase mutations improve polymerase activity and increase innate immune activation
The influenza A virus RNA polymerase plays a major role in the adaptation of avian influenza virus strains to mammalian cells. Several major host-adaptive mutations induce a strong activation of innate immune responses which correlates with the improvement in viral replication.
One of the most well-studied polymerase adaptations is a E-to-K mutation at residue 627 of PB2 [118]. This mutation is located in the PB2 627-domain (named after the 627 mutation; Fig. 5) and able to improve the activity of avian-adapted influenza A virus polymerases in mammalian cells [31]. This improvement results in higher viral loads, extra-respiratory spread and enhanced virulence of the E627K-containing avian viral strains in mice [119,120,121,122,123]. In the majority of cases, the increased virulence of the 627K-containing avian viruses is also accompanied by an overproduction of pro-inflammatory cytokines, persistent neutrophil infiltration and delayed lymphocyte recruitment, which are all hallmarks of the cytokine storm [119,120,121,122,123,124].

Host-adaptive immunostimulatory mutations. A surface representation of the replicating-encapsidating polymerase dimer of the ICV polymerase in the complex with chicken ANP32A (PDB 6XZR). PB2 residues 614 and 649 of ICV are highlighted in red and correspond to residues 591 and 627 of IAV shown in parenthesis
Recent experiments have shown that a lysine at position 627 is essential for a stable interaction of the viral RNA polymerase with mammalian host-cell protein ANP32A [50]. ANP32A had previously been proposed to be essential for the synthesis of vRNA from a cRNA template [53], but recent structural and biochemical evidence suggests that it brings together the replicating and encapsidating polymerases in a viral replicase complex that can synthesise both vRNA or cRNA [48, 49]. The ANP32A-supported dimer shows that the basic K627, but not the acidic E627, can efficiently interact with the C-terminal low-complexity acidic region of mammalian ANP32A (Fig. 5) [48]. By contrast, avian ANP32A homologs contain an additional exon, and can interact with the polymerase dimer when the polymerases in the dimer contain an E627 [50, 125].
In some avian influenza virus strains, a Q591K mutation in the PB2 627 domain [G590S and Q591R in pdm09 (H1N1)] can compensate for the absence of the E627K signature and support the activity of avian-adapted polymerases in mammalian cells [126, 127]. Like PB2 residue 627, residue 591 is located at the binding site for the C-terminal region of ANP32A (Fig. 5) and introduction of a basic amino acid at this site is thought to improve the interaction between the polymerase and mammalian ANP32A [48]. By stabilising the replicase dimer, the Q591K mutation increases the replication of the avian H7N9 and H9N2 viruses in murine lungs, inducing strong inflammatory responses and enhancing virulence as a result [122, 123, 128].
PB2 mutation D701N has also been associated with improved replication and higher levels of innate immune activation [31, 123, 128]. However, unlike the mutations discussed above, amino acid 701 resides in the PB2 NLS and promotes replication by improving nuclear import of PB2 and vRNPs [129,130,131].
An emerging role of the Mid-link domain of PB2 in innate immune activation
To explore the distribution of published polymerase mutations that affect innate immune responses and identify potentially novel clusters of interest, we mapped those mutations to the polymerase subunits (Fig. 6, Supplementary Table 2). The PB2 subunit contained the majority of the identified immunostimulatory or immunoinhibitory mutations (Fig. 6). In particular, a number of these mutations cluster in the Mid-link domain of PB2 (residues 247–320/482–538). This clustering indicates that Mid-link might play an important role in polymerase’s function and innate immune recognition of the viral infection, yet this region has presently not been assigned a specific role in viral genome replication or transcription. What could the role of the Mid-link domain and its mutations be?

Immunostimulatory and immunoinhibitory mutations in the influenza A virus RNA polymerase. Polymerase mutations that affect innate immune activation (Supplementary Table 2) were mapped to the polymerase subunits of the influenza A virus. Mutations that induced higher innate immune activation in comparison to the background strain are shown in red and those that decreased innate immune activation are shown in blue. Mutations that did not affect innate immune responses in comparison to the background virus strain are shown in grey. Combinations of mutations are marked with Greek symbols. The structural domains in each subunit are indicated in different colours. Additionally, functional regions involved in importin binding [131], RNA Pol II interactions [117] (RNA Pol II binding sites differ for influenza B and C viruses [44]), formation of polymerase dimers [48, 56] and ANP32A-binding [48] are highlighted in the bottom half of each segment
The PB2 Mid-link domain might play a key role in stabilizing the conformational rearrangements of the RNA polymerase as it transitions from one state to another or in encapsidation of the nascent RNA strand. It forms extensive, transient interactions with the flexible domains of PA and PB2 in various conformations of the viral polymerase. In addition, analysis of the ANP32A-supported replicase suggests that the Mid-link domain may play a role in nascent strand egress or encapsidation. In the dimer, residues 252–273 and 519–523 (influenza C virus PB2 residues 259–280 and 538–542, respectively) of the replicating polymerase face the encapsidating polymerase but are not directly interacting with it. Instead, these residues face a groove that separates the two polymerases in the dimer and which the emerging 5′ terminus of the nascent strand must bridge to reach the promoter binding pocket of the encapsidating polymerase (Fig. 7A). It is tempting to speculate that these residues of the Mid-link domain play a role in NP recruitment or the encapsidation of the nascent strand [48].

