
Abstract
For a long time, PLS3 (plastin 3, also known as T-plastin or fimbrin) has been considered a rather inconspicuous protein, involved in F-actin-binding and -bundling. However, in recent years, a plethora of discoveries have turned PLS3 into a highly interesting protein involved in many cellular processes, signaling pathways, and diseases. PLS3 is localized on the X-chromosome, but shows sex-specific, inter-individual and tissue-specific expression variability pointing towards skewed X-inactivation. PLS3 is expressed in all solid tissues but usually not in hematopoietic cells. When escaping X-inactivation, PLS3 triggers a plethora of different types of cancers. Elevated PLS3 levels are considered a prognostic biomarker for cancer and refractory response to therapies. When it is knocked out or mutated in humans and mice, it causes osteoporosis with bone fractures; it is the only protein involved in actin dynamics responsible for osteoporosis. Instead, when PLS3 is upregulated, it acts as a highly protective SMN-independent modifier in spinal muscular atrophy (SMA). Here, it seems to counteract reduced F-actin levels by restoring impaired endocytosis and disturbed calcium homeostasis caused by reduced SMN levels. In contrast, an upregulation of PLS3 on wild-type level might cause osteoarthritis. This emphasizes that the amount of PLS3 in our cells must be precisely balanced; both too much and too little can be detrimental. Actin-dynamics, regulated by PLS3 among others, are crucial in a lot of cellular processes including endocytosis, cell migration, axonal growth, neurotransmission, translation, and others. Also, PLS3 levels influence the infection with different bacteria, mycosis, and other pathogens.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The ever-growing number of diseases, in which PLS3 is involved, highlights the importance of this F-actin-binding and -bundling protein, with a broad spectrum of cellular pathways (Fig. 1). It seems that the expression of PLS3 is tightly regulated since knockout or mutations cause osteoporosis, while overexpression seems to trigger osteoarthritis and various types of cancer. Instead in several neuromuscular disorders, such as spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS) and CHP1-associated ataxia, PLS3 overexpression acts as a protective modifier. In this review, we present the current knowledge on the gene expression and protein function, the various cellular functions, in which PLS3 is involved and the disorders associated with PLS3 levels (decreased or increased) (Fig. 2).

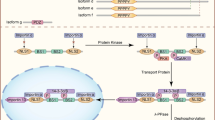
PLS3-associated disorders and main cell types involved. a PLS3 is involved in a variety of diseases, which associate with the PLS3 levels in a cell. Low protein abundance leads to osteoporosis, while increased levels are associated with cancer and osteoarthritis. In some neurodegenerative diseases, where F-actin levels are reduced, PLS3 overexpression acts protective. Green letters imply a protective role of PLS3 while red letters highlight PLS3 as disease driving protein in the depicted disorders. PLS3 plastin 3, SMA spinal muscular atrophy, ALS amyotrophic lateral sclerosis. b PLS3 fulfills distinct functions within different cell types. Osteoclasts, osteoblasts, osteocytes and chondrocytes are the target cells within the bone disease spectrum, which are influenced when PLS3 is dysregulated. In some neurodegenerative disorders, where motor neurons (e.g., SMA) or Purkinje cells (e.g., ataxia) are affected, overexpression of PLS3 showed a protective effect. Involvement of PLS3 in cancer is highly divers and includes different kinds of solid tissues as well as hematopoietic and lymphatic cancers. The figure was created with BioRender.com

Cellular function of PLS3. The main function of PLS3 is its role in F-actin-binding and -bundling activity and, thus, in F-actin dynamics. Consequently, PLS3 is involved in endocytosis, cell motility, cell adhesion, mechanotransduction, pathogen infection, Ca2+ homeostasis, exocytosis, vesicle trafficking, axonal local translation and others. PLS3 protein is depicted as red circle. ER endoplasmic reticulum, ECM extra cellular matrix. The figure was created with BioRender.com
Gene and expression
In 1980, Bretscher & Weber identified fimbrin (later renamed to plastin), a 68 kD protein isolated from small intestine of the chicken [1]. Plastins are a family of actin-binding and -bundling proteins consisting of three tissue-specific paralogs PLS1, PLS2, and PLS3 localized in humans on chromosomes 3, 13 and X, respectively [2]. The three isoforms share approximately 70% nucleotide sequence identity [3]. PLS1 (plastin 1, I-plastin) is expressed in the intestine, colon, kidney and hair cell stereocilia of the inner ear, PLS2 (plastin 2, L-plastin) in hematopoietic cell lineages and many types of cancer cells, and PLS3 (plastin 3, T-plastin), the most abundant isoform, in cells from solid tissues [2, 3]. In lower organisms, such as yeast, only one isoform is present, known as fimbrin or Sac6. Before 2008, papers referred to fimbrin or T-plastin, and only after that, the correct name of plastin 3 has been established in publications.
The expression of PLS3 is of utmost interest in the context of several medical conditions, such as spinal muscular atrophy (SMA), osteoporosis, osteoarthritis as well as a large number of cancers. While the regulation of the PLS3 expression in different tissues is still elusive, several mechanisms have been proposed, which do not necessarily exclude each other (Table 1). Early reports stated that PLS3 expression is limited to solid tissues [4]. However, elevated expression levels were found in blood samples in about 5% of healthy individuals [5]. PLS3 shows high expression in the fetal and adult spinal cord and PC12 cells during neuronal differentiation, where it accumulates in growth cones [5]. PLS3 mRNA is enriched and locally transcribed in the axonal compartment of motor neurons [6].
PLS3 is located in both, humans and mice, on the X-chromosome next to the macrosatellite DXZ4 [7]. In women, approximately 15% of genes escape the X-chromosomal inactivation (XCI) and are, therefore, biallelically expressed [8]. Another 15% of genes variably escape XCI in a tissue-specific manner [8,9,10]. PLS3 is a facultative escape gene in humans but not in mice [11]. The macrosatellite DXZ4 is essential for XCI and has a highly variable repeat number of 50–100 copies of a 3 kb repeat monomer [12]. Interestingly, on the inactive X-chromosome, DXZ4 is hypomethylated and binds to the transcriptional regulator protein CCTF-binding factor (CTCF), both features of chromatin regulation [13]. It has been hypothesized that the copy number of DXZ4 may modulate the escape of genes in its molecular neighborhood and thereby the expression level of PLS3 in females [14].
Accordingly, in SMN1-deleted asymptomatic women of spinal muscular atrophy (SMA) discordant families, the PLS3 expression is elevated in blood and Epstein–Barr Virus transformed lymphoblastoid cells but not in their SMA-affected siblings, indicating a sex-specific protective effect (see more details in Sect. 3.6.1) [5]. Moreover, mutations or knockout of PLS3 mainly affect men or male mice, while women or female mice are either very mildly or not affected, further supporting the sex specificity of PLS3 expression [15, 16].
Function
The main function of PLS3 is in F-actin-binding and -bundling. Consequently, PLS3 is involved in all processes dependent on F-actin dynamics such as cell motility, focal adhesion, cell division, endocytosis, neurotransmission, vesicle trafficking, axonal local translation, intracellular calcium PLS3-dependent processes, pathogen infection and others (Fig. 2).
Protein structure
The three plastin isoforms seem to have different effects on the actin cytoskeleton organization depending on the cell type [17]. Plastins are monomeric proteins with a single polypeptide chain composed of two tandem repeats of actin-binding domains (ABD1 and ABD2) [18]. Each of these ABDs encompasses a pair of ~ 125 residue calponin homology (CH) domains (ABD1: CH1 and CH2, ABD2: CH3 and CH4) [19]. Each of the CH domains consists of four major α-helices. While three α-helices form a loose bundle of helices, the fourth is orientated perpendicular to the major bundles [20]. At their N-terminal end, plastins possess a Ca2+-binding regulatory domain (RD) of approximately 100 amino acids. The RD contains two EF-hands and a calmodulin-binding motif (CBM) [3, 21, 22]. The RD is connected with the ABD core via a linker [23]. PLS3 consists of 630 amino acids and the molecular mass of the protein is 70.8 kDa. In humans, the homology between PLS1 and PLS2 is 73%, 75% between PLS1 and PLS3, and 79% between PLS2 and PLS3 [3]. Comparisons of the plastin isoforms between humans and mice show 94% identity for PLS1, 96% for PLS2, and 99% for PLS3 [3].
Plastins cross-link actin filaments into bundles, which are higher-order assemblies, through the tandem pair in their ABDs [24, 25]. So far, only the crystal structures of the ABD1 and ABD2 actin-binding cores are resolved [24, 26]. A homology-based model of the PLS3 actin-binding core has been generated using the Phyre2 web portal for protein modeling, prediction, and analysis [23]. The full-length crystal structures of all plastins remain to be determined.
F-actin dynamics/ cell motility
The main function of plastins is the binding and bundling of F-actin [3, 23, 27, 28]. F-actin is a polymer composed of globular (G-actin) subunits. These microfilaments are the main components of the cytoskeleton [29]. The actin cytoskeleton is a complex network whose dynamic formation influences numerous fundamental physiological cellular processes, such as focal adhesion, cell motility, endo- as well as exocytosis, mechanotransduction and cell division [30]. The F-actin network underlies a tight regulation and must be assembled, maintained, and disassembled in 3D at the correct time and place, and with proper filament organization and dynamics [31]. Thereby, the dynamic formation of the actin cytoskeleton is dependent on various F-actin-binding and -bundling proteins [32].
Plastins are involved in the assembly and organization of the actin cytoskeleton through their two ABDs. Binding of each ABD to two separate actin filaments promotes the formation of a bundle with a center-to-center distance of ~ 120 Å between the filaments [28]. However, the exact mechanism by which the ABD1 and ABD2 bind and bundle actin is poorly understood. It is assumed that both ABDs differently interact with actin [26]. Actin-binding is mediated through ABD1, while bundling is facilitated through both ABDs [33]. Furthermore, the plastin isoforms differentially interact with F-actin resulting in distinct F-actin organization [17, 20]. It has been suggested that PLS3 binding via both ABD1 and ABD2 is essential at the leading edge and focal adhesions [23]. The F-actin-binding and -bundling activity of the ABD domains is Ca2+ dependent, as the binding of Ca2+ by EF-hands inhibits the interaction with F-actin [33, 34].
The ABD cores form a compact and rather globular structure in an antiparallel arrangement induced by the contact between the CH1 and CH4 domains at the N- and C-terminal ends [3, 26]. This conformational structure seems to be highly dynamic and dependent on the presence of Ca2+ [35]. The conformational plasticity of CH2, located within the structurally polymorphic ABD1, influences the diverse functions of different actin assemblies [22]. This indicates that the structural plasticity is related to the function of the plastin isoform. Furthermore, the crystal structure indicates that the two ABDs harbor different binding characteristics with F-actin [20, 33].
As an F-actin-binding and -bundling protein, PLS3 plays an essential role in cell motility [36]. The movement of a cell is activated by adhesion to the extracellular matrix, executed through membrane remodeling at the leading edge and controlled by the actin cytoskeleton [37, 38]. PLS3 promotes membrane protrusion and cell migration to overcome gaps in the extracellular matrix and when adhesion is spatially gapped [39]. In the skin, PLS3 influences the basement membrane assembly [40]. Furthermore, PLS3 is involved in membrane trafficking under hypoxia [41]. All these functions arise from the interaction with the actin cytoskeleton. It has been speculated that PLS3 serves as a mechanical link between the actin polymerization network at the front of the cell and the myosin motor activity in the cell body [36]. PLS3 modulates the actin dynamics and generates force independent of cross-bridge formation mediated by actin related protein 2/3 complex (Arp2/3) [42]. The Arp2/3 complex facilitates the actin polymerization which is essential to generate pushing forces capable of deforming the cell membrane [31].
Influence of intracellular calcium on PLS3-dependent processes
The crucial role of Ca2+ for F-actin-bundling by plastins has been a subject of research for a long time [43]. Especially, in terms of PLS2, studies focused on its Ca2+-dependent functions regarding actin-bundling and specifically T-cell activation and motility [34, 44], reviewed in Babich, Burkhardt [45], Morley [46].
Interestingly, comparisons of the three plastin isoforms show a much lower amino acid identity at the N-terminal EF-hand domains than the higher conserved actin-binding domains. Thus, several studies compared the plastin isoforms regarding their Ca2+ sensitivities and Ca2+-dependent functions [33, 47,48,49].
EF-hands of both PLS2 and PLS3 were shown to consist of alpha-helix-rich sequences and underlie conformational changes upon Ca2+-binding [47]. PLS3 has a lower sensitivity for Ca2+ as indicated by only a slight change of the EF-hands’ secondary structure in contrast to PLS2. Also, higher concentrations of Ca2+ are required for structural changes of the EF-hand motifs of PLS3 [47]. Congruently, monitoring the disassembly of plastin/F-actin bundles induced by Ca2+, PLS3 has been proven to be less sensitive to Ca2+ than both PLS1 and PLS2 [33]. Further analysis has shown a difference in affinity to Ca2+ within the two EF-hands of PLS3 (one high and one low affinity site) in contrast to PLS2 comprising two high-affinity sites [47].
