
Abstract
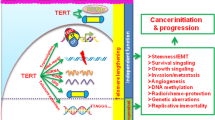
Reactivation of telomerase is a major hallmark observed in 90% of all cancers. Yet paradoxically, enhanced telomerase activity does not correlate with telomere length and cancers often possess short telomeres; suggestive of supplementary non-canonical roles that telomerase might play in the development of cancer. Moreover, studies have shown that aberrant expression of shelterin proteins coupled with their release from shortening telomeres can further promote cancer by mechanisms independent of their telomeric role. While targeting telomerase activity appears to be an attractive therapeutic option, this approach has failed in clinical trials due to undesirable cytotoxic effects on stem cells. To circumvent this concern, an alternative strategy could be to target the molecules involved in the non-canonical functions of telomeric proteins. In this review, we will focus on emerging evidence that has demonstrated the non-canonical roles of telomeric proteins and their impact on tumorigenesis. Furthermore, we aim to address current knowledge gaps in telomeric protein functions and propose future research approaches that can be undertaken to achieve this.
Similar content being viewed by others
Introduction
Telomeres are repetitive DNA sequences located at the end of linear chromosomes that protect chromosomes from DNA loss which occur after each cell division due to end replication problem [1, 2] or when cells experience heightened oxidative stress [3]. Maintenance of telomeres is essential to prevent loss of genetic information, chromatin instability, senescence, and apoptosis. Telomere length maintenance is regulated by the telomerase complex, a holoenzyme expressed in proliferating cells like germ cells, stem cells and some of the immune cells, but remains mostly inactive in terminally differentiated somatic cells [4,5,6].
Aside from the telomerase complex, another protein complex essential to the function of telomeres is shelterin. Each component of shelterin has distinct functions and serves to protect telomeres by inhibiting specific DNA damage repair pathways [7]. Shelterin is critical in maintaining genome integrity, and dysregulation in any of its components can lead to genomic instability [7]. The shelterin proteins play highly dynamic roles in the development and progression of cancer, where conflicting studies have shown that they are both upregulated and/or downregulated in different cancer types at both transcriptional and translational levels.
Moreover, the interactions and interdependence among these proteins further complicate understanding of how each may contribute to development and progression of human malignancies. In addition, though changes in expression levels of shelterin proteins have been shown to affect telomere length, it has been difficult to distinguish if this occurs due to their influence on telomerase activity or by other molecular mechanisms. Furthermore, studies have shown that the development of cancer has also been attributed to non-telomeric roles of these shelterin and telomerase proteins.
However, though cancer cells possess high-telomerase activity, their telomere length remains short yet sufficient to protect chromosomes [8]. In most instances, telomerase activity and telomere length are correlated in cancer cells. However, telomerase only extends telomeres until they are no longer at the critical length that triggers apoptosis, thus ensuring immortality for the cancer cells [9]. Live cell imaging results revealed that in cancer cells, telomerase forms short dynamic interactions with telomeres during S phase of the cell cycle, suggesting a less stable association and hence low processivity [10, 11]. Therefore, if the increase in telomerase activity in cancer cells is not simply to provide unrestrained lengthening of telomeres, could these telomerase molecules be playing other non-canonical roles in tumorigenesis? Over the years, many inhibitors/drugs such as reverse transcriptase inhibitors, telomere disrupting agents, and immunotherapy targeting the TERT peptide were suggested to suppress telomerase and/or telomere maintenance mechanisms. Although these approaches had the desired effect in in vitro and animal studies, they failed in clinical trials due to cytotoxic effects on the stem cell compartment [12,13,14,15]. Since the results of abolishing telomerase activity in cancer have proven to be less than ideal, perhaps a better strategy would involve targeting the molecules or pathways that are involved in the non-canonical/non-telomeric roles of these telomeric proteins that have been implicated in malignancies. Therefore, it is essential to determine the exact molecular mechanisms of telomeric proteins in cancer and in telomere homeostasis to generate effective therapeutic approaches against cancer and diseases related with aging.