Mid-link domain mutations. A A surface representation of the encapsidating-replicating polymerase dimer of ICV (PDB 6XZR). The Mid-link domain of PB2 is shown in violet. The movement of the 5′ end of the nascent RNA from the product exit channel of the replicating polymerase to the 5′ binding site of the encapsidating polymerase is shown with a yellow arrow. Mid-link residues, whose alteration affects innate immune activation, are shown as red spheres (residue numbering is as in ICV, corresponding IAV amino acids are shown in parenthesis). B Localisation of the IAV Mid-link residues 249, 292 and 503 (shown in red) on the structure of the human H3N2 influenza A virus RNA polymerase (PDB 6RR7)
Mid-link domain might also play a role in adaptation as several mutations in the Mid-link domain were shown to improve polymerase activity and the replication of avian influenza virus strains in mammalian cells. For instance, the well-documented adaptive mutation Q591K in the 627 domain occurs together with the Mid-link mutation D253N in an avian H9N2 virus [132]. In this strain, D253N improves polymerase activity, enhances viral replication in mice, and stimulates interferon and pro-inflammatory cytokine production [132]. Similarly, Mid-link mutation T271A in the RNA polymerase of an avian H7N9 isolate partially compensates for the lack of a PB2 E627K mutation in mammalian cells, improving polymerase activity and viral replication, while at the same time increasing pro-inflammatory cytokine expression [128]. Moreover, in H5N1 isolates, a M283L mutation was shown to increase polymerase activity, viral replication and innate immune activation in mice, whereas a M283I mutation decreased these properties [133]. Similarly, the reverse I283M mutation in the combination with K526R in the avian H5N8 virus significantly upregulated innate immune activation in murine lungs [134]. Thus, mutations in the Mid-link domain might induce innate immune activation by improving polymerase activity, similar to the other adaptive mutations.
The Mid-link domain could also have a stand-alone immunomodulatory function. Several mutations in this region supressed innate immune activation despite improving polymerase activity. Two of them, V249A and I503V, arose in a recombinant A/PR/8/34 (H1N1) virus, which contained a dysfunctional NS1 of the bat influenza A virus [135]. The mutations were able to compensate for the absence of innate immune modulation by NS1, reducing IFN induction, despite simultaneously increasing viral replication [135]. The I292V mutation in the avian H9N2 virus, which increases binding to MAVS, also suppressed innate immune activation despite higher activity of the mutant polymerase [86]. All three mutations reside in the solvent-exposed region of the Mid-link domain (Fig. 7B) and may thus be able to inhibit innate immune activation by interacting with the components of the host innate immune system.
Conclusions and outstanding questions
The influenza A virus RNA polymerase plays multiple roles in the innate immune response to influenza virus infection. Not only can the subunits of the RNA polymerase affect innate immune activation by interfering with the components of the cellular signalling pathways, the enzyme can also produce immunostimulatory RNA species. These activities of the RNA polymerase are not unique to influenza A viruses, as RNA polymerases of viruses belonging to Flaviviridae, Picornaviridae and Coronaviridae families are also known to specifically target and inhibit innate immune signalling [136,137,138,139], while production of aberrant RNA species has been described for the majority of RNA viruses as well [140].
Despite recent advances in our understanding of the structure and the immunostimulatory and immunoinhibitory activities of the influenza A virus polymerase, many fundamental questions about the molecular mechanisms involved remain unanswered. These include, but are not limited to, (i) how does RIG-I gain access to viral RNA (and the viral RNA termini in particular) in the context of a fully assembled RNP; (ii) what is the molecular mechanism underlying the generation of DVGs and mvRNAs; (iii) why do aberrant viral RNAs trigger innate immune responses more efficiently than full-length viral RNA segments; (iv) does aberrant RNA synthesis confer any evolutionary advantage, and (v) are the immunosuppressive and enzymatic activities of the viral polymerase separated in space and time? To find answers to those questions, existing and novel RNA polymerase mutants can be used. Screening approaches in combination with next generation sequencing have proven to be particularly powerful for the identification of such immunostimulatory RNA polymerase mutants [33, 141]. However, biochemical and molecular research is still needed to better understand how such mutations affect polymerase function.
The immunomodulatory or -stimulatory activity of the viral polymerase could also guide the development of novel antiviral treatments. Even though current drugs targeting the influenza virus polymerase primarily focus on blocking its activity, targeting its immunomodulatory function could potentially have an added benefit of activating the host’s natural immune defence during infection. However, care must be taken to not over-stimulate the innate immune response. Additionally, DVG-containing influenza viruses or cloned DVGs have been proposed as both influenza-specific and a broad-spectrum antiviral treatment due to their interfering and immunostimulatory activity [106, 142].
The knowledge of the processes by which influenza virus polymerase stimulates or inhibits innate immune responses can also be used in rational vaccine design. Several PB1, PB2 and NP mutations in the current live-attenuated influenza vaccine confer its cold-adapted, attenuated and temperature-sensitive phenotype [143], while alterations in NS1 protein have been explored as a novel approach to improve efficacy of the live-attenuated vaccines [144, 145]. Recent studies also showed that addition of immunostimulatory polymerase mutations, for instance in combination with mutations or deletions in NS1, improves vaccine immunogenicity and protection against infection [141, 146]. DVG-containing (interfering) vaccines were also shown to protect mice and ferrets from severe influenza infection [147, 148]. However, the presence of DVGs in the live-attenuated vaccines has been suggested to reduce their immunogenicity by interfering with the replication of the vaccine strain [149, 150], suggesting that a careful balance may need to be found for some of the above approaches. Understanding the viral and host molecular determinants of DVG production can therefore help to regulate aberrant polymerase activity of the vaccine strains. In this way, the multifunctional role of the viral RNA polymerase in innate immune activation represents an important area of future research.
Data availability
Not applicable.
Code availability
Not applicable.