Generally, the presence of Ca2+ reduces the ability to bundle F-actin in all three isoforms [33]. Further studies on the functional linkage between the EF-hands and the linker region CBM revealed inhibition of rapid proteolytic cleavage at the CBM upon Ca2+ stimulation, emphasizing a Ca2+-dependent linkage of these two regions in all three human isoforms. Focusing on the impact of Ca2+ on the ABDs, inequality is found: one ABD, most likely ABD1, binds F-actin independent of the presence of Ca2+; whereas at the other ABD, most likely ABD2, F-actin-binding is inhibited by Ca2+ bound by the EF-hands [33].
To gain further insight into the process of Ca2+-binding to the EF-hands as well as into kinetic and structural details on Ca2+-regulated domains of plastins, three biophysical methods have been used: surface plasmon resonance studies, isothermal titration calorimetry studies, and nuclear magnetic resonance spectroscopy [48]. In terms of Ca2+-regulated function, a crucial role of the regulatory helix 5 (H5) linker region localized between the second EF-hand motif and the first CH1 domain of ABD1 has been suggested, which may even differ between the plastin isoforms. Precisely, the H5 region of PLS3 (TPL-EF-H5 construct) is not displaced by the bee venom peptide melittin, unlike a possible displacement in PLS2 (LPL-EF-H5 construct) [48].
Another approach assessed the Ca2+-coordination structures of PLS2 and PLS3 and their synthetic peptide analogs by Fourier transform infrared spectroscopy [49]. Based on the results, an association between the aggregation tendency of the two Ca2+ binding sites of PLS3 and its lower sensitivity to Ca2+ was suggested [49].
The importance of well-regulated Ca2+ homeostasis for the proper function of PLS3 is underlined by several findings of disturbed Ca2+ regulation associated with loss of its functionality, shown in PLS3 rescue in SMA models [50] or osteogenesis imperfecta (OI)-associated PLS3 mutations [23].
Endocytosis
The role of PLS3 in endocytosis has been first described in budding yeast [51]. Knockout of the PLS3 ortholog, Sac6, causes massive impairment of endocytosis; especially of the receptor-mediated internalization of the pheromone α-factor [51]. Actin-bundling is a crucial process required for proper endocytosis in which Sac6-mediated bundling of actin filaments forms a framework in the early stages of this process [52]. Actin organization in cortical actin patches, which are dynamic actin structures within the inner faced layer of the cell membrane in yeast, dependent on actin-bundling proteins Sac6 and Scp1 (ortholog of human SM22). Loss of both proteins results in tremendous defects in patch biogenesis (increased patch lifetime), while Sac6 loss alone causes random movement or patch disassembly, with the absence of fast movements [52]. In filamentous fungi, Ashbya gossypii and Aspergillus nidulans, the polarized hyphal tip growth and endocytosis are Sac6/fimbrin A dependent, and their lack causes reduced rates of endocytic uptake [53, 54]. This highlights the importance of PLS3 orthologs as actin regulators and the requirement of functional actin dynamics to fulfill proper extension growth across species. Additionally, Sac6 along with other actin-binding and -bundling proteins (such as Abp85) are required for the uptake of the maltose transporter in yeast [55]. Moreover, fimbrin cross-linkers, twisting the actin filaments, provide approximately one-sixth of the energy required for endocytosis [56, 57]. Importantly, Arp2/3-actin networks are crucial to generate the force during membrane invaginations. Those networks are disrupted in Sac6-deficient cells and consequently result in disturbed endocytosis [56].
In fission yeast, it has been proposed that fimbrin selectively regulates the access to actin filaments for other actin-binding proteins, such as the tropomyosin Cdc8p [58]. Accordingly, in fimbrin-deficient cells, mislocalization of tropomyosin to actin patches results in increased patch lifespan and decreased motility [58]. Fimbrin is also able to displace tropomyosin from actin filaments lowering the inhibition of myosin I by tropomyosin and thereby ensuring motor activity [59].
The role of PLS3 in endocytosis caught first attention in human studies when PLS3 has been identified as a protective modifier of SMA [5]. In the context of SMA, both F-actin levels and Ca2+- homoeostasis are reduced, two processes crucial for endocytosis, but restored by PLS3 overexpression [5, 60,61,62,63,64]. Importantly, endocytosis is a key process in neurons as they rely on constant refilling of the recycling vesicle pool in the presynapse. Apart from other reasons, endocytosis is a main cellular process disturbed in SMA, contributing to disturbed function of neuromuscular junctions (NMJs) [61, 65,66,67,68].
Another approach assessed defects in endocytic pathways in a SMA model and hypothesize that PLS3 increases the expression of endocytic proteins by supporting the availability of ribonuclear protein granules containing the required components for their translation [68]. Local axonal translation is dependent on transport of mRNAs and microRNAs along the axons to the growth cone of polarized neurons. Thereby, mRNAs and microRNAs not only hitchhike on late endosomes and lysosomes but they also act as hubs for local translation, finally contributing to the function and maintenance of neurons and neuronal circuits [69, 70]. Congruently, PLS3 directly interacts with activated RAB5, an early endosomal protein, and thereby regulates endocytic activity in mammalian cells [71]. It is, therefore, tempting to postulate that PLS3 overexpression may restore impaired vesicle trafficking in SMA and consequently local translation.
Signaling
The dynamic regulation of F-actin assembly and the interaction with the actin cytoskeleton is regulated by the coordinated activation of actin assembly factors through different signaling cascades [31]. It is accepted that PLS3 is involved in these signal transduction pathways [72]. Early studies show that the up- or down-regulation of PLS3 influences different signaling pathways. Downregulation of PLS3 has been shown to inhibit the p38 mitogen-activated protein kinase (MAPK) signaling pathway in MDA-MB-231 cells, which mediates apoptosis [73]. In keratinocytes, PLS3 is involved in the calcineurin/nuclear factor of activated T-cell (NFAT) pathway, which is a major regulator of cell migration [74]. In osteocytes, PLS3 levels regulate the expression of RELA proto-oncogene, NFΚκB subunit. PLS3 interacts with NFκB repressing factor (NKRF) facilitating its translocation into the nucleus, and thus the transcription of nuclear factor-activated T cells c1 (NFATC1) and an important factor in osteoclastogenesis [16]. Through the PI3K/AKT signaling pathway, PLS3 regulates tumor progression by promoting the proliferation and migration of cancer cells [75].
Several studies have demonstrated that PLS2 can be phosphorylated at positions Ser 5 and Ser7 during leukocyte activation by various stimuli [3]. Phosphorylation of Ser5 is mediated by cAMP-dependent protein kinase A, while the relevant kinase for Ser7 is still controversially discussed. In osteoclasts, phosphorylation of PLS2 on Ser5 and Ser7 increases the F-actin-bundling capacity [76] and the avidity for cellular F-actin and F-actin-binding activity [77]. In macrophages, PLS2 is phosphorylated exclusively on Ser5 by stimulation with bacterial lipopolysaccharide [78]. Ser5 is only found in PLS2 and not conserved in other plastin isoforms. To date, no phosphorylation of PLS3 is known and further studies are required [72].
Disease involvement
PLS3 levels require a tight balancing in the cell. Aberrantly increased levels are associated with cancer and osteoarthritis. Instead in several neurodegenerative conditions associated with decreased F-actin levels, PLS3 overexpression acts protective. Instead, deletions or loss-of-function mutations in PLS3 cause osteoporosis in humans and mice. Finally, S-nitrosylation of PLS3 associates with thoracic aortic dissection (Fig. 1). Moreover, PLS3 is involved in infection and pathogen entry.
Cancer
Malignancies in solid tissues
A rising number of biomedical studies investigated PLS3 in the context of the most common malignancies (colorectal, prostate, breast, gastric, and lung cancer) and explored its potential use as a biomarker. Metastases in distant organs are the most common cause of death in these solid tissue tumors. The cell migration and invasion of tumor cells are driven by the modulation of the actin cytoskeleton. PLS3 is highly expressed during epithelial–mesenchymal transition (EMT) in circulating tumor cells (CTCs) in colorectal cancer. Elevated levels of PLS3 during EMT in CTCs negatively regulate expression levels of epithelial genes, and positively regulate the levels of mesenchymal genes and thereby increase the invasiveness of colorectal cancer [79]. Several mechanisms that could explain the regulation of PLS3 in CTCs had been suggested. Chromosomal instability, a hallmark of cancer formation, can trigger a copy number gain of Xq23 leading to PLS3 overexpression [79, 80]. A study in colorectal cancer cells states that PLS3 is a downstream target of Lamin A, which is a risk factor for this type of cancer. PLS3 in turn downregulates E-cadherin leading to increased invasiveness during EMT [81]. Furthermore, genetic polymorphisms modifying PLS3 levels have been discussed. The intronic polymorphism rs871773 C > T is associated with a low recurrence time of colorectal cancer, while the rs6643869 polymorphism correlates with the time of tumor recurrence in women and has been identified in CTCs [82,83,84].
PLS3 overexpression is found in two-third of CTCs from breast cancer patients in different stages of EMT and is associated with poor overall survival, especially in patients with luminal type A or triple-negative breast cancer [85]. Silencing of PLS3 in MDA-MB-231 triple-negative breast cancer cells increases the sensitivity towards the anti-cancer agent paclitaxel [73]. A recent study has published a gene panel consisting of seven genes (PLS3, MGB1, HER2, CK19, CDH1, CDH2, and VIM) that can be used to discriminate EMT stages in CTCs tested by qPCR [86]. For prostate cancer, a gene panel with 14 genes has been developed including PLS3, VIM, and CDH2, and in 9.5% of patients an increased expression of PLS3 has been found [87]. Interestingly, LOXL1 downregulates CDH1 and upregulates VIM, CDH2, PLS3, and SNAI2 in gastric cancer cells, where expression of LOXL1 correlates with EMT [88]. In gastric cancer, increased expression of PLS3 indicates a poor prognosis and is associated with cancer differentiation, the depth of tumor invasion and EMT [89]. Chromatin immunoprecipitation (ChIP) assays in patients with gastric cancer have shown that the transcription factor ZNF471 acts as a tumor suppressor gene. ZNF471 binds the PLS3 promoter and suppresses its expression. Methylation of the CpG-methylation site of the ZNF471 promoter is a useful prognostic marker for overall survival in gastric cancer patients [90]. High PLS3 expression is also a feature of pancreatic cancer cells. Here, PLS3 acts as an oncogene and its overexpression triggers the activation of the PI3K/AKT signaling pathway leading to cancer progression [75]. In non-small-cell lung cancer (NSCLC), increased PLS3 plasma levels are a predictor of poor survival. PLS3 expression is of therapeutic value and can predict the responsiveness to treatment with Nivolumab—a PD-1 monoclonal antibody—in NSCLC patients [91]. Moreover, UV-light- and cisplatin-resistant tumors show elevated PLS3 expression [92, 93]. Downregulation of PLS3 in human liver cancer cells increased the sensitivity of these cells to cisplatin [94]. Cells lacking PLS3 expression are sensitive to DNA damage and silencing of PLS3 led to increased damage by UV light. By this, expression of PLS3 could be used as a therapeutic marker in irradiation therapy [94]. Together, the mentioned studies suggest an association of increased PLS3 expression during EMT and a high prognostic potential of PLS3 as a biomarker in malignancies of solid tissues. The expression levels of PLS3 represent a useful marker for cancer prognosis especially as part of a gene panel.
Malignancies of the hematopoietic and lymphatic system
Elevated PLS3 expression is a negative prognostic marker for acute myeloid leukemia (AML), the most common acute leukemia in adults, while knockdown of PLS3 increases survival in vivo [95]. In Sézary Syndrome (SS), an aggressive, rare form of cutaneous T-cell lymphomas (CTCL), circulating CD4+ T-cells show increased expression of PLS3, TWIST1 and GATA6 compared to normal CD4+ T-cells [96, 97]. The promoter regions of all three genes have been found to be hypomethylated in SS CD4+ T cells indicating an epigenetic regulation of the expression levels. This is of large interest, since all three genes are located on different chromosomes [98,99,100,101]. Therefore, PLS3 has been suggested as a biomarker for SS CD4+ T cells and it is associated with an unfavorable disease outcome [102,103,104].
Bone disorders
Osteoporosis
Dynamic regulation of the actin cytoskeleton by actin-binding and -bundling proteins like PLS3 [3, 20] is particularly crucial in the musculoskeletal system to instantly adapt to environmental changes through mechanotransduction [105, 106]. Mechanotransduction is the conversion of mechanical signals into cellular response, which is assumed to play an essential role in several pathologies of the musculoskeletal system, such as osteoporosis [107, 108] and osteoarthritis [109,110,111]. In chicken, fimbrin is detected in the dendrites of osteocytes [105, 112], which play a key role in mechanotransduction [113]. Therefore, PLS3 could influence the mechanical signal transformation [108].
In 2013, pathogenic variants in PLS3 have been reported to be associated with osteoporosis including fractures in men and mild osteoporosis in women [108]. Moreover, PLS3 showed a significant association with osteoporosis in women upon menopause, suggesting that mutations in PLS3 not only associate with the monogenic but also the complex genetic trait of osteoporosis [108]. Osteoporosis is a multifactorial disease that is dependent on hormonal, environmental as well as genetic factors [114, 115]. Low bone mineral density (BMD) seems to be genetically determined in 50–85% of cases [114, 116,117,118]. This emphasizes the genetic component as a determining factor for BMD and its relating fracture risk. Primary (hereditary) osteoporosis usually becomes symptomatic in childhood and is, therefore, referred to as early-onset osteoporosis. The clinical features are characterized by low BMD (age normalized average score (Z-score < 2.0)), occurring vertebral compression fractures (VCFs), or low-trauma fracture history [119, 120]. The most common form of monogenic osteoporosis is OI [120], which additionally affects extraskeletal features like blue sclera, joint hypermobility, and deafness [15, 114, 121]. Around 85–90% [15, 114] of OI cases are linked to a dysregulation in type I collagen, although the list of OI-associated genes is increasing [15, 114, 122,123,124,125].