In this review, we will discuss the recent findings of telomere-associated proteins in cancer, with a particular focus on their non-canonical function, away from their traditional telomeric roles. We will propose novel research approaches to gain a better understanding of the underlying molecular mechanisms and pathways by which these telomeric proteins function, so as to generate better targeted therapeutic strategies.
The telomerase complex
The catalytic core of the human telomerase complex consists of two integral subunits, a reverse transcriptase TERT, and a specific RNA component TERC [4]. TERC is a long non-coding RNA that serves as a template for the addition of telomeric repeats to telomeres by TERT [16,17,18]. In vitro, these two components have been shown to be sufficient for driving telomerase activity [19]. However, in vivo, other proteins such as the H/ACA complex proteins (Dyskerin, NOP10, NHP2 and GAR1) and TCAB1, which bind directly or indirectly to TERC, are required for the assembly, recruitment and physiological functioning of active telomerase [20]. While it has been shown that these proteins form part of the telomerase complex, the crystal structure of active telomerase has been difficult to elucidate. Recently, with the advances in cryogenic electron microscopy, the structure of human telomerase bound to its DNA substrate has been determined [21]. In vivo, the active telomerase complex assumes a bilobal structure composed of 10 protein subunits surrounding a centrally located TERC. One lobe of the complex is the catalytic core, formed by the binding of the telomerase RNA-binding and reverse transcriptase domains of TERT to the template/pseudoknot (t/PK) and conserved regions 4/5 (CR4/5) of TERC, respectively. The other lobe is formed by TERC bound to TCAB1 via a CAB box motif [22, 23], and two H/ACA complexes, each made up of Dyskerin, NOP10, NHP2 and GAR1 [21].
The H/ACA complex interacts with the box H/ACA domain on TERC, which leads to its accumulation in the nucleus [24] and assists in the proper folding and assembly of the active telomerase complex [24, 25]. Dyskerin, NHP2 and NOP10, first form a trimer, which is recruited to the site of RNA transcription [26] and is required for the accumulation of TERC and overall stability of the telomerase complex [27]. This recruitment is mediated by specific assembly factors SHQ1 and NAF1 [26, 28]. SHQ1 acts as a chaperone that stabilizes the newly synthesized Dyskerin prior to its binding to NHP2 and NOP10 [28]. The remaining member of the H/ACA complex, GAR1, which binds directly to Dyskerin, is only required for the proper functioning of active telomerase [26]. Two other factors essential to the assembly of the H/ACA complex on TERC are Reptin and Pontin. These AAA + ATPases interact directly with Dyskerin and TERT to bring about the proper assembly and stabilization of the complex [29]. In particular, Reptin and Pontin are required for the release of SHQ1 from Dyskerin [30], which then allows it to bind to the other H/ACA complex proteins and TERC. TCAB1 is required for the localization of the telomerase complex to Cajal bodies of the nucleus [23], which in turn brings the complex into close proximity with telomeres [23, 31]. The accumulation of TERT and other telomerase complex molecules in Cajal bodies have also been found to prompt the extension of telomeres during S phase of the cell cycle [31].
Role of TERT in cancer
Telomerase regulates telomere length and prevents telomere erosion associated with the activation of DNA damage response [32]. Mutations in the telomerase complex are mainly associated with aging syndromes caused by accelerated telomere shortening. On the other hand, overexpression of TERT and TERC are associated with cancer progression as they enhance cell proliferation [33]. Given that in most somatic cells, telomerase activity is limited by TERT expression, cancer cells thus require reactivation of TERT expression to restore telomerase activity. Despite reacquiring telomerase activity, it is regularly observed that cancer cells have short telomere lengths that are just sufficient to protect the information encoded by genomic DNA [10, 11]. Therefore, if the role of reactivated TERT in cancer is not limited to