References
WHO (2021) Influenza—estimating burden of disease. https://www.euro.who.int/en/health-topics/communicable-diseases/influenza/seasonal-influenza/burden-of-influenza. Accessed 5 April 2021
Kuiken T, Taubenberger JK (2008) Pathology of human influenza revisited. Vaccine 26(Suppl 4):D59-66. https://doi.org/10.1016/j.vaccine.2008.07.025
Short KR, Veeris R, Leijten LM, van den Brand JM, Jong VL, Stittelaar K, Osterhaus A, Andeweg A, van Riel D (2017) Proinflammatory cytokine responses in extra-respiratory tissues during severe influenza. J Infect Dis 216(7):829–833. https://doi.org/10.1093/infdis/jix281
de Wit E, Siegers JY, Cronin JM, Weatherman S, van den Brand JM, Leijten LM, van Run P, Begeman L, van den Ham HJ, Andeweg AC, Bushmaker T, Scott DP, Saturday G, Munster VJ, Feldmann H, van Riel D (2018) 1918 H1N1 influenza virus replicates and induces proinflammatory cytokine responses in extrarespiratory tissues of ferrets. J Infect Dis 217(8):1237–1246. https://doi.org/10.1093/infdis/jiy003
Honce R, Wohlgemuth N, Meliopoulos VA, Short KR, Schultz-Cherry S (2020) Influenza in high-risk hosts-lessons learned from animal models. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a038604
Mettelman RC, Thomas PG (2021) Human susceptibility to influenza infection and severe disease. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a038711
Liu G, Park HS, Pyo HM, Liu Q, Zhou Y (2015) Influenza A virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol 89(11):6067–6079. https://doi.org/10.1128/JVI.00232-15
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S, (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089):101–105. https://doi.org/10.1038/nature04734
Liu G, Lu Y, Thulasi Raman SN, Xu F, Wu Q, Li Z, Brownlie R, Liu Q, Zhou Y (2018) Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat Commun 9(1):3199. https://doi.org/10.1038/s41467-018-05745-w
Pang IK, Pillai PS, Iwasaki A (2013) Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Natl Acad Sci USA 110(34):13910–13915. https://doi.org/10.1073/pnas.1303275110
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76(1):16–32. https://doi.org/10.1128/MMBR.05015-11
Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T (2014) Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14(1):57–69. https://doi.org/10.1016/s1473-3099(13)70286-x
Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, Hartmann R, Wack A (2020) Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369(6504):712–717. https://doi.org/10.1126/science.abc2061
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, do CamHa BVQ, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J (2006) Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12(10):1203–1207. https://doi.org/10.1038/nm1477
Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y (2007) Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445(7125):319–323. https://doi.org/10.1038/nature05495
Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, Garcia-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, Aicher LD, Rosenzweig ER, Murali-Krishna K, Clark EA, Kotur MS, Fornek JL, Proll S, Palermo RE, Sabourin CL, Katze MG (2009) Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci USA 106(9):3455–3460. https://doi.org/10.1073/pnas.0813234106
Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, Ren Y, Wang J, Dong Y, Bao M, Li L, Zhou M, Yuan S, Sun J, Zhu Z, Chen L, Li Q, Zhang Z, Zhang X, Lu S, Doherty PC, Kedzierska K, Xu J (2014) Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci USA 111(2):769–774. https://doi.org/10.1073/pnas.1321748111
Berri F, Le VB, Jandrot-Perrus M, Lina B, Riteau B (2014) Switch from protective to adverse inflammation during influenza: viral determinants and hemostasis are caught as culprits. Cell Mol Life Sci 71(5):885–898. https://doi.org/10.1007/s00018-013-1479-x
Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, ReiseSousa C (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140(3):397–408. https://doi.org/10.1016/j.cell.2010.01.020
Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, Garcia-Sastre A (2018) Influenza. Nat Rev Dis Primers 4(1):3. https://doi.org/10.1038/s41572-018-0002-y
Te Velthuis AJW, Grimes JM, Fodor E (2021) Structural insights into RNA polymerases of negative-sense RNA viruses. Nat Rev Microbiol 19(5):303–318. https://doi.org/10.1038/s41579-020-00501-8
Fodor E, Te Velthuis AJW (2019) Structure and function of the influenza virus transcription and replication machinery. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a038398
Killip MJ, Fodor E, Randall RE (2015) Influenza virus activation of the interferon system. Virus Res 209:11–22. https://doi.org/10.1016/j.virusres.2015.02.003
Te Velthuis AJW, Long JC, Bauer DLV, Fan RLY, Yen H-L, Sharps J, Siegers JY, Killip MJ, French H, Oliva-Martin MJ, Randall RE, de Wit E, van Riel D, Poon LLM, Fodor E (2018) Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat Microbiol 3(11):1234–1242. https://doi.org/10.1038/s41564-018-0240-5
Nogales A, Martinez-Sobrido L, Topham DJ, DeDiego ML (2018) Modulation of innate immune responses by the influenza A NS1 and PA-X proteins. Viruses. https://doi.org/10.3390/v10120708
Cheung PH, Lee TT, Chan CP, Jin DY (2020) Influenza A virus PB1-F2 protein: an ambivalent innate immune modulator and virulence factor. J Leukoc Biol 107(5):763–771. https://doi.org/10.1002/JLB.4MR0320-206R