Up to date, 27 mutations in PLS3 are associated with early-onset osteoporosis (Table 2; Fig. 3). Nevertheless, rare variants also indicate OI traits [108, 126, 127], which is why PLS3 was also included as a genetic cause for OI according to Van Dijk, Sillence [121]. However, several studies have reported variants that lead to severe skeletal abnormalities among women, resembling the osteoporotic phenotype of men [108, 115, 128,129,130]. This huge variation in heterozygous women is suggested to be caused by X-inactivation of the mutant allele or PLS3 escaping X-inactivation which is why women are less severely affected [15, 126, 129, 131, 132]. Even though bone morphometry was very heterogeneous [16, 126, 133,134,135,136], most male patients showed peripheral fractures, low BMD, VCFs, especially in the thoracic spine, and low bone turnover rate, while only a few developed extraskeletal OI traits [108, 126,127,128, 131, 137,138,139,140], developmental delay [15, 127] or neuromuscular abnormalities, like waddling gait [108, 126, 131, 141]. So far, no specific biomarkers have been identified, which can distinguish genetic factors of osteoporosis, although microRNAs are getting more and more popular as functional markers [120, 142]. This makes it difficult to diagnose PLS3 mutations, although genetic analysis would be important to assess bone fragility risk within families. Thus, to find genetic causes of bone disorders, it is recommended to use gene panel screening containing known monogenic osteoporosis genes in fracture-prone children to identify genetic factors influencing bone health and to reveal family risk [114, 143,144,145].

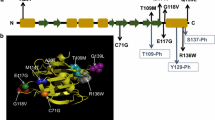
Schematic representation of the PLS3 gene and corresponding protein domains. Illustrated are the causative mutations for osteoporosis and the resulting amino acid changes that were previously published. All depicted mutations were modified according to HGMD Professional (NM_005032.7 (GRCh38)). References and clinical characterization are shown in Table 2. Note, RNA containing frameshift and nonsense variants (except for the last exon 16), most likely undergo nonsense-mediated mRNA decay and thus no protein is produced. E EF-hand motifs, CBM calmodulin-binding motif, RD regulatory domain, CH calponin-homology domain, ABD actin-binding domain, Core actin-binding core domain, SNP single-nucleotide polymorphism, CNV copy number variation, NA not available; ★ = PLS3 isoform including 18 exons, accession No.: NG_012518. The figure was created with BioRender.com
In summary, most of the osteoporosis-related PLS3 variants are frameshift [108, 122, 135, 137, 138, 144] or nonsense mutations resulting in premature termination codons [108, 128, 130, 140], which are followed by mRNA decay [108, 128, 131, 135]. Next to that, partial deletions within PLS3 were identified [126, 140, 146]. Surprisingly, no phenotypic differences were seen between mutations causing deletion or truncation of PLS3 protein or missense or in frame insertion mutations resulting in mutated PLS3 protein, most likely because all types of mutations cause a loss of function of PLS3 [108, 128,129,130,131, 135, 140, 147].
Studies of missense mutations in full-length PLS3 report impairments in F-actin-bundling ability or defects in Ca2+ sensitivity, which lead to localization shifts within the cell and can also disturb Ca2+-dependent structural rearrangements necessary for actin dynamics [23]. Impaired actin dynamics caused by mutations within the ABD2 of PLS3 result in inefficient F-actin-bundling properties, without influencing actin-binding by ABD1 [23]. In turn, this specific mutation interferes with PLS3′s actin-cytoskeletal association and thus, its localization at the lamellipodia and leading edge [23]. Considering these findings, it could be hypothesized that the proper intracellular localization of PLS3 is dependent on its actin-bundling function [23]. Similarly, in another study, a PLS3 mutation, disrupting the ABD interface, results in conformational changes and defective actin-binding and -bundling function [140].
A cellular mislocalization and impaired Ca2+-dependent actin-associated distribution of PLS3 were also related to mutations influencing the Ca2+ sensitivity, causing either hypo- or hypersensitivity to Ca2+. Misregulated PLS3 cycling could thereby result in impaired bone homeostasis and might explain the osteoporotic phenotype in these patients [23].
To analyze the proposed functions of PLS3 regarding mechanotransduction [5, 15, 108, 120, 143, 144, 148], Ca2+ regulation [23, 33, 149], osteoblastic bone mineralization [15, 120, 126, 131, 134, 143, 144, 150], osteoclastogenesis [16] and vesicular trafficking [15, 16, 61, 151], various PLS3 mutations have been investigated and mouse model studies established to specifically investigate the effect of PLS3 loss or overexpression [16].
It is well established that PLS3 is highly abundant in the dendrites of osteocytes, which not only control mechanosensing but also regulate osteoblast and osteoclast activity, and an impairment of this cell system might explain the resulting osteoporotic phenotype [136, 152]. Indeed, in osteocytes, mutations in PLS3 interfere with cellular signaling, resulting in imbalanced bone homeostasis [121, 129]. In line with this, data acquired from PLS3 mutation carriers showed increased levels of apoptotic osteocytes together with abnormal gene expression of osteocyte-related genes [153].
Because PLS3 mutations cause childhood-onset osteoporosis associated with low BMD, it has been further hypothesized that the bone-forming osteoblasts might be disturbed by PLS3 loss [129, 143]. In healthy bone conditions, PLS3 abundance increases upon osteoblast differentiation [131, 154]. However, in some patients with PLS3 mutations, matrix mineralization and osteoblast number are decreased [126, 129, 131, 155]. The resulting imbalanced levels of osteoblasts and osteoclasts, favoring bone resorption, might result in osteoporosis.
For differentiation and bone formation, osteoblasts rely on high Ca2+ concentrations, which is in part regulated by the interaction of the Ca2+-binding proteins PLS3 and PLS2. Through their interaction, bound Ca2+ is released to increase the intracellular Ca2+ level when the extracellular Ca2+ level is low [149]. However, mutations in Ca2+ binding domains (EF-hands) of PLS3 weakened the interaction of PLS3 with PLS2, as revealed by PLS2-TRAP co-immunoprecipitation and western blot analysis, and thus might contribute to the osteoporotic phenotype [149].
To enable further analyses, a ubiquitous Pls3 KO mouse model has been generated that is comparable to loss-of-function mutations observed in OI patients [16]. In 3-month-old Pls3 KO animals reduced cortical and trabecular thickness and decreased bone parameters is seen. In line with humans, the osteoporotic phenotype in male mice is more pronounced than in females [16]. Importantly, the NFκB pathway, an essential pathway for osteoclastogenesis, is strongly influenced upon PLS3 loss [16]. NFκB signaling is initiated through binding of receptor activator of NFκB ligand (RANK-L) to its receptor RANK on the surface of osteoclasts, activating the transcription of nuclear factor-activated T cells c1 (Nfatc1) [156]. Besides, Nfatc1 expression is negatively regulated by the NFκB-repressing factor, NKRF. As PLS3 binds to NKRF, Pls3 KO results in insufficient repression of Nfatc1 nuclear translocation, and therefore in increased osteoclast differentiation [16]. Moreover, podosomes, specific F-actin ring structures in osteoclasts, important for resorptive activity, migration, and adhesion, are structurally disturbed in Pls3 KO mice, which might be due to increased depolymerization of F-actin and, thus, instability of the cytoskeleton [16].
When investigating the effects of elevated PLS3 levels in mice carrying homozygously a human PLS3 transgene under an ubiquitously expressing promotor and integrated into the Rosa26 locus on chromosome 6, a thickening of the cortical bone is seen in 3-month-old female PLS3 OE mice compared to the controls [16]. This led to increased stiffness and breaking force in the females, which might be due to impaired bone renewal [16, 116]. In contrast to Pls3 KO, PLS3 OE might cause increased translocation of NKRF to the nucleus, which in turn results in inhibition of Nfatc1 transcription and consequently inhibits osteoclastogenesis [16]. These PLS3 effects might be additionally influenced by impaired intracellular vesicle dynamics [61]. Similar to the Pls3 KO, also in PLS3 OE mice defective podosome formation is detected, probably caused by reduced F-actin disassembly [16]. In summary, it is assumed that an optimal amount of PLS3 is essential to sustain bone health. Neither removal of PLS3, resulting in bone abnormalities, nor an increased level of PLS3, leading to a hyperosteotic phenotype, seems to be beneficial for bone homeostasis but rather detrimental.
Osteoarthritis
In 2015, increased PLS3 levels have been reported in chondrocytes from patients with osteoarthritis compared to healthy individuals [157]. Osteoarthritis is a multifactorial disease that affects the whole joint and is defined by degeneration of the articular cartilage, synovial inflammation and increased bone mass in the subchondral bone [158]. This is in line with the observed elevated cortical thickness in PLS3 OE mice, which might be due to inhibited osteoclastogenesis [16]. Furthermore, the RANKL–RANK pathway plays a crucial role not only in osteoarthritis in general [159,160,161], but seems to be stressed in osteoarthritis patients showing increased PLS3 levels [157]. Further, differentially upregulated pathways in osteoarthritis patients are related to the actin cytoskeleton, endocytosis, TGF-β, MAPK, TNF-α, WNT and general metabolism processes [157]. As mentioned before, an interference of the mechanotransduction process is a crucial factor associated with the pathogenesis of osteoarthritis, where also the impact of PLS3 on the actin cytoskeleton is suggested to play an essential role [109,110,111]. Recent findings have shown that patients with varus gonarthrosis-induced osteoarthritis show higher levels of PLS3 in the medial compartment compared to the less affected lateral knee compartment. Besides, PLS3 was also predominantly found at the direct force-facing contact side of the respective joints [162]. These data might confirm previous assumptions that PLS3 seems to be involved in the mechanotransduction system and its enhanced level is associated with osteoarthritis. So far, less is known about the effect of PLS3 deletions on cartilage. Another study has shown no effect on the intervertebral disc of osteoporosis patients linked to loss of function and PLS3 deletions [148]. The influence of PLS3 overexpression, PLS3 loss, and PLS3 mutations on articular cartilage itself remains elusive and the underlying mechanism affecting cartilage health still needs to be explored.
Since about 5% of the general population show increased PLS3 expression in blood [5], it would be interesting to study if the increased expression of PLS3 in blood is associated with an increase of PLS3 in cartilage, that seems to be associated with osteoarthritis, and thus can act as a biomarker for osteoarthritis as well [162].
Thoracic aortic dissection
In terms of its role in diseases, PLS3 has also been shown to be involved in the causative mechanisms of thoracic aortic dissection in mice [163]. In more detail, S-nitrosylation of PLS3, induced by angiotensin II and mediated by inducible nitric oxide synthase, promoted the formation of a complex of PLS3 with cofilin, which depolymerizes actin and facilitates actin dynamics, and plectin, which links various cytoskeletal proteins and is important in formation of cell junctions in endothelial cells. This process was detected to aggravate the development of thoracic aortic dissection via pathological angiogenesis as well as disruption of the adherent junctions at the endothelial barrier [163].
Neurodegeneration
PLS3 is highly abundant in certain areas of the brain especially in the hippocampus but also in the spinal cord [164]. It is strongly upregulated during the development and maturation of motor neurons pointing towards an important role in these processes [66]. PLS3 is an important interaction partner of several proteins or protein complexes involved in neurodegeneration disorders such as SMA [5, 61], ataxia [66], amyotrophic lateral sclerosis (ALS) [68], and Charcot–Marie–Tooth (CMT) [165] (Fig. 1).
Spinal muscular atrophy
One of the most unexpected findings was the discovery that PLS3 overexpression acts as a protective modifier of SMA [5]. SMA is primarily a neurodegenerative motor neuron disorder and the most common cause of genetic death in infancy. It is caused by deletions and functional loss of SMN1, while the copy gene SMN2 strongly modulates the disease severity [166].
Rarely, individuals carrying a homozygous deletion of SMN1 and three or four SMN2 copies are fully asymptomatic, in contrast to approximately 99% of individuals with such a genotype who develop SMA. This suggests that protective modifier(s) in the genome of these subjects counteract the detrimental effect of reduced SMN levels [5, 65, 167]. PLS3 is the first human SMA protective modifier identified by transcriptome analysis of differentially expressed genes. All asymptomatic, but none of the symptomatic siblings of six discordant families showed an up to ~ 40-fold upregulation of PLS3 in lymphoblastoid cell lines (where PLS3 usually is not expressed). Instead, the same individuals show no difference in PLS3 expression in fibroblasts, suggesting a tissue-specific regulation [5]. Generation of iPSCs from fibroblasts of two discordant families showed that the expression of PLS3 is highly elevated in asymptomatic individuals but not in symptomatic siblings [167]. This is also found in motor neurons (MNs) differentiated from iPSCs [167]. The mechanism behind is still not fully understood.
Moreover, three additional SMA protective modifiers, coronin-1C (CORO1C), a Ca2+-dependent protein involved in actin dynamics, and two Ca2+ sensor proteins, neurocalcin delta (NCALD) and calcineurin-like EF-hand protein 1 (CHP1) have been identified [61, 65, 66]. Similar to PLS3, CORO1C acts protective when upregulated, whereas NCALD and CHP1 act protective when being downregulated as shown in various animal models [61, 65, 66]. Importantly CORO1C and CHP1 interact with PLS3. All modifiers are involved in endocytosis and restore reduced endocytosis in SMA (see section of 2.4).
PLS3 overexpression acts protective not only in humans with SMN1 mutations but also in zebrafish, worm, fly and mouse SMA models [5, 61, 64, 168,169,170]. In all systems, PLS3 overexpression ameliorates or counteracts the major hallmarks of SMA pathology. Reduced SMN level impairs F-actin dynamics, which might be due to a disturbed transport of β-actin mRNA along the axons [63]. F-actin dynamics is pivotal for cellular integrity and is involved in cellular shape, migration, vesicular trafficking, RNA translation, and endocytosis, among others [171]. In highly polarized MNs, all these processes are particularly crucial. In severely affected SMA models, numerous F-actin-dependent processes including axonal growth, axonal connectivity at the NMJ, neurotransmission, F-actin caging, synaptic vesicle recycling as well as proprioceptive input at MN somata are reduced [60, 64, 172,173,174]. All these processes were either restored or ameliorated by PLS3 overexpression as shown across various SMA animal models [61, 64].
Unexpectedly, while impaired NMJ function and motoric abilities are ameliorated in severely affected mice, survival is prolonged by only a few days [64]. To generate a more comparable situation as found in discordant SMA families (where 3–4 SMN2 copies are present, but never only 2 copies as in the severe SMA mouse model), an intermediate-like SMA mouse phenotype has been generated by subcutaneous injection of a low dose of SMN-ASOs (Nusinersen, 30 µg at P2 and P3) [61]. This slightly increased SMN level rescues inner organ function and doubles the survival rate. These mice still die due to MN loss with 1 month of age. In contrast, additional overexpression of human PLS3, from a homozygous PLS3 transgene [64], rescues MN and NMJ function and significantly prolongs survival (60% survive > 250 days; 30% > 400 days), clearly proving the protective effect of PLS3 overexpression in an intermediate SMA mouse model [61]. Besides, a gene therapy approach using adenovirus-associated virus (AAV9)-PLS3, in combination with low dose SMN-ASO, ameliorates the SMA phenotype in mice and prolongs survival [175, 176]. These experiments provide strong evidence that PLS3 is a genuine protective modifier and highlight the power of combinatorial therapies in SMA.
Exo- and endocytosis are crucial for neurotransmission in neurons. To properly maintain neurotransmission, endocytic uptake is essential to replenish the recycling pool, which supplies vesicles to the readily releasable pool (RRP) [177]. At NMJs of severely affected SMA mice, the organization and number of docked vesicles on the presynaptic site are significantly reduced causing a decrease of neurotransmitter release. Moreover, the RRP size is significantly reduced in SMA, and the depletion and refilling time constants of this pool tend to be slower [64, 172, 178]. F-actin is essential in all types of endocytosis [179] and its inhibition reduces endocytosis in neurons under high frequency stimulation [180]. Indeed, reduced SMN levels dramatically decreased endocytosis in vitro, as well as the FM1-43 uptake at NMJ level in SMA mice. Instead, PLS3 overexpression fully rescued endocytosis to similar levels as observed in controls [61]. Moreover, endocytic uptake of FM1-43 in the presynaptic terminal of NMJs upon electrical stimulation was significantly reduced in SMA and fully restored to control levels in SMA-PLS3 OE mice [61].
Chp1-associated ataxia
PLS3 is a genuine interaction partner of CHP1, a Ca2+ sensor protein [66]. CHP1 is ubiquitously expressed, but particularly abundant in neuronal tissues. CHP1 is a negative regulator of calcineurin, the most important phosphatase dephosphorylating the dephosphins involved in endocytosis [181]. Biallelic mutations in CHP1 cause autosomal recessive spastic ataxia (SPAX9; MIM 618438) in humans and mice [182,183,184]. Moreover, PLS3 overexpression in homozygous Chp1 mutant mice delays but does not ameliorate the ataxic phenotype at an early disease stage by preventing axon degeneration of Purkinje neurons [185]. PLS3 overexpression increases membrane targeting of NHE1 [185], an important binding partner of CHP1 [186] that is also associated with ataxia when mutated, at an early disease stage [187]. Thus, PLS3 overexpression has a moderate protective effect on ataxia caused by Chp1 depletion and demonstrates its potential as a cross-disease modifier [185].
SOD1-associated amyotrophic lateral sclerosis
PLS3 overexpression in a C. elegans model of ALS, carrying the dominantly inherited G85R SOD1 mutant, proved to be beneficial [68]. PLS3 increased locomotion rate, ameliorated pharyngeal pumping defects and counteracted sensitivity to paralysis by aldicarb. Contrary, on a wild-type C. elegans background, PLS3 overexpression had a detrimental effect on locomotion rate, strengthening the observation that the balance of F-actin is essential and its overload in a cell or organisms may lead to the opposite effect [68]. Since ALS and SMA share a lot of common pathological features, as well as molecular and cellular commonalities, further investigation of a beneficial impact of PLS3 on other ALS-involved genes, e.g., C9ORF72 would be very interesting.
Infection and pathogen entry
Plastins are conserved from yeast to mammals. Within all species, they fulfill functions in actin dynamics rendering rapid actin rearrangements upon pathogen challenges in the host cell while harboring distinct functions within the pathogen itself. Pathogens such as Salmonella typhimurium, Shigella flexneri or Trichomonas vaginalis enter the host cells by remodeling the actin cytoskeleton [188,189,190,191]. S. tysphimurium can even enter nonphagocytic cells like endothelial cells, upon the stimulation of CDC42 and RAC-1, which activate signaling of the pathogen protein SipA and thereby increase PLS3′s bundling activity [188]. At the contact point of host cell and pathogen, this process triggers actin rearrangements while leading to the formation of nucleation zones. PLS3 is recruited to these zones and stabilizes F-actin filaments. Arising from those zones, elongated actin filaments form protrusions surrounding the pathogen [188, 189, 191]. Nucleated actin filaments are the source of G-actin through active depolymerization to fuel the leading tips for the construction of the protrusion [191]. Finally, the pathogen is indulged in a vacuolic structure and released into the cytoplasm of the host cell. During infection with the Hepatitis C virus (HCV), PLS3 has been shown to regulate pathogen replication. Knockdown of PLS3 results in decreased viral replication and is, therefore, a potential target for HCV therapy [192].
Moreover, fimbrin facilitates active actin-cytoskeleton organizational changes to, e.g., enable amoebic movements and protrusion formation [190]. In Rickettsia, PLS3 together with profilin, capping protein, and cofilin determine the actin tail length required for motility during infection. In this species, actin bundles underlying tail formation resemble the organization of cell protrusions [193].
In the fungal pathogen, Candida albicans, the PLS3 ortholog Sac6 associates with oxidative stress responses. Sac6 negatively regulates cytosol–nucleus transport of the key transcription factor Cap1 and thereby the expression of oxidative stress response genes [194].
Biomarker
Differential expression of PLS3 is a frequent characteristic of CTCs as well as primary tumors, while usually being absent from the hematopoietic system. This renders the protein as well as the mRNA as immaculate biomarker candidates. In a recent study, a peptide library has been screened for affinity to PLS3-overexpressing cancer cells. They identify the peptide TP1 (KVKSDRVC) and develop a fluorescein isothiocyanate-labeled TP1 molecule that is able to identify PLS3-overexpressing CTCs in peripheral blood [195].
With regard to SMA, PLS3 is probably of limited use as a biomarker since it is only expressed in a minority of the human population in the hematopoietic system [5]. A study tested the expression of six putative protein biomarkers (COMP, DPP4, CLEC3B, SPP1, VTN, and AHSG) in mice with or without overexpression of a human PLS3 transgene. The expression level of PLS3 did neither affect the amount of SMN nor did the other putative biomarkers, supporting the hypothesis that PLS3 acts as an independent protective modifier in SMA [196].
One pitfall of PLS3 is that its differential expression is linked to multiple different diseases. Comorbidity of other PLS3-associated medical conditions should always be considered when an analysis of PLS3 as a biomarker in peripheral blood is performed. In addition to that, as an X-linked gene, a sex-specific expression bias towards women has been observed [5, 197].
Conclusion
Despite PLS3 not being essential for the survival of a cell or organism, both conditions overexpression or knockdown of PLS3 can influence many cellular processes either causing, facilitating or rescuing certain pathological phenotypes. While the full knockout in humans and mice causes a disruption of the bone remodeling cells and, thus, osteoporosis, its overexpression in some neurodegenerative disorders such as SMA or ALS, where F-actin levels are decreased, acts as a protective modifier. However, in wild-type condition, overexpression of PLS3 is detrimental and causes osteoarthritis or facilitates cancer. PLS3 overexpression is strongly correlated with several cancer types, and has been recommended for use as biomarker in cancer and poor prognosis for survival. PLS3 has an important function in F-actin-binding and -bundling and as such is involved in a plethora of cellular processes dependent on F-actin dynamics. These include cell migration and growth, axonal and neurite outgrowth of polarized cells, axonal local translation, endocytosis, influence of intracellular calcium on PLS3-dependent processes, mechanotransduction, signaling, infection with pathogens and others. This ever-growing knowledge on PLS3 is crucial to wisely use PLS3 overexpression or PLS3 reduction as a therapeutic target.