Mok KP, Wong CHK, Cheung CY, Chan MC, Lee SMY, Nicholls JM, Guan Y, Peiris JSM (2009) Viral genetic determinants of H5N1 influenza viruses that contribute to cytokine dysregulation. J Infect Dis 200(7):1104–1112. https://doi.org/10.1086/605606
Forero A, Tisoncik-Go J, Watanabe T, Zhong G, Hatta M, Tchitchek N, Selinger C, Chang J, Barker K, Morrison J, Berndt JD, Moon RT, Josset L, Kawaoka Y, Katze MG (2015) The 1918 influenza virus PB2 protein enhances virulence through the disruption of inflammatory and Wnt-mediated signaling in mice. J Virol 90(5):2240–2253. https://doi.org/10.1128/JVI.02974-15
Li OTW, Chan MCW, Leung CSW, Chan RWY, Guan Y, Nicholls JM, Poon LLM (2009) Full factorial analysis of mammalian and avian influenza polymerase subunits suggests a role of an efficient polymerase for virus adaptation. PLoS ONE 4(5):e5658. https://doi.org/10.1371/journal.pone.0005658
Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A (2005) Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310(5745):77–80. https://doi.org/10.1126/science.1119392
Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J (2005) The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc Natl Acad Sci USA 102(51):18590–18595. https://doi.org/10.1073/pnas.0507415102
Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, McCullers JA, Ludwig S, Ehrhardt C (2014) Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun 5:5645. https://doi.org/10.1038/ncomms6645
Russell AB, Elshina E, Kowalsky JR, Te Velthuis AJW, Bloom JD (2019) Single-cell virus sequencing of influenza infections that trigger innate immunity. J Virol. https://doi.org/10.1128/JVI.00500-19
Perez-Cidoncha M, Killip MJ, Oliveros JC, Asensio VJ, Fernandez Y, Bengoechea JA, Randall RE, Ortin J (2014) An unbiased genetic screen reveals the polygenic nature of the influenza virus anti-interferon response. J Virol 88(9):4632–4646. https://doi.org/10.1128/JVI.00014-14
Hulme KD, Karawita AC, Pegg C, Bunte MJ, Bielefeldt-Ohmann H, Bloxham CJ, Van den Hoecke S, Setoh YX, Vrancken B, Spronken M, Steele LE, Verzele NA, Upton KR, Khromykh AA, Chew KY, Sukkar M, Phipps S, Short KR (2021) A paucigranulocytic asthma host environment promotes the emergence of virulent influenza viral variants. Elife. https://doi.org/10.7554/eLife.61803
Pflug A, Guilligay D, Reich S, Cusack S (2014) Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516(7531):355–360. https://doi.org/10.1038/nature14008
Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J (2012) The structure of native influenza virion ribonucleoproteins. Science 338(6114):1634–1637. https://doi.org/10.1126/science.1228172
Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA (2012) Organization of the influenza virus replication machinery. Science 338(6114):1631–1634. https://doi.org/10.1126/science.1227270
te Velthuis AJ (2014) Common and unique features of viral RNA-dependent polymerases. Cell Mol Life Sci 71(22):4403–4420. https://doi.org/10.1007/s00018-014-1695-z
Te Velthuis AJ, Fodor E (2016) Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol 14(8):479–493. https://doi.org/10.1038/nrmicro.2016.87
Wandzik JM, Kouba T, Karuppasamy M, Pflug A, Drncova P, Provaznik J, Azevedo N, Cusack S (2020) A structure-based model for the complete transcription cycle of influenza polymerase. Cell 181(4):877-893 e821. https://doi.org/10.1016/j.cell.2020.03.061
Pflug A, Lukarska M, Resa-Infante P, Reich S, Cusack S (2017) Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res 234:103–117. https://doi.org/10.1016/j.virusres.2017.01.013
Resa-Infante P, Gabriel G (2013) The nuclear import machinery is a determinant of influenza virus host adaptation. BioEssays 35(1):23–27. https://doi.org/10.1002/bies.201200138
Walker AP, Fodor E (2019) Interplay between influenza virus and the host RNA polymerase II transcriptional machinery. Trends Microbiol 27(5):398–407. https://doi.org/10.1016/j.tim.2018.12.013
Krischuns T, Lukarska M, Naffakh N, Cusack S (2021) Influenza virus RNA-dependent RNA polymerase and the host transcriptional apparatus. Annu Rev Biochem. https://doi.org/10.1146/annurev-biochem-072820-100645
Poon LL, Pritlove DC, Fodor E, Brownlee GG (1999) Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J Virol 73(4):3473–3476. https://doi.org/10.1128/JVI.73.4.3473-3476.1999
Hutchinson EC, Fodor E (2012) Nuclear import of the influenza A virus transcriptional machinery. Vaccine 30(51):7353–7358. https://doi.org/10.1016/j.vaccine.2012.04.085
Carrique L, Fan H, Walker AP, Keown JR, Sharps J, Staller E, Barclay WS, Fodor E, Grimes JM (2020) Host ANP32A mediates the assembly of the influenza virus replicase. Nature 587(7835):638–643. https://doi.org/10.1038/s41586-020-2927-z
Nilsson-Payant BE, tenOever BR, te Velthuis AJW (2021) The host factor ANP32A is required for influenza A virus vRNA and cRNA synthesis. bioRxiv. https://doi.org/10.1101/2021.04.30.442228
Long JS, Giotis ES, Moncorge O, Frise R, Mistry B, James J, Morisson M, Iqbal M, Vignal A, Skinner MA, Barclay WS (2016) Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 529(7584):101–104. https://doi.org/10.1038/nature16474
Staller E, Sheppard CM, Neasham PJ, Mistry B, Peacock TP, Goldhill DH, Long JS, Barclay WS (2019) ANP32 proteins are essential for influenza virus replication in human cells. J Virol. https://doi.org/10.1128/JVI.00217-19
Chen KY, Santos Afonso ED, Enouf V, Isel C, Naffakh N (2019) Influenza virus polymerase subunits co-evolve to ensure proper levels of dimerization of the heterotrimer. PLoS Pathog 15(10):e1008034. https://doi.org/10.1371/journal.ppat.1008034
Sugiyama K, Kawaguchi A, Okuwaki M, Nagata K (2015) pp32 and APRIL are host cell-derived regulators of influenza virus RNA synthesis from cRNA. Elife. https://doi.org/10.7554/eLife.08939
Jorba N, Coloma R, Ortin J (2009) Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5(5):e1000462. https://doi.org/10.1371/journal.ppat.1000462
York A, Hengrung N, Vreede FT, Huiskonen JT, Fodor E (2013) Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc Natl Acad Sci USA 110(45):E4238-4245. https://doi.org/10.1073/pnas.1315068110
Fan H, Walker AP, Carrique L, Keown JR, Serna Martin I, Karia D, Sharps J, Hengrung N, Pardon E, Steyaert J, Grimes JM, Fodor E (2019) Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 573(7773):287–290. https://doi.org/10.1038/s41586-019-1530-7
Oymans J, Te Velthuis AJW (2018) A mechanism for priming and realignment during influenza A virus replication. J Virol. https://doi.org/10.1128/JVI.01773-17
Peacock TP, Sheppard CM, Staller E, Barclay WS (2019) Host determinants of influenza RNA synthesis. Annu Rev Virol 6(1):215–233. https://doi.org/10.1146/annurev-virology-092917-043339
Iwasaki A, Pillai PS (2014) Innate immunity to influenza virus infection. Nat Rev Immunol 14(5):315–328. https://doi.org/10.1038/nri3665
Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, Ragan KB, Ishizuka T, Crawford JC, Tummers B, Rodriguez DA, Xue J, Peri S, Kaiser WJ, Lopez CB, Xu Y, Upton JW, Thomas PG, Green DR, Balachandran S (2020) Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180(6):1115-1129 e1113. https://doi.org/10.1016/j.cell.2020.02.050
Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M Jr (2008) Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82(1):335–345. https://doi.org/10.1128/JVI.01080-07
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA 101(15):5598–5603. https://doi.org/10.1073/pnas.0400937101
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303(5663):1529–1531. https://doi.org/10.1126/science.1093616
Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM (2011) Structural insights into RNA recognition by RIG-I. Cell 147(2):409–422. https://doi.org/10.1016/j.cell.2011.09.023
Kolakofsky D, Kowalinski E, Cusack S (2012) A structure-based model of RIG-I activation. RNA 18(12):2118–2127. https://doi.org/10.1261/rna.035949.112
Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147(2):423–435. https://doi.org/10.1016/j.cell.2011.09.039
Patel JR, Jain A, Chou YY, Baum A, Ha T, Garcia-Sastre A (2013) ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 14(9):780–787. https://doi.org/10.1038/embor.2013.102
Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T (2009) Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science 323(5917):1070–1074. https://doi.org/10.1126/science.1168352
Peisley A, Wu B, Yao H, Walz T, Hur S (2013) RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell 51(5):573–583. https://doi.org/10.1016/j.molcel.2013.07.024
Peisley A, Wu B, Xu H, Chen ZJ, Hur S (2014) Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509(7498):110–114. https://doi.org/10.1038/nature13140
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446(7138):916–920. https://doi.org/10.1038/nature05732
Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, Hur S (2014) Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol Cell 55(4):511–523. https://doi.org/10.1016/j.molcel.2014.06.010
Murphy K, Weaver C (2017) Janeway’s immunobiology, 9th edn. Garland Science, New York, pp 1–904
Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crepin T, Hart D, Lunardi T, Nanao M, Ruigrok RW, Cusack S (2014) Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516(7531):361–366. https://doi.org/10.1038/nature14009
Anchisi S, Guerra J, Mottet-Osman G, Garcin D (2016) Mismatches in the influenza A virus RNA panhandle prevent retinoic acid-inducible gene I (RIG-I) sensing by impairing RNA/RIG-I complex formation. J Virol 90(1):586–590. https://doi.org/10.1128/JVI.01671-15
Weber M, Sediri H, Felgenhauer U, Binzen I, Banfer S, Jacob R, Brunotte L, Garcia-Sastre A, Schmid-Burgk JL, Schmidt T, Hornung V, Kochs G, Schwemmle M, Klenk H-D, Weber F (2015) Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17(3):309–319. https://doi.org/10.1016/j.chom.2015.01.005
Li W, Chen H, Sutton T, Obadan A, Perez DR (2014) Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J Virol 88(18):10432–10447. https://doi.org/10.1128/JVI.01383-14
Liedmann S, Hrincius ER, Anhlan D, McCullers JA, Ludwig S, Ehrhardt C (2014) New virulence determinants contribute to the enhanced immune response and reduced virulence of an influenza A virus A/PR8/34 variant. J Infect Dis 209(4):532–541. https://doi.org/10.1093/infdis/jit463
Patel RC, Sen GC (1998) PACT, a protein activator of the interferon-induced protein kinase. PKR EMBO J 17(15):4379–4390. https://doi.org/10.1093/emboj/17.15.4379
Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY (2011) The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9(4):299–309. https://doi.org/10.1016/j.chom.2011.03.007
Chan C-P, Yuen C-K, Cheung P-HH, Fung S-Y, Lui P-Y, Chen H, Kok K-H, Jin D-Y (2018) Antiviral activity of double-stranded RNA-binding protein PACT against influenza A virus mediated via suppression of viral RNA polymerase. FASEB J Off Publ Fed Am Soc Exp Biol 32(8):4380–4393. https://doi.org/10.1096/fj.201701361R