Abbreviations
- ABD1:
-
Actin-binding domain 1
- ABD2:
-
Actin-binding domain 2
- ALS:
-
Amyotrophic lateral sclerosis
- AML:
-
Acute myeloid leukemia
- Arp2/3:
-
Actin related protein 2/3 complex
- BMD:
-
Bone mineral density
- CBM:
-
Calmodulin-binding motif
- CH:
-
Calponin-homology
- CHP1:
-
Calcineurin EF-hand protein 1
- CMT:
-
Charcot–Marie–Tooth
- CORO1C:
-
Coronin-1C
- CTC:
-
Circulating tumor cell
- CTCL:
-
Cutaneous T-cell lymphoma
- EMT:
-
Epithelial–mesenchymal transition
- F-actin:
-
Filamentous actin
- G-actin:
-
Globular actin
- HCV:
-
Hepatitis C virus
- iPSC:
-
Induced pluripotent stem cell
- MAPK:
-
P38 mitogen-activated protein kinase
- MN:
-
Motor neuron
- NCALD:
-
Neurocalcin delta
- NFATC1:
-
Nuclear factor-activated T cells c1
- NFκB:
-
Nuclear factor 'kappa-light-chain-enhancer' of activated B cells
- NKRF:
-
NFκB repressing factor
- NMJ:
-
Neuromuscular junction
- NSCLC:
-
Non-small-cell lung cancer
- OI:
-
Osteogenesis imperfecta
- PLS1:
-
Plastin 1
- PLS2:
-
Plastin 2
- PLS3:
-
Plastin 3
- RELA:
-
RELA proto-oncogene, NFκB subunit
- RD:
-
Regulatory domain
- SMA:
-
Spinal muscular atrophy
- SMN1:
-
Survival of motor neuron protein 1
- SMN2:
-
Survival of motor neuron protein 2
- SS:
-
Sézary syndrome
- VCF:
-
Vertebral compression fracture
- XCI:
-
X-chromosomal inactivation
References
Bretscher A, Weber K (1980) Fimbrin, a new microfilament-associated protein present in microvilli and other cell surface structures. J Cell Biol 86(1):335–340
Lin CS, Park T, Chen ZP, Leavitt J (1993) Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J Biol Chem 268(4):2781–2792
Shinomiya H (2012) Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int J Cell Biol 2012:1
Lin CS, Lau A, Huynh T, Lue TF (1999) Differential regulation of human T-plastin gene in leukocytes and non-leukocytes: identification of the promoter, enhancer, and CpG island. DNA Cell Biol 18(1):27–37. https://doi.org/10.1089/104454999315592
Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320(5875):524–527. https://doi.org/10.1126/science.1155085
Briese M, Saal L, Appenzeller S, Moradi M, Baluapuri A, Sendtner M (2016) Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res 44(4):e33. https://doi.org/10.1093/nar/gkv1027
Horakova AH, Moseley SC, McLaughlin CR, Tremblay DC, Chadwick BP (2012) The macrosatellite DXZ4 mediates CTCF-dependent long-range intrachromosomal interactions on the human inactive X chromosome. Hum Mol Genet 21(20):4367–4377. https://doi.org/10.1093/hmg/dds270
Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434(7031):400–404. https://doi.org/10.1038/nature03479
Balaton BP, Cotton AM, Brown CJ (2015) Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol Sex Differ 6:35. https://doi.org/10.1186/s13293-015-0053-7
Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, Kong X, Hurst LD (2013) Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol 30(12):2588–2601. https://doi.org/10.1093/molbev/mst148
Yang F, Babak T, Shendure J, Disteche CM (2010) Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res 20(5):614–622. https://doi.org/10.1101/gr.103200.109
Giacalone J, Friedes J, Francke U (1992) A novel GC-rich human macrosatellite VNTR in Xq24 is differentially methylated on active and inactive X chromosomes. Nat Genet 1(2):137–143. https://doi.org/10.1038/ng0592-137
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7):1665–1680. https://doi.org/10.1016/j.cell.2014.11.021
Horakova AH, Calabrese JM, McLaughlin CR, Tremblay DC, Magnuson T, Chadwick BP (2012) The mouse DXZ4 homolog retains Ctcf binding and proximity to Pls3 despite substantial organizational differences compared to the primate macrosatellite. Genome Biol 13(8):R70. https://doi.org/10.1186/gb-2012-13-8-r70
Besio R, Chow CW, Tonelli F, Marini JC, Forlino A (2019) Bone biology: insights from osteogenesis imperfecta and related rare fragility syndromes. FEBS J 286(15):3033–3056. https://doi.org/10.1111/febs.14963
Neugebauer J, Heilig J, Hosseinibarkooie S, Ross BC, Mendoza-Ferreira N, Nolte F, Peters M, Holker I, Hupperich K, Tschanz T, Grysko V, Zaucke F, Niehoff A, Wirth B (2018) Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum Mol Genet 27(24):4249–4262. https://doi.org/10.1093/hmg/ddy318
Arpin M, Friederich E, Algrain M, Vernel F, Louvard D (1994) Functional differences between L- and T-plastin isoforms. J Cell Biol 127(6 Pt 2):1995–2008
de Arruda MV, Watson S, Lin CS, Leavitt J, Matsudaira P (1990) Fimbrin is a homologue of the cytoplasmic phosphoprotein plastin and has domains homologous with calmodulin and actin gelation proteins. J Cell Biol 111(3):1069–1079
Banuelos S, Saraste M, Djinovic Carugo K (1998) Structural comparisons of calponin homology domains: implications for actin binding. Structure 6(11):1419–1431. https://doi.org/10.1016/s0969-2126(98)00141-5
Delanote V, Vandekerckhove J, Gettemans J (2005) Plastins: versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol Sin 26(7):769–779
Lebart MC, Mejean C, Roustan C, Benyamin Y (1993) Further characterization of the alpha-actinin-actin interface and comparison with filamin-binding sites on actin. J Biol Chem 268(8):5642–5648
Zhang R, Chang M, Zhang M, Wu Y, Qu X, Huang S (2016) The structurally plastic CH2 domain is linked to distinct functions of fimbrins/plastins. J Biol Chem 291(34):17881–17896. https://doi.org/10.1074/jbc.M116.730069
Schwebach CL, Kudryashova E, Zheng W, Orchard M, Smith H, Runyan LA, Egelman EH, Kudryashov DS (2020) Osteogenesis imperfecta mutations in plastin 3 lead to impaired calcium regulation of actin bundling. Bone Res 8:21. https://doi.org/10.1038/s41413-020-0095-2
Goldsmith SC, Pokala N, Shen W, Fedorov AA, Matsudaira P, Almo SC (1997) The structure of an actin-crosslinking domain from human fimbrin. Nat Struct Biol 4(9):708–712
Hanein D, Volkmann N, Goldsmith S, Michon AM, Lehman W, Craig R, DeRosier D, Almo S, Matsudaira P (1998) An atomic model of fimbrin binding to F-actin and its implications for filament crosslinking and regulation. Nat Struct Biol 5(9):787–792
Klein MG, Shi W, Ramagopal U, Tseng Y, Wirtz D, Kovar DR, Staiger CJ, Almo SC (2004) Structure of the actin crosslinking core of fimbrin. Structure 12(6):999–1013. https://doi.org/10.1016/j.str.2004.04.010
Karpova TS, Tatchell K, Cooper JA (1995) Actin filaments in yeast are unstable in the absence of capping protein or fimbrin. J Cell Biol 131(6 Pt 1):1483–1493. https://doi.org/10.1083/jcb.131.6.1483
Bretscher A (1981) Fimbrin is a cytoskeletal protein that crosslinks F-actin in vitro. Proc Natl Acad Sci USA 78(11):6849–6853. https://doi.org/10.1073/pnas.78.11.6849
Pollard TD, Cooper JA (2009) Actin, a central player in cell shape and movement. Science 326(5957):1208–1212. https://doi.org/10.1126/science.1175862
Hurtley SM (1998) Cell biology of the cytoskeleton. Science 279(5350):459. https://doi.org/10.1126/science.279.5350.459a
Suarez C, Kovar DR (2016) Internetwork competition for monomers governs actin cytoskeleton organization. Nat Rev Mol Cell Biol 17(12):799–810. https://doi.org/10.1038/nrm.2016.106
dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ (2003) Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83(2):433–473. https://doi.org/10.1152/physrev.00026.2002
Schwebach CL, Agrawal R, Lindert S, Kudryashova E, Kudryashov DS (2017) The roles of actin-binding domains 1 and 2 in the calcium-dependent regulation of actin filament bundling by human plastins. J Mol Biol 429(16):2490–2508. https://doi.org/10.1016/j.jmb.2017.06.021
Namba Y, Ito M, Zu Y, Shigesada K, Maruyama K (1992) Human T cell L-plastin bundles actin filaments in a calcium-dependent manner. J Biochem 112(4):503–507. https://doi.org/10.1093/oxfordjournals.jbchem.a123929
Shinomiya H, Shinjo M, Fengzhi L, Asano Y, Kihara H (2007) Conformational analysis of the leukocyte-specific EF-hand protein p65/L-plastin by X-ray scattering in solution. Biophys Chem 131(1–3):36–42. https://doi.org/10.1016/j.bpc.2007.09.001
Xue F, Janzen DM, Knecht DA (2010) Contribution of filopodia to cell migration: a mechanical link between protrusion and contraction. Int J Cell Biol 2010:507821. https://doi.org/10.1155/2010/507821
Lambrechts A, Van Troys M, Ampe C (2004) The actin cytoskeleton in normal and pathological cell motility. Int J Biochem Cell Biol 36(10):1890–1909. https://doi.org/10.1016/j.biocel.2004.01.024
Lauffenburger DA, Horwitz AF (1996) Cell migration: a physically integrated molecular process. Cell 84(3):359–369. https://doi.org/10.1016/s0092-8674(00)81280-5
Garbett D, Bisaria A, Yang C, McCarthy DG, Hayer A, Moerner WE, Svitkina TM, Meyer T (2020) T-Plastin reinforces membrane protrusions to bridge matrix gaps during cell migration. Nat Commun 11(1):4818. https://doi.org/10.1038/s41467-020-18586-3
Dor-On E, Raviv S, Cohen Y, Adir O, Padmanabhan K, Luxenburg C (2017) T-plastin is essential for basement membrane assembly and epidermal morphogenesis. Sci Signal. https://doi.org/10.1126/scisignal.aal3154
Wottawa M, Naas S, Bottger J, van Belle GJ, Mobius W, Revelo NH, Heidenreich D, von Ahlen M, Zieseniss A, Krohnert K, Lutz S, Lenz C, Urlaub H, Rizzoli SO, Katschinski DM (2017) Hypoxia-stimulated membrane trafficking requires T-plastin. Acta Physiol (Oxf) 221(1):59–73. https://doi.org/10.1111/apha.12859
Giganti A, Plastino J, Janji B, Van Troys M, Lentz D, Ampe C, Sykes C, Friederich E (2005) Actin-filament cross-linking protein T-plastin increases Arp2/3-mediated actin-based movement. J Cell Sci 118(Pt 6):1255–1265
Lin CS, Shen W, Chen ZP, Tu YH, Matsudaira P (1994) Identification of I-plastin, a human fimbrin isoform expressed in intestine and kidney. Mol Cell Biol 14(4):2457–2467
Ishida H, Jensen KV, Woodman AG, Hyndman ME, Vogel HJ (2017) The calcium-dependent switch helix of L-plastin regulates actin bundling. Sci Rep 7:40662. https://doi.org/10.1038/srep40662
Babich A, Burkhardt JK (2013) Coordinate control of cytoskeletal remodeling and calcium mobilization during T-cell activation. Immunol Rev 256(1):80–94. https://doi.org/10.1111/imr.12123
Morley SC (2013) The actin-bundling protein L-plastin supports T-cell motility and activation. Immunol Rev 256(1):48–62. https://doi.org/10.1111/imr.12102
Miyakawa T, Shinomiya H, Yumoto F, Miyauchi Y, Tanaka H, Ojima T, Kato YS, Tanokura M (2012) Different Ca(2)(+)-sensitivities between the EF-hands of T- and L-plastins. Biochem Biophys Res Commun 429(3–4):137–141
Khassan O, Jensen KV, Woodman AG, Vogel HJ, Ishida H (2019) Characterization of the EF-hand calcium-binding domains of human plastins. Methods Mol Biol 1929:245–260. https://doi.org/10.1007/978-1-4939-9030-6_16
Nara M, Morii H, Shimizu T, Shinomiya H, Furuta Y, Miyazono K, Miyakawa T, Tanokura M (2019) Characterization of the Ca(2+)-coordination structures of L- and T-plastins in combination with their synthetic peptide analogs by FTIR spectroscopy. Sci Rep 9(1):4217. https://doi.org/10.1038/s41598-019-40889-9
Lyon AN, Pineda RH, le Hao T, Kudryashova E, Kudryashov DS, Beattie CE (2014) Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum Mol Genet 23(8):1990–2004. https://doi.org/10.1093/hmg/ddt595
Kubler E, Riezman H (1993) Actin and fimbrin are required for the internalization step of endocytosis in yeast. Embo J 12(7):2855–2862
Gheorghe DM, Aghamohammadzadeh S, Smaczynska-de R II, Allwood EG, Winder SJ, Ayscough KR (2008) Interactions between the yeast SM22 homologue Scp1 and actin demonstrate the importance of actin bundling in endocytosis. J Biol Chem 283(22):15037–15046. https://doi.org/10.1074/jbc.M710332200
Jorde S, Walther A, Wendland J (2011) The Ashbya gossypii fimbrin SAC6 is required for fast polarized hyphal tip growth and endocytosis. Microbiol Res 166(3):137–145. https://doi.org/10.1016/j.micres.2010.09.003
Upadhyay S, Shaw BD (2008) The role of actin, fimbrin and endocytosis in growth of hyphae in Aspergillus nidulans. Mol Microbiol 68(3):690–705. https://doi.org/10.1111/j.1365-2958.2008.06178.x
Penalver E, Ojeda L, Moreno E, Lagunas R (1997) Role of the cytoskeleton in endocytosis of the yeast maltose transporter. Yeast 13(6):541–549. https://doi.org/10.1002/(SICI)1097-0061(199705)13:6%3c541::AID-YEA112%3e3.0.CO;2-4