Zeng Y, Xu S, Wei Y, Zhang X, Wang Q, Jia Y, Wang W, Han L, Chen Z, Wang Z, Zhang B, Chen H, Lei C-Q, Zhu Q (2021) The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog 17(2):e1009300. https://doi.org/10.1371/journal.ppat.1009300
Iwai A, Shiozaki T, Kawai T, Akira S, Kawaoka Y, Takada A, Kida H, Miyazaki T (2010) Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J Biol Chem 285(42):32064–32074. https://doi.org/10.1074/jbc.M110.112458
Graef KM, Vreede FT, Lau Y-F, McCall AW, Carr SM, Subbarao K, Fodor E (2010) The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol 84(17):8433–8445. https://doi.org/10.1128/JVI.00879-10
Zhao Z, Yi C, Zhao L, Wang S, Zhou L, Hu Y, Zou W, Chen H, Jin M (2014) PB2-588I enhances 2009 H1N1 pandemic influenza virus virulence by increasing viral replication and exacerbating PB2 inhibition of beta interferon expression. J Virol 88(4):2260–2267. https://doi.org/10.1128/JVI.03024-13
Gao W, Zu Z, Liu J, Song J, Wang X, Wang C, Liu L, Tong Q, Wang M, Sun H, Sun Y, Liu J, Chang K-C, Pu J (2019) Prevailing I292V PB2 mutation in avian influenza H9N2 virus increases viral polymerase function and attenuates IFN-beta induction in human cells. J Gen Virol 100(9):1273–1281. https://doi.org/10.1099/jgv.0.001294
Patel D, Schultz LW, Umland TC (2013) Influenza A polymerase subunit PB2 possesses overlapping binding sites for polymerase subunit PB1 and human MAVS proteins. Virus Res 172(1–2):75–80. https://doi.org/10.1016/j.virusres.2012.12.003
Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146(3):448–461. https://doi.org/10.1016/j.cell.2011.06.041
Vazquez C, Horner SM (2015) MAVS coordination of antiviral innate immunity. J Virol 89(14):6974–6977. https://doi.org/10.1128/JVI.01918-14
Sun N, Jiang L, Ye M, Wang Y, Wang G, Wan X, Zhao Y, Wen X, Liang L, Ma S, Liu L, Bu Z, Chen H, Li C (2020) TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 11(12):894–914. https://doi.org/10.1007/s13238-020-00734-6
Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G (2006) Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25(14):3257–3263. https://doi.org/10.1038/sj.emboj.7601220
Yi C, Zhao Z, Wang S, Sun X, Zhang D, Sun X, Zhang A, Jin M (2017) Influenza A virus PA antagonizes interferon-beta by interacting with interferon regulatory factor 3. Front Immunol 8:1051. https://doi.org/10.3389/fimmu.2017.01051
Sanchez-Aparicio MT, Ayllon J, Leo-Macias A, Wolff T, Garcia-Sastre A (2017) Subcellular localizations of RIG-I, TRIM25, and MAVS complexes. J Virol. https://doi.org/10.1128/JVI.01155-16
Weinberg SE, Sena LA, Chandel NS (2015) Mitochondria in the regulation of innate and adaptive immunity. Immunity 42(3):406–417. https://doi.org/10.1016/j.immuni.2015.02.002
Carr SM, Carnero E, Garcia-Sastre A, Brownlee GG, Fodor E (2006) Characterization of a mitochondrial-targeting signal in the PB2 protein of influenza viruses. Virology 344(2):492–508. https://doi.org/10.1016/j.virol.2005.08.041
Long JCD, Fodor E (2016) The PB2 subunit of the influenza A virus RNA polymerase is imported into the mitochondrial matrix. J Virol 90(19):8729–8738. https://doi.org/10.1128/JVI.01384-16
Woodfin BM, Kazim AL (1993) Interaction of the amino-terminus of an influenza virus protein with mitochondria. Arch Biochem Biophys 306(2):427–430. https://doi.org/10.1006/abbi.1993.1533
Alnaji FG, Brooke CB (2020) Influenza virus DI particles: defective interfering or delightfully interesting? PLoS Pathog 16(5):e1008436. https://doi.org/10.1371/journal.ppat.1008436
Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, tenOever BR (2010) Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci USA 107(25):11525–11530. https://doi.org/10.1073/pnas.1001984107
Umbach JL, Yen HL, Poon LL, Cullen BR (2010) Influenza A virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. MBio. https://doi.org/10.1128/mBio.00204-10
Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, Dwyer DE, Freiberg M, Horban A, Losso M, Lynfield R, Wentworth DN, Holmes EC, Davey R, Wentworth DE, Ghedin E, Group IFS, Group IFS (2013) Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol 87(14):8064–8074. https://doi.org/10.1128/JVI.00240-13
Jennings PA, Finch JT, Winter G, Robertson JS (1983) Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 34(2):619–627. https://doi.org/10.1016/0092-8674(83)90394-X
Cheong HK, Cheong C, Lee YS, Seong BL, Choi BS (1999) Structure of influenza virus panhandle RNA studied by NMR spectroscopy and molecular modeling. Nucleic Acids Res 27(5):1392–1397. https://doi.org/10.1093/nar/27.5.1392
Baum A, Sachidanandam R, Garcia-Sastre A (2010) Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci USA 107(37):16303–16308. https://doi.org/10.1073/pnas.1005077107
Linehan MM, Dickey TH, Molinari ES, Fitzgerald ME, Potapova O, Iwasaki A, Pyle AM (2018) A minimal RNA ligand for potent RIG-I activation in living mice. Sci Adv 4(2):e1701854. https://doi.org/10.1126/sciadv.1701854
Easton AJ, Scott PD, Edworthy NL, Meng B, Marriott AC, Dimmock NJ (2011) A novel broad-spectrum treatment for respiratory virus infections: influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 29(15):2777–2784. https://doi.org/10.1016/j.vaccine.2011.01.102
Tapia K, Kim WK, Sun Y, Mercado-Lopez X, Dunay E, Wise M, Adu M, Lopez CB (2013) Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9(10):e1003703. https://doi.org/10.1371/journal.ppat.1003703
Boussier J, Munier S, Achouri E, Crescenzo-Chaigne B, Behillil S, Enouf V, van der Werf S, Naffakh N (2020) RNA-seq accuracy and reproducibility for the mapping and quantification of influenza defective viral genomes. RNA. https://doi.org/10.1261/rna.077529.120
Alnaji FG, Reiser WK, Velthuis A, Brooke CB (2021) Influenza A virus defective viral genomes are inefficiently packaged into virions relative to wild-type genomic RNAs. bioRxiv. https://doi.org/10.1101/2021.05.13.444068
Liu G, Lu Y, Liu Q, Zhou Y (2019) Inhibition of ongoing influenza A virus replication reveals different mechanisms of RIG-I activation. J Virol. https://doi.org/10.1128/JVI.02066-18
Nilsson-Payant BE, Blanco-Melo D, Uhl S, Escudero-Perez B, Olschewski S, Thibault P, Panis M, Rosenthal M, Munoz-Fontela C, Lee B, tenOever BR (2021) Reduced nucleoprotein availability impairs negative-sense RNA virus replication and promotes host recognition. J Virol. https://doi.org/10.1128/JVI.02274-20
Odagiri T, Tobita K (1990) Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc Natl Acad Sci USA 87(15):5988–5992. https://doi.org/10.1073/pnas.87.15.5988
Fodor E, Mingay LJ, Crow M, Deng T, Brownlee GG (2003) A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase promotes the generation of defective interfering RNAs. J Virol 77(8):5017–5020. https://doi.org/10.1128/jvi.77.8.5017-5020.2003
Vasilijevic J, Zamarreno N, Oliveros JC, Rodriguez-Frandsen A, Gomez G, Rodriguez G, Perez-Ruiz M, Rey S, Barba I, Pozo F, Casas I, Nieto A, Falcon A (2017) Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13(10):e1006650. https://doi.org/10.1371/journal.ppat.1006650
Naito T, Mori K, Ushirogawa H, Takizawa N, Nobusawa E, Odagiri T, Tashiro M, Ohniwa RL, Nagata K, Saito M (2017) Generation of a genetically stable high-fidelity influenza vaccine strain. J Virol. https://doi.org/10.1128/JVI.01073-16
Cheung PP, Watson SJ, Choy KT, Fun Sia S, Wong DD, Poon LL, Kellam P, Guan Y, Malik Peiris JS, Yen HL (2014) Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat Commun 5:4794. https://doi.org/10.1038/ncomms5794
Lukarska M, Fournier G, Pflug A, Resa-Infante P, Reich S, Naffakh N, Cusack S (2017) Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 541(7635):117–121. https://doi.org/10.1038/nature20594
Subbarao EK, London W, Murphy BR (1993) A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J Virol 67(4):1761–1764. https://doi.org/10.1128/JVI.67.4.1761-1764.1993
Shinya K, Hamm S, Hatta M, Ito H, Ito T, Kawaoka Y (2004) PB2 amino acid at position 627 affects replicative efficiency, but not cell tropism, of Hong Kong H5N1 influenza A viruses in mice. Virology 320(2):258–266. https://doi.org/10.1016/j.virol.2003.11.030
Fornek JL, Gillim-Ross L, Santos C, Carter V, Ward JM, Cheng LI, Proll S, Katze MG, Subbarao K (2009) A single-amino-acid substitution in a polymerase protein of an H5N1 influenza virus is associated with systemic infection and impaired T-cell activation in mice. J Virol 83(21):11102–11115. https://doi.org/10.1128/JVI.00994-09
Tian J, Qi W, Li X, He J, Jiao P, Zhang C, Liu G-Q, Liao M (2012) A single E627K mutation in the PB2 protein of H9N2 avian influenza virus increases virulence by inducing higher glucocorticoids (GCs) level. PLoS ONE 7(6):e38233. https://doi.org/10.1371/journal.pone.0038233
Wang C, Lee HHY, Yang ZF, Mok CKP, Zhang Z (2016) PB2-Q591K mutation determines the pathogenicity of avian H9N2 influenza viruses for mammalian species. PLoS ONE 11(9):e0162163. https://doi.org/10.1371/journal.pone.0162163
Li W, Lee HHY, Li RF, Zhu HM, Yi G, Peiris JSM, Yang ZF, Mok CKP (2017) The PB2 mutation with lysine at 627 enhances the pathogenicity of avian influenza (H7N9) virus which belongs to a non-zoonotic lineage. Sci Rep 7(1):2352. https://doi.org/10.1038/s41598-017-02598-z
Yu M, Zhang K, Qi W, Huang Z, Ye J, Ma Y, Liao M, Ning Z (2014) Expression pattern of NLRP3 and its related cytokines in the lung and brain of avian influenza virus H9N2 infected BALB/c mice. Virol J 11:229. https://doi.org/10.1186/s12985-014-0229-5
Domingues P, Hale BG (2017) Functional insights into ANP32A-dependent influenza A virus polymerase host restriction. Cell Rep 20(11):2538–2546. https://doi.org/10.1016/j.celrep.2017.08.061
Mehle A, Doudna JA (2009) Adaptive strategies of the influenza virus polymerase for replication in humans. Proc Natl Acad Sci USA 106(50):21312–21316. https://doi.org/10.1073/pnas.0911915106
Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, Sakai-Tagawa Y, Ito M, Ozawa M, Watanabe T, Sakabe S, Li C, Kim JH, Myler PJ, Phan I, Raymond A, Smith E, Stacy R, Nidom CA, Lank SM, Wiseman RW, Bimber BN, O’Connor DH, Neumann G, Stewart LJ, Kawaoka Y (2010) Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6(8):e1001034. https://doi.org/10.1371/journal.ppat.1001034
Mok CKP, Lee HHY, Lestra M, Nicholls JM, Chan MCW, Sia SF, Zhu H, Poon LLM, Guan Y, Peiris JSM (2014) Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel A/H7N9 influenza virus in mammalian hosts. J Virol 88(6):3568–3576. https://doi.org/10.1128/JVI.02740-13
Gabriel G, Herwig A, Klenk HD (2008) Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog 4(2):e11. https://doi.org/10.1371/journal.ppat.0040011
Sediri H, Schwalm F, Gabriel G, Klenk HD (2015) Adaptive mutation PB2 D701N promotes nuclear import of influenza vRNPs in mammalian cells. Eur J Cell Biol 94(7–9):368–374. https://doi.org/10.1016/j.ejcb.2015.05.012
Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RW, Daigle N, Ellenberg J, Cusack S, Simorre JP, Hart DJ (2007) Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat Struct Mol Biol 14(3):229–233. https://doi.org/10.1038/nsmb1212