Planade J, Belbahri R, Boiero Sanders M, Guillotin A, du Roure O, Michelot A, Heuvingh J (2019) Mechanical stiffness of reconstituted actin patches correlates tightly with endocytosis efficiency. PLoS Biol 17(10):e3000500. https://doi.org/10.1371/journal.pbio.3000500
Ma R, Berro J (2018) Structural organization and energy storage in crosslinked actin assemblies. PLoS Comput Biol 14(5):e1006150. https://doi.org/10.1371/journal.pcbi.1006150
Skau CT, Kovar DR (2010) Fimbrin and tropomyosin competition regulates endocytosis and cytokinesis kinetics in fission yeast. Curr Biol 20(16):1415–1422. https://doi.org/10.1016/j.cub.2010.06.020
Clayton JE, Sammons MR, Stark BC, Hodges AR, Lord M (2010) Differential regulation of unconventional fission yeast myosins via the actin track. Curr Biol 20(16):1423–1431. https://doi.org/10.1016/j.cub.2010.07.026
Ruiz R, Casanas JJ, Torres-Benito L, Cano R, Tabares L (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30(3):849–857
Hosseinibarkooie S, Peters M, Torres-Benito L, Rastetter RH, Hupperich K, Hoffmann A, Mendoza-Ferreira N, Kaczmarek A, Janzen E, Milbradt J, Lamkemeyer T, Rigo F, Bennett CF, Guschlbauer C, Buschges A, Hammerschmidt M, Riessland M, Kye MJ, Clemen CS, Wirth B (2016) The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet 99(3):647–665. https://doi.org/10.1016/j.ajhg.2016.07.014
Gabanella F, Pisani C, Borreca A, Farioli-Vecchioli S, Ciotti MT, Ingegnere T, Onori A, Ammassari-Teule M, Corbi N, Canu N, Monaco L, Passananti C, Di Certo MG (2016) SMN affects membrane remodelling and anchoring of the protein synthesis machinery. J Cell Sci 129(4):804–816. https://doi.org/10.1242/jcs.176750
Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, Sendtner M (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163(4):801–812. https://doi.org/10.1083/jcb.200304128
Ackermann B, Krober S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, Wunderlich TF, Riessland M, Tabares L, Wirth B (2013) Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 22(7):1328–1347. https://doi.org/10.1093/hmg/dds540
Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Lohr H, Bradler C, Grysko V, Dimitriadi M, Hosseinibarkooie S, Torres-Benito L, Peters M, Upadhyay A, Biglari N, Krober S, Holker I, Garbes L, Gilissen C, Hoischen A, Nurnberg G, Nurnberg P, Walter M, Rigo F, Bennett CF, Kye MJ, Hart AC, Hammerschmidt M, Kloppenburg P, Wirth B (2017) Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet 100(2):297–315. https://doi.org/10.1016/j.ajhg.2017.01.005
Janzen E, Mendoza-Ferreira N, Hosseinibarkooie S, Schneider S, Hupperich K, Tschanz T, Grysko V, Riessland M, Hammerschmidt M, Rigo F, Bennett CF, Kye MJ, Torres-Benito L, Wirth B (2018) CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 141(8):2343–2361. https://doi.org/10.1093/brain/awy167
Dimitriadi M, Derdowski A, Kalloo G, Maginnis MS, O’Hern P, Bliska B, Sorkac A, Nguyen KC, Cook SJ, Poulogiannis G, Atwood WJ, Hall DH, Hart AC (2016) Decreased function of survival motor neuron protein impairs endocytic pathways. Proc Natl Acad Sci USA 113(30):E4377-4386. https://doi.org/10.1073/pnas.1600015113
Walsh MB, Janzen E, Wingrove E, Hosseinibarkooie S, Muela NR, Davidow L, Dimitriadi M, Norabuena EM, Rubin LL, Wirth B, Hart AC (2020) Genetic modifiers ameliorate endocytic and neuromuscular defects in a model of spinal muscular atrophy. BMC Biol 18(1):127. https://doi.org/10.1186/s12915-020-00845-w
Cioni JM, Lin JQ, Holtermann AV, Koppers M, Jakobs MAH, Azizi A, Turner-Bridger B, Shigeoka T, Franze K, Harris WA, Holt CE (2019) Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176(1–2):56–72. https://doi.org/10.1016/j.cell.2018.11.030
Corradi E, Dalla Costa I, Gavoci A, Iyer A, Roccuzzo M, Otto TA, Oliani E, Bridi S, Strohbuecker S, Santos-Rodriguez G, Valdembri D, Serini G, Abreu-Goodger C, Baudet ML (2020) Axonal precursor miRNAs hitchhike on endosomes and locally regulate the development of neural circuits. EMBO J 39(6):e102513. https://doi.org/10.15252/embj.2019102513
Hagiwara M, Shinomiya H, Kashihara M, Kobayashi K, Tadokoro T, Yamamoto Y (2011) Interaction of activated Rab5 with actin-bundling proteins, L- and T-plastin and its relevance to endocytic functions in mammalian cells. Biochem Biophys Res Commun 407(3):615–619. https://doi.org/10.1016/j.bbrc.2011.03.082
Lin CS, Lau A, Lue TF (1998) Analysis and mapping of plastin phosphorylation. DNA Cell Biol 17(12):1041–1046. https://doi.org/10.1089/dna.1998.17.1041
Ma Y, Lai W, Zhao M, Yue C, Shi F, Li R, Hu Z (2019) Plastin 3 down-regulation augments the sensitivity of MDA-MB-231 cells to paclitaxel via the p38 MAPK signalling pathway. Artif Cells Nanomed Biotechnol 47(1):685–695. https://doi.org/10.1080/21691401.2019.1576707
Brun C, Demeaux A, Guaddachi F, Jean-Louis F, Oddos T, Bagot M, Bensussan A, Jauliac S, Michel L (2014) T-plastin expression downstream to the calcineurin/NFAT pathway is involved in keratinocyte migration. PLoS ONE 9(9):e104700. https://doi.org/10.1371/journal.pone.0104700
Xin Z, Li D, Mao F, Du Y, Wang X, Xu P, Li Z, Qian J, Yao J (2020) PLS3 predicts poor prognosis in pancreatic cancer and promotes cancer cell proliferation via PI3K/AKT signaling. J Cell Physiol 235(11):8416–8423. https://doi.org/10.1002/jcp.29685
Chellaiah MA, Ma T, Majumdar S (2018) L-plastin phosphorylation regulates the early phase of sealing ring formation by actin bundling process in mouse osteoclasts. Exp Cell Res 372(1):73–82. https://doi.org/10.1016/j.yexcr.2018.09.014
Janji B, Giganti A, De Corte V, Catillon M, Bruyneel E, Lentz D, Plastino J, Gettemans J, Friederich E (2006) Phosphorylation on Ser5 increases the F-actin-binding activity of L-plastin and promotes its targeting to sites of actin assembly in cells. J Cell Sci 119(Pt 9):1947–1960
Shinomiya H, Hagi A, Fukuzumi M, Mizobuchi M, Hirata H, Utsumi S (1995) Complete primary structure and phosphorylation site of the 65-kDa macrophage protein phosphorylated by stimulation with bacterial lipopolysaccharide. J Immunol 154(7):3471–3478
Sugimachi K, Yokobori T, Iinuma H, Ueda M, Ueo H, Shinden Y, Eguchi H, Sudo T, Suzuki A, Maehara Y, Mori M, Mimori K (2014) Aberrant expression of plastin-3 via copy number gain induces the epithelial-mesenchymal transition in circulating colorectal cancer cells. Ann Surg Oncol 21(11):3680–3690. https://doi.org/10.1245/s10434-013-3366-y
Schvartzman JM, Sotillo R, Benezra R (2010) Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10(2):102–115. https://doi.org/10.1038/nrc2781
Willis ND, Cox TR, Rahman-Casans SF, Smits K, Przyborski SA, van den Brandt P, van Engeland M, Weijenberg M, Wilson RG, de Bruine A, Hutchison CJ (2008) Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 3(8):e2988. https://doi.org/10.1371/journal.pone.0002988
Ning Y, Gerger A, Zhang W, Hanna DL, Yang D, Winder T, Wakatsuki T, Labonte MJ, Stintzing S, Volz N, Sunakawa Y, Stremitzer S, El-Khoueiry R, Lenz HJ (2014) Plastin polymorphisms predict gender- and stage-specific colon cancer recurrence after adjuvant chemotherapy. Mol Cancer Ther 13(2):528–539. https://doi.org/10.1158/1535-7163.MCT-13-0646
Lyberopoulou A, Aravantinos G, Efstathopoulos EP, Nikiteas N, Bouziotis P, Isaakidou A, Papalois A, Marinos E, Gazouli M (2015) Mutational analysis of circulating tumor cells from colorectal cancer patients and correlation with primary tumor tissue. PLoS ONE 10(4):e0123902. https://doi.org/10.1371/journal.pone.0123902
Szkandera J, Winder T, Stotz M, Weissmueller M, Langsenlehner T, Pichler M, Samonigg H, Renner W, Gerger A, Absenger G (2013) A common gene variant in PLS3 predicts colon cancer recurrence in women. Tumour Biol 34(4):2183–2188. https://doi.org/10.1007/s13277-013-0754-7
Ueo H, Sugimachi K, Gorges TM, Bartkowiak K, Yokobori T, Muller V, Shinden Y, Ueda M, Ueo H, Mori M, Kuwano H, Maehara Y, Ohno S, Pantel K, Mimori K (2015) Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br J Cancer 112(9):1519–1526. https://doi.org/10.1038/bjc.2015.132
Markiewicz A, Topa J, Nagel A, Skokowski J, Seroczynska B, Stokowy T, Welnicka-Jaskiewicz M, Zaczek AJ (2019) Spectrum of epithelial-mesenchymal transition phenotypes in circulating tumour cells from early breast cancer patients. Cancers (Basel). https://doi.org/10.3390/cancers11010059
Markou A, Lazaridou M, Paraskevopoulos P, Chen S, Swierczewska M, Budna J, Kuske A, Gorges TM, Joosse SA, Kroneis T, Zabel M, Sedlmayr P, Alix-Panabieres C, Pantel K, Lianidou ES (2018) Multiplex gene expression profiling of in vivo isolated circulating tumor cells in high-risk prostate cancer patients. Clin Chem 64(2):297–306. https://doi.org/10.1373/clinchem.2017.275503
Hu Q, Masuda T, Kuramitsu S, Tobo T, Sato K, Kidogami S, Nambara S, Ueda M, Tsuruda Y, Kuroda Y, Ito S, Oki E, Mori M, Mimori K (2020) Potential association of LOXL1 with peritoneal dissemination in gastric cancer possibly via promotion of EMT. PLoS ONE 15(10):e0241140. https://doi.org/10.1371/journal.pone.0241140
Kurashige J, Yokobori T, Mima K, Sawada G, Takahashi Y, Ueo H, Takano Y, Matsumura T, Uchi R, Eguchi H, Sudo T, Sugimachi K, Mori M, Baba H, Mimori K (2019) Plastin3 is associated with epithelial-mesenchymal transition and poor prognosis in gastric cancer. Oncol Lett 17(2):2393–2399. https://doi.org/10.3892/ol.2018.9819
Cao L, Wang S, Zhang Y, Wong KC, Nakatsu G, Wang X, Wong S, Ji J, Yu J (2018) Zinc-finger protein 471 suppresses gastric cancer through transcriptionally repressing downstream oncogenic PLS3 and TFAP2A. Oncogene 37(26):3601–3616. https://doi.org/10.1038/s41388-018-0220-5
Kuriyama K, Yokobori T, Sohda M, Nakazawa N, Yajima T, Naruse I, Kuwano H, Shirabe K, Kaira K, Saeki H (2021) Plasma plastin-3: a tumor marker in patients with non-small-cell lung cancer treated with nivolumab. Oncol Lett 21(1):11. https://doi.org/10.3892/ol.2020.12272
Hisano T, Ono M, Nakayama M, Naito S, Kuwano M, Wada M (1996) Increased expression of T-plastin gene in cisplatin-resistant human cancer cells: identification by mRNA differential display. FEBS Lett 397(1):101–107
Higuchi Y, Kita K, Nakanishi H, Wang XL, Sugaya S, Tanzawa H, Yamamori H, Sugita K, Yamaura A, Suzuki N (1998) Search for genes involved in UV-resistance in human cells by mRNA differential display: increased transcriptional expression of nucleophosmin and T-plastin genes in association with the resistance. Biochem Biophys Res Commun 248(3):597–602. https://doi.org/10.1006/bbrc.1998.8978
Ikeda H, Sasaki Y, Kobayashi T, Suzuki H, Mita H, Toyota M, Itoh F, Shinomura Y, Tokino T, Imai K (2005) The role of T-fimbrin in the response to DNA damage: silencing of T-fimbrin by small interfering RNA sensitizes human liver cancer cells to DNA-damaging agents. Int J Oncol 27(4):933–940
Velthaus A, Cornils K, Hennigs JK, Grub S, Stamm H, Wicklein D, Bokemeyer C, Heuser M, Windhorst S, Fiedler W, Wellbrock J (2019) The actin binding protein plastin-3 is involved in the pathogenesis of acute myeloid leukemia. Cancers Basel. https://doi.org/10.3390/cancers11111663
van Doorn R, Dijkman R, Vermeer MH, Out-Luiting JJ, van der Raaij-Helmer EM, Willemze R, Tensen CP (2004) Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sezary syndrome identified by gene expression analysis. Cancer Res 64(16):5578–5586. https://doi.org/10.1158/0008-5472.CAN-04-1253
Wang Y, Su M, Zhou LL, Tu P, Zhang X, Jiang X, Zhou Y (2011) Deficiency of SATB1 expression in Sezary cells causes apoptosis resistance by regulating FasL/CD95L transcription. Blood 117(14):3826–3835. https://doi.org/10.1182/blood-2010-07-294819
Wong HK, Gibson H, Hake T, Geyer S, Frederickson J, Marcucci G, Caligiuri MA, Porcu P, Mishra A (2015) Promoter-specific hypomethylation is associated with overexpression of PLS3, GATA6, and TWIST1 in the Sezary syndrome. J Invest Dermatol 135(8):2084–2092. https://doi.org/10.1038/jid.2015.116
Moerman-Herzog AM, Acheampong DA, Brooks AG, Blair SM, Hsu PC, Wong HK (2019) Transcriptome analysis of Sezary syndrome and lymphocytic-variant hypereosinophilic syndrome T cells reveals common and divergent genes. Oncotarget 10(49):5052–5069. https://doi.org/10.18632/oncotarget.27120