Mok CK, Yen HL, Yu MY, Yuen KM, Sia SF, Chan MC, Qin G, Tu WW, Peiris JS (2011) Amino acid residues 253 and 591 of the PB2 protein of avian influenza virus A H9N2 contribute to mammalian pathogenesis. J Virol 85(18):9641–9645. https://doi.org/10.1128/JVI.00702-11
Chen S, Xie Y, Su X, Xue J, Wang X, Du Y, Qin T, Peng D, Liu X (2020) Substitutions in the PB2 methionine 283 residue affect H5 subtype avian influenza virus virulence. Transbound Emerg Dis 67(6):2554–2563. https://doi.org/10.1111/tbed.13601
Chen S, Wang X, Su X, Miao X, Qin T, Peng D, Liu X (2021) Deep sequencing of the transcriptome from murine lung infected with H5N8 subtype avian influenza virus with combined substitutions I283M and K526R in PB2 gene. Infect Genet Evol 87:104672. https://doi.org/10.1016/j.meegid.2020.104672
Aydillo T, Ayllon J, Pavlisin A, Martinez-Romero C, Tripathi S, Mena I, Moreira-Soto A, Vicente-Santos A, Corrales-Aguilar E, Schwemmle M, Garcia-Sastre A (2018) Specific mutations in the PB2 protein of influenza A virus compensate for the lack of efficient interferon antagonism of the NS1 protein of bat influenza A-like viruses. J Virol. https://doi.org/10.1128/JVI.02021-17
Kuo RL, Chen CJ, Wang RYL, Huang HI, Lin YH, Tam EH, Tu WJ, Wu SE, Shih SR (2019) Role of enteroviral RNA-dependent RNA polymerase in regulation of MDA5-mediated beta interferon activation. J Virol. https://doi.org/10.1128/JVI.00132-19
Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, Garcia-Sastre A (2016) Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19(6):882–890. https://doi.org/10.1016/j.chom.2016.05.009
Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A (2009) NS5 of dengue virus mediates STAT2 binding and degradation. J Virol 83(11):5408–5418. https://doi.org/10.1128/JVI.02188-08
Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, Li L, Ren L, Lei X, Xiang Z, Wang J (2021) SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol 18(4):945–953. https://doi.org/10.1038/s41423-020-00619-y
Vignuzzi M, Lopez CB (2019) Defective viral genomes are key drivers of the virus-host interaction. Nat Microbiol 4(7):1075–1087. https://doi.org/10.1038/s41564-019-0465-y
Du Y, Xin L, Shi Y, Zhang TH, Wu NC, Dai L, Gong D, Brar G, Shu S, Luo J, Reiley W, Tseng YW, Bai H, Wu TT, Wang J, Shu Y, Sun R (2018) Genome-wide identification of interferon-sensitive mutations enables influenza vaccine design. Science 359(6373):290–296. https://doi.org/10.1126/science.aan8806
Dimmock NJ, Easton AJ (2015) Cloned defective interfering influenza RNA and a possible pan-specific treatment of respiratory virus diseases. Viruses 7(7):3768–3788. https://doi.org/10.3390/v7072796
Jin H, Lu B, Zhou H, Ma C, Zhao J, Yang C-f, Kemble G, Greenberg H (2003) Multiple amino acid residues confer temperature sensitivity to human influenza virus vaccine strains (FluMist) derived from cold-adapted a/ann arbor/6/60. Virology 306(1):18–24. https://doi.org/10.1016/s0042-6822(02)00035-1
Talon J, Salvatore M, O’Neill RE, Nakaya Y, Zheng H, Muster T, Garcia-Sastre A, Palese P (2000) Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci USA 97(8):4309–4314. https://doi.org/10.1073/pnas.070525997
Mossler C, Groiss F, Wolzt M, Wolschek M, Seipelt J, Muster T (2013) Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine 31(52):6194–6200. https://doi.org/10.1016/j.vaccine.2013.10.061
Ghorbani A, Abundo MC, Ji H, Taylor KJM, Ngunjiri JM, Lee CW (2020) Viral subpopulation screening guides in designing a high interferon-inducing live attenuated influenza vaccine by targeting rare mutations in NS1 and PB2 proteins. J Virol. https://doi.org/10.1128/JVI.01722-20
Noble S, McLain L, Dimmock NJ (2004) Interfering vaccine: a novel antiviral that converts a potentially virulent infection into one that is subclinical and immunizing. Vaccine 22(23–24):3018–3025. https://doi.org/10.1016/j.vaccine.2004.02.013
Mann A, Marriott AC, Balasingam S, Lambkin R, Oxford JS, Dimmock NJ (2006) Interfering vaccine (defective interfering influenza A virus) protects ferrets from influenza, and allows them to develop solid immunity to reinfection. Vaccine 24(20):4290–4296. https://doi.org/10.1016/j.vaccine.2006.03.004
Frensing T, Pflugmacher A, Bachmann M, Peschel B, Reichl U (2014) Impact of defective interfering particles on virus replication and antiviral host response in cell culture-based influenza vaccine production. Appl Microbiol Biotechnol 98(21):8999–9008. https://doi.org/10.1007/s00253-014-5933-y
Gould PS, Easton AJ, Dimmock NJ (2017) Live attenuated influenza vaccine contains substantial and unexpected amounts of defective viral genomic RNA. Viruses. https://doi.org/10.3390/v9100269
Acknowledgements
We thank Dr Valeria Lulla for suggestions and advice.
Funding
Aartjan J.W. te Velthuis is supported by joint Wellcome Trust and Royal Society Grant 206579/Z/17/Z, and the National Institutes of Health Grant R21AI147172. Elizaveta Elshina is supported by Engineering and Physical Sciences Research Council scholarship EP/S515322/1.
Author information
Authors and Affiliations
Contributions
EE wrote manuscript, compiled tables and created figures. AJWtV wrote manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elshina, E., te Velthuis, A.J.W. The influenza virus RNA polymerase as an innate immune agonist and antagonist. Cell. Mol. Life Sci. 78, 7237–7256 (2021). https://doi.org/10.1007/s00018-021-03957-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03957-w