Henn A, Michel L, Fite C, Deschamps L, Ortonne N, Ingen-Housz-Oro S, Marinho E, Beylot-Barry M, Bagot M, Laroche L, Crickx B, Maubec E (2015) Sezary syndrome without erythroderma. J Am Acad Dermatol 72(6):1003–1009. https://doi.org/10.1016/j.jaad.2014.11.015
Su MW, Dorocicz I, Dragowska WH, Ho V, Li G, Voss N, Gascoyne R, Zhou Y (2003) Aberrant expression of T-plastin in Sezary cells. Cancer Res 63(21):7122–7127
Begue E, Jean-Louis F, Bagot M, Jauliac S, Cayuela JM, Laroche L, Parquet N, Bachelez H, Bensussan A, Courtois G, Michel L (2012) Inducible expression and pathophysiologic functions of T-plastin in cutaneous T-cell lymphoma. Blood 120(1):143–154. https://doi.org/10.1182/blood-2011-09-379156
Boonk SE, Zoutman WH, Putter H, Ram-Wolff C, Felcht M, Klemke CD, Ranki A, Quaglino P, Whittaker S, Bagot M, Willemze R, Vermeer MH (2017) Increased expression of PLS3 correlates with better outcome in sezary syndrome. J Invest Dermatol 137(3):754–757. https://doi.org/10.1016/j.jid.2016.10.025
Tang N, Gibson H, Germeroth T, Porcu P, Lim HW, Wong HK (2010) T-plastin (PLS3) gene expression differentiates Sezary syndrome from mycosis fungoides and inflammatory skin diseases and can serve as a biomarker to monitor disease progression. Br J Dermatol 162(2):463–466. https://doi.org/10.1111/j.1365-2133.2009.09587.x
Kamioka H, Sugawara Y, Honjo T, Yamashiro T, Takano-Yamamoto T (2004) Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J Bone Miner Res 19(3):471–478
Wang N, Tytell JD, Ingber DE (2009) Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol 10(1):75–82. https://doi.org/10.1038/nrm2594
Mulvihill BM, Prendergast PJ (2010) Mechanobiological regulation of the remodelling cycle in trabecular bone and possible biomechanical pathways for osteoporosis. Clin Biomech (Bristol, Avon) 25(5):491–498. https://doi.org/10.1016/j.clinbiomech.2010.01.006
van Dijk FS, Zillikens MC, Micha D, Riessland M, Marcelis CL, de Die-Smulders CE, Milbradt J, Franken AA, Harsevoort AJ, Lichtenbelt KD, Pruijs HE, Rubio-Gozalbo ME, Zwertbroek R, Moutaouakil Y, Egthuijsen J, Hammerschmidt M, Bijman R, Semeins CM, Bakker AD, Everts V, Klein-Nulend J, Campos-Obando N, Hofman A, te Meerman GJ, Verkerk AJ, Uitterlinden AG, Maugeri A, Sistermans EA, Waisfisz Q, Meijers-Heijboer H, Wirth B, Simon ME, Pals G (2013) PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med 369(16):1529–1536. https://doi.org/10.1056/NEJMoa1308223
Vincent TL (2013) Targeting mechanotransduction pathways in osteoarthritis: a focus on the pericellular matrix. Curr Opin Pharmacol 13(3):449–454. https://doi.org/10.1016/j.coph.2013.01.010
Leong DJ, Hardin JA, Cobelli NJ, Sun HB (2011) Mechanotransduction and cartilage integrity. Ann NY Acad Sci 1240:32–37. https://doi.org/10.1111/j.1749-6632.2011.06301.x
Martinez-Moreno D, Jimenez G, Galvez-Martin P, Rus G, Marchal JA (1865) Cartilage biomechanics: a key factor for osteoarthritis regenerative medicine. Biochim Biophys Acta Mol Basis Dis 6:1067–1075. https://doi.org/10.1016/j.bbadis.2019.03.011
Tanaka-Kamioka K, Kamioka H, Ris H, Lim SS (1998) Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner Res 13(10):1555–1568
Weinbaum S, Duan Y, Thi MM, You L (2011) An integrative review of mechanotransduction in endothelial, epithelial (renal) and dendritic cells (osteocytes). Cell Mol Bioeng 4(4):510–537. https://doi.org/10.1007/s12195-011-0179-6
Makitie RE, Kampe AJ, Taylan F, Makitie O (2017) Recent discoveries in monogenic disorders of childhood bone fragility. Curr Osteoporos Rep 15(4):303–310. https://doi.org/10.1007/s11914-017-0388-6
Shao C, Wang YW, He JW, Fu WZ, Wang C, Zhang ZL (2019) Genetic variants in the PLS3 gene are associated with osteoporotic fractures in postmenopausal Chinese women. Acta Pharmacol Sin 40(9):1212–1218. https://doi.org/10.1038/s41401-019-0219-7
Rocha-Braz MG, Ferraz-de-Souza B (2016) Genetics of osteoporosis: searching for candidate genes for bone fragility. Arch Endocrinol Metab 60(4):391–401. https://doi.org/10.1590/2359-3997000000178
Soroko SB, Barrett-Connor E, Edelstein SL, Kritz-Silverstein D (1994) Family history of osteoporosis and bone mineral density at the axial skeleton: the Rancho Bernardo Study. J Bone Miner Res 9(6):761–769. https://doi.org/10.1002/jbmr.5650090602
Krall EA, Dawson-Hughes B (1993) Heritable and life-style determinants of bone mineral density. J Bone Miner Res 8(1):1–9. https://doi.org/10.1002/jbmr.5650080102
Bishop N, Arundel P, Clark E, Dimitri P, Farr J, Jones G, Makitie O, Munns CF, Shaw N (2014) Fracture prediction and the definition of osteoporosis in children and adolescents: the ISCD 2013 Pediatric Official Positions. J Clin Densitom 17(2):275–280. https://doi.org/10.1016/j.jocd.2014.01.004
Makitie RE, Costantini A, Kampe A, Alm JJ, Makitie O (2019) New insights into monogenic causes of osteoporosis. Front Endocrinol (Lausanne) 10:70. https://doi.org/10.3389/fendo.2019.00070
Van Dijk FS, Sillence DO (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164A(6):1470–1481. https://doi.org/10.1002/ajmg.a.36545
Collet C, Ostertag A, Ricquebourg M, Delecourt M, Tueur G, Isidor B, Guillot P, Schaefer E, Javier RM, Funck-Brentano T, Orcel P, Laplanche JL, Cohen-Solal M (2018) Primary osteoporosis in young adults: genetic basis and identification of novel variants in causal genes. JBMR Plus 2(1):12–21. https://doi.org/10.1002/jbm4.10020
Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B (2014) What is new in genetics and osteogenesis imperfecta classification? J Pediatr (Rio J) 90(6):536–541. https://doi.org/10.1016/j.jped.2014.05.003
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387(10028):1657–1671. https://doi.org/10.1016/S0140-6736(15)00728-X
Etich J, Rehberg M, Eckes B, Sengle G, Semler O, Zaucke F (2020) Signaling pathways affected by mutations causing osteogenesis imperfecta. Cell Signal 76:109789. https://doi.org/10.1016/j.cellsig.2020.109789
Kampe AJ, Costantini A, Levy-Shraga Y, Zeitlin L, Roschger P, Taylan F, Lindstrand A, Paschalis EP, Gamsjaeger S, Raas-Rothschild A, Hovel M, Jiao H, Klaushofer K, Grasemann C, Makitie O (2017) PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J Bone Miner Res 32(12):2394–2404. https://doi.org/10.1002/jbmr.3233
Nishi E, Masuda K, Arakawa M, Kawame H, Kosho T, Kitahara M, Kubota N, Hidaka E, Katoh Y, Shirahige K, Izumi K (2016) Exome sequencing-based identification of mutations in non-syndromic genes among individuals with apparently syndromic features. Am J Med Genet A 170(11):2889–2894. https://doi.org/10.1002/ajmg.a.37826
Kampe AJ, Costantini A, Makitie RE, Jantti N, Valta H, Mayranpaa M, Kroger H, Pekkinen M, Taylan F, Jiao H, Makitie O (2017) PLS3 sequencing in childhood-onset primary osteoporosis identifies two novel disease-causing variants. Osteoporos Int 28(10):3023–3032. https://doi.org/10.1007/s00198-017-4150-9
Laine CM, Wessman M, Toiviainen-Salo S, Kaunisto MA, Mayranpaa MK, Laine T, Pekkinen M, Kroger H, Valimaki VV, Valimaki MJ, Lehesjoki AE, Makitie O (2015) A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res 30(3):510–518. https://doi.org/10.1002/jbmr.2355
Wang L, Bian X, Cheng G, Zhao P, Xiang X, Tian W, Li T, Zhai Q (2020) A novel nonsense variant in PLS3 causes X-linked osteoporosis in a Chinese family. Ann Hum Genet 84(1):92–96. https://doi.org/10.1111/ahg.12344
Fahiminiya S, Majewski J, Al-Jallad H, Moffatt P, Mort J, Glorieux FH, Roschger P, Klaushofer K, Rauch F (2014) Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Miner Res 29(8):1805–1814. https://doi.org/10.1002/jbmr.2208
Connallon T, Clark AG (2013) Sex-differential selection and the evolution of X inactivation strategies. PLoS Genet 9(4):e1003440. https://doi.org/10.1371/journal.pgen.1003440
Valimaki VV, Makitie O, Pereira R, Laine C, Wesseling-Perry K, Maatta J, Kirjavainen M, Viljakainen H, Valimaki MJ (2017) Teriparatide treatment in patients with WNT1 or PLS3 mutation-related early-onset osteoporosis: a pilot study. J Clin Endocrinol Metab 102(2):535–544. https://doi.org/10.1210/jc.2016-2423
Yorgan TA, Sari H, Rolvien T, Windhorst S, Failla AV, Kornak U, Oheim R, Amling M, Schinke T (2020) Mice lacking plastin-3 display a specific defect of cortical bone acquisition. Bone 130:115062. https://doi.org/10.1016/j.bone.2019.115062
Hu J, Li LJ, Zheng WB, Zhao DC, Wang O, Jiang Y, Xing XP, Li M, Xia W (2020) A novel mutation in PLS3 causes extremely rare X-linked osteogenesis imperfecta. Mol Genet Genomic Med 8:e1525. https://doi.org/10.1002/mgg3.1525
Treurniet S, Eekhoff EMW, Schmidt FN, Micha D, Busse B, Bravenboer N (2020) A clinical perspective on advanced developments in bone biopsy assessment in rare bone disorders. Front Endocrinol (Lausanne) 11:399. https://doi.org/10.3389/fendo.2020.00399
Costantini A, Krallis P, Kampe A, Karavitakis EM, Taylan F, Makitie O, Doulgeraki A (2018) A novel frameshift deletion in PLS3 causing severe primary osteoporosis. J Hum Genet 63(8):923–926. https://doi.org/10.1038/s10038-018-0472-5
Kannu P, Mahjoub A, Babul-Hirji R, Carter MT, Harrington J (2017) PLS3 Mutations in X-linked osteoporosis: clinical and bone characteristics of two novel mutations. Horm Res Paediatr 88(3–4):298–304. https://doi.org/10.1159/000477242
Cao YJ, Zhang H, Zhang ZL (2019) Novel mutations in the Wnt1, Tmem38b, P4hb, and Pls3 genes in four unrelated chinese families with osteogenesis imperfecta. Endocr Pract 25(3):230–241. https://doi.org/10.4158/EP-2018-0443
Chen T, Wu H, Zhang C, Feng J, Chen L, Xie R, Wang F, Chen X, Zhou H, Sun H, Xiao F (2018) Clinical, genetics, and bioinformatic characterization of mutations affecting an essential region of PLS3 in patients with BMND18. Int J Endocrinol 2018:8953217. https://doi.org/10.1155/2018/8953217
Prihodova L, Nagyova I, Rosenberger J, Roland R, Majernikova M, Groothoff JW, van Dijk JP (2015) Social participation after kidney transplantation as a predictor of graft loss and mortality over 10 years: a longitudinal study. Transplantation 99(3):568–575. https://doi.org/10.1097/TP.0000000000000347
Makitie RE, Hackl M, Weigl M, Frischer A, Kampe A, Costantini A, Grillari J, Makitie O (2020) Unique, gender-dependent serum microRNA Profile in PLS3 gene-related osteoporosis. J Bone Miner Res. https://doi.org/10.1002/jbmr.4097
Kampe AJ, Makitie RE, Makitie O (2015) New genetic forms of childhood-onset primary osteoporosis. Horm Res Paediatr 84(6):361–369. https://doi.org/10.1159/000439566
Balasubramanian M, Fratzl-Zelman N, O’Sullivan R, Bull M, Fa Peel N, Pollitt RC, Jones R, Milne E, Smith K, Roschger P, Klaushofer K, Bishop NJ (2018) Novel PLS3 variants in X-linked osteoporosis: exploring bone material properties. Am J Med Genet A 176(7):1578–1586. https://doi.org/10.1002/ajmg.a.38830
Bardai G, Ward LM, Trejo P, Moffatt P, Glorieux FH, Rauch F (2017) Molecular diagnosis in children with fractures but no extraskeletal signs of osteogenesis imperfecta. Osteoporos Int 28(7):2095–2101. https://doi.org/10.1007/s00198-017-4031-2
Lv F, Ma M, Liu W, Xu X, Song Y, Li L, Jiang Y, Wang O, Xia W, Xing X, Qiu Z, Li M (2017) A novel large fragment deletion in PLS3 causes rare X-linked early-onset osteoporosis and response to zoledronic acid. Osteoporos Int 28(9):2691–2700. https://doi.org/10.1007/s00198-017-4094-0
Costantini A, Skarp S, Kampe A, Makitie RE, Pettersson M, Mannikko M, Jiao H, Taylan F, Lindstrand A, Makitie O (2018) Rare copy number variants in array-based comparative genomic hybridization in early-onset skeletal fragility. Front Endocrinol (Lausanne) 9:380. https://doi.org/10.3389/fendo.2018.00380
Makitie RE, Niinimaki T, Suo-Palosaari M, Kampe A, Costantini A, Toiviainen-Salo S, Niinimaki J, Makitie O (2020) PLS3 mutations cause severe age and sex-related spinal pathology. Front Endocrinol (Lausanne) 11:393. https://doi.org/10.3389/fendo.2020.00393
Wang L, Zhai Q, Zhao P, Xiang X, Zhang X, Tian W, Li T (2018) Functional analysis of p.Ala253_Leu254insAsn mutation in PLS3 responsible for X-linked osteoporosis. Clin Genet 93(1):178–181. https://doi.org/10.1111/cge.13081
Thouverey C, Malinowska A, Balcerzak M, Strzelecka-Kiliszek A, Buchet R, Dadlez M, Pikula S (2011) Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics 74(7):1123–1134. https://doi.org/10.1016/j.jprot.2011.04.005
Hussein NJ, Mbimba T, Al-Adlaan AA, Ansari MY, Jaber FA, McDermott S, Kasumov T, Safadi FF (2020) A novel regulatory role of TRAPPC9 in L-plastin-mediated osteoclast actin ring formation. J Cell Biochem 121(1):284–298. https://doi.org/10.1002/jcb.29168
Pathak JL, Bravenboer N, Klein-Nulend J (2020) The osteocyte as the new discovery of therapeutic options in rare bone diseases. Front Endocrinol. https://doi.org/10.3389/fendo.2020.00405
Wesseling-Perry K, Makitie RE, Valimaki VV, Laine T, Laine CM, Valimaki MJ, Pereira RC, Makitie O (2017) Osteocyte protein expression is altered in low-turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocrinol Metab 102(7):2340–2348. https://doi.org/10.1210/jc.2017-00099
Kim JM, Kim J, Kim YH, Kim KT, Ryu SH, Lee TG, Suh PG (2013) Comparative secretome analysis of human bone marrow-derived mesenchymal stem cells during osteogenesis. J Cell Physiol 228(1):216–224. https://doi.org/10.1002/jcp.24123
Makitie RE, Kampe A, Costantini A, Alm JJ, Magnusson P, Makitie O (2020) Biomarkers in WNT1 and PLS3 osteoporosis: altered concentrations of DKK1 and FGF23. J Bone Miner Res 35(5):901–912. https://doi.org/10.1002/jbmr.3959
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3(6):889–901
Tsolis KC, Bei ES, Papathanasiou I, Kostopoulou F, Gkretsi V, Kalantzaki K, Malizos K, Zervakis M, Tsezou A, Economou A (2015) Comparative proteomic analysis of hypertrophic chondrocytes in osteoarthritis. Clin Proteomics 12(1):12. https://doi.org/10.1186/s12014-015-9085-6
Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, Carr AJ (2015) Osteoarthritis. Lancet 386(9991):376–387. https://doi.org/10.1016/S0140-6736(14)60802-3
Kong DC, Zheng TS, Zhang M, Wang DD, Du SH, Li X, Fang JH, Cao XJ (2013) Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS ONE 8:7. https://doi.org/10.1371/journal.pone.0069403
Allen JL, Cooke ME, Alliston T (2012) ECM stiffness primes the TGFbeta pathway to promote chondrocyte differentiation. Mol Biol Cell 23(18):3731–3742. https://doi.org/10.1091/mbc.E12-03-0172
Wu Q, Zhu M, Rosier RN, Zuscik MJ, O’Keefe RJ, Chen D (2010) Beta-catenin, cartilage, and osteoarthritis. Ann N Y Acad Sci 1192:344–350. https://doi.org/10.1111/j.1749-6632.2009.05212.x
Mählich D, Glasmacher A, Müller I, Oppermann J, Grevenstein D, Eysel P, Heilig J, Wirth B, Zaucke F, Niehoff A (2021) Expression and localization of thrombospondins, plastin 3, and STIM1 in different cartilage compartments of the osteoarthritic varus knee. Int J Mol Sci 22:1–19. https://doi.org/10.3390/ijms22063073
Pan L, Lin Z, Tang X, Tian J, Zheng Q, Jing J, Xie L, Chen H, Lu Q, Wang H, Li Q, Han Y, Ji Y (2020) S-Nitrosylation of plastin-3 exacerbates thoracic aortic dissection formation via endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 40(1):175–188. https://doi.org/10.1161/ATVBAHA.119.313440
Girard F, Venail J, Schwaller B, Celio MR (2015) The EF-hand Ca(2+)-binding protein super-family: a genome-wide analysis of gene expression patterns in the adult mouse brain. Neuroscience 294:116–155. https://doi.org/10.1016/j.neuroscience.2015.02.018
Hosseinibarkooie S (2016) Identification and characterization of molecular pathways underlying rescue function of Plastin 3 in Spinal Muscular Atrophy. PhD thesis, University of Cologne
Wirth B (2021) Spinal muscular atrophy: in the challenge lies a solution. Trends Neurosci 44(4):306–322. https://doi.org/10.1016/j.tins.2020.11.009
Heesen L, Peitz M, Torres-Benito L, Holker I, Hupperich K, Dobrindt K, Jungverdorben J, Ritzenhofen S, Weykopf B, Eckert D, Hosseini-Barkooie SM, Storbeck M, Fusaki N, Lonigro R, Heller R, Kye MJ, Brustle O, Wirth B (2016) Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell Mol Life Sci 73(10):2089–2104. https://doi.org/10.1007/s00018-015-2084-y
Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S (2008) Modeling spinal muscular atrophy in Drosophila. PLoS ONE 3(9):e3209. https://doi.org/10.1371/journal.pone.0003209
Dimitriadi M, Sleigh JN, Walker A, Chang HC, Sen A, Kalloo G, Harris J, Barsby T, Walsh MB, Satterlee JS, Li C, Van Vactor D, Artavanis-Tsakonas S, Hart AC (2010) Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet 6(10):e1001172. https://doi.org/10.1371/journal.pgen.1001172
Hao LT, Wolman M, Granato M, Beattie CE (2012) Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci 32(15):5074–5084
Doherty GJ, McMahon HT (2009) Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. https://doi.org/10.1146/annurev.biochem.78.081307.110540
Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marce M, Griffin JW, Rich MM, Sumner CJ (2009) Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29(3):842–851
Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O’Donovan MJ (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69(3):453–467
Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M (2007) Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 179(1):139–149. https://doi.org/10.1083/jcb.200703187
Kaifer KA, Villalon E, Osman EY, Glascock JJ, Arnold LL, Cornelison DDW, Lorson CL (2017) Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2(5):e89970. https://doi.org/10.1172/jci.insight.89970
Alrafiah A, Karyka E, Coldicott I, Iremonger K, Lewis KE, Ning K, Azzouz M (2018) Plastin 3 promotes motor neuron axonal growth and extends survival in a mouse model of spinal muscular atrophy. Mol Ther Methods Clin Dev 9:81–89. https://doi.org/10.1016/j.omtm.2018.01.007
Sudhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547
Ruiz R, Tabares L (2014) Neurotransmitter release in motor nerve terminals of a mouse model of mild spinal muscular atrophy. J Anat 224(1):74–84. https://doi.org/10.1111/joa.12038
Wu XS, Lee SH, Sheng J, Zhang Z, Zhao WD, Wang D, Jin Y, Charnay P, Ervasti JM, Wu LG (2016) Actin is crucial for all kinetically distinguishable forms of endocytosis at synapses. Neuron 92(5):1020–1035. https://doi.org/10.1016/j.neuron.2016.10.014
Watanabe S, Rost BR, Camacho-Perez M, Davis MW, Sohl-Kielczynski B, Rosenmund C, Jorgensen EM (2013) Ultrafast endocytosis at mouse hippocampal synapses. Nature 504(7479):242–247. https://doi.org/10.1038/nature12809
Cousin MA, Robinson PJ (2001) The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci 24(11):659–665
Liu Y, Zaun HC, Orlowski J, Ackerman SL (2013) CHP1-mediated NHE1 biosynthetic maturation is required for Purkinje cell axon homeostasis. J Neurosci 33(31):12656–12669. https://doi.org/10.1523/JNEUROSCI.0406-13.2013
Mendoza-Ferreira N, Coutelier M, Janzen E, Hosseinibarkooie S, Lohr H, Schneider S, Milbradt J, Karakaya M, Riessland M, Pichlo C, Torres-Benito L, Singleton A, Zuchner S, Brice A, Durr A, Hammerschmidt M, Stevanin G, Wirth B (2018) Biallelic CHP1 mutation causes human autosomal recessive ataxia by impairing NHE1 function. Neurol Genet 4(1):e209. https://doi.org/10.1212/NXG.0000000000000209
Wubetu GY, Utsunomiya T, Ishikawa D, Ikemoto T, Yamada S, Morine Y, Iwahashi S, Saito Y, Arakawa Y, Imura S, Arimochi H, Shimada M (2014) Branched chain amino acid suppressed insulin-initiated proliferation of human cancer cells through induction of autophagy. Anticancer Res 34(9):4789–4796
Janzen E, Wolff L, Mendoza-Ferreira N, Hupperich K, Delle Vedove A, Hosseinibarkooie S, Kye MJ, Wirth B (2019) PLS3 overexpression delays ataxia in Chp1 mutant mice. Front Neurosci 13:993. https://doi.org/10.3389/fnins.2019.00993
Pang T, Su X, Wakabayashi S, Shigekawa M (2001) Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J Biol Chem 276(20):17367–17372. https://doi.org/10.1074/jbc.M100296200
Guissart C, Li X, Leheup B, Drouot N, Montaut-Verient B, Raffo E, Jonveaux P, Roux AF, Claustres M, Fliegel L, Koenig M (2015) Mutation of SLC9A1, encoding the major Na(+)/H(+) exchanger, causes ataxia-deafness Lichtenstein-Knorr syndrome. Hum Mol Genet 24(2):463–470. https://doi.org/10.1093/hmg/ddu461
Galan JE, Zhou D (2000) Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc Natl Acad Sci USA 97(16):8754–8761. https://doi.org/10.1073/pnas.97.16.8754
Zhou D, Mooseker MS, Galan JE (1999) An invasion-associated salmonella protein modulates the actin-bundling activity of plastin. Proc Natl Acad Sci USA 96(18):10176–10181. https://doi.org/10.1073/pnas.96.18.10176
Kusdian G, Woehle C, Martin WF, Gould SB (2013) The actin-based machinery of Trichomonas vaginalis mediates flagellate-amoeboid transition and migration across host tissue. Cell Microbiol 15(10):1707–1721. https://doi.org/10.1111/cmi.12144
Adam T, Arpin M, Prevost MC, Gounon P, Sansonetti PJ (1995) Cytoskeletal rearrangements and the functional role of T-plastin during entry of Shigella flexneri into HeLa cells. J Cell Biol 129(2):367–381. https://doi.org/10.1083/jcb.129.2.367
Yoo YH, Yun J, Yoon CN, Lee JS (2015) Chemical proteomic identification of T-plastin as a novel host cell response factor in HCV infection. Sci Rep 5:9773. https://doi.org/10.1038/srep09773
Serio AW, Jeng RL, Haglund CM, Reed SC, Welch MD (2010) Defining a core set of actin cytoskeletal proteins critical for actin-based motility of Rickettsia. Cell Host Microbe 7(5):388–398. https://doi.org/10.1016/j.chom.2010.04.008
Zhang B, Yu Q, Wang Y, Xiao C, Li J, Huo D, Zhang D, Jia C (1863) Li M (2016) The Candida albicans fimbrin Sac6 regulates oxidative stress response (OSR) and morphogenesis at the transcriptional level. Biochim Biophys Acta 9:2255–2266. https://doi.org/10.1016/j.bbamcr.2016.06.002
Shi F, Ma Y, Qian Y, Wang Y, Wang Z, Zhao M, Hu Z (2019) A novel peptide probe for identification of PLS3-expressed cancer cells. Anal Chem 91(15):9640–9647. https://doi.org/10.1021/acs.analchem.9b01061
Strathmann EA, Peters M, Hosseinibarkooie S, Rigo FW, Bennett CF, Zaworski PG, Chen KS, Nothnagel M, Wirth B (2018) Evaluation of potential effects of Plastin 3 overexpression and low-dose SMN-antisense oligonucleotides on putative biomarkers in spinal muscular atrophy mice. PLoS ONE 13(9):e0203398. https://doi.org/10.1371/journal.pone.0203398
Stratigopoulos G, Lanzano P, Deng L, Guo J, Kaufmann P, Darras B, Finkel R, Tawil R, McDermott MP, Martens W, Devivo DC, Chung WK (2010) Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch Neurol 67(10):1252–1256. https://doi.org/10.1001/archneurol.2010.239
Acknowledgements
We thank Jutta Becker, Sophie Kaspar and Marlen Lauffer, who helped to cross-check the PLS3 variants and HGVS-conform nomenclature. We are grateful to the entire Wirth group members, who contributed currently and in past to research on PLS3.
Funding
Open Access funding enabled and organized by Project DEAL. Research on PLS3 and protective modifiers in SMA, ataxia and ALS, osteoporosis and osteoarthritis was supported by the German Research Foundation [Wi 945/17-1 to BW; FOR2722 to BW & AN (project ID 407176282), SFB 1451 to BW (project-ID 431549029-A01), and GRK1960 to BW (project ID 233886668], the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 956185 (SMABEYOND) to BW, Center for Molecular Medicine Cologne to BW (project No C18) and Köln Fortune MD candidate fellowship (to CV).
Author information
Authors and Affiliations
Contributions
BW proposed and selected the publications, compiled all parts of the review, and wrote the neurodegenerative part, abstract and conclusion; LW wrote the endocytosis and infection part and designed Figures 1and 2; EAS wrote the gene expression, cancer and biomarker parts, and Table 1, IM and DM wrote the osteoarthritis and osteoporosis parts and Table 2, and designed figure 3; CV wrote the calcium homeostasis part; AN wrote the protein structure, F-actin/cell motility and signaling parts. All co-authors read and corrected the entire manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wolff, L., Strathmann, E.A., Müller, I. et al. Plastin 3 in health and disease: a matter of balance. Cell. Mol. Life Sci. 78, 5275–5301 (2021). https://doi.org/10.1007/s00018-021-03843-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03843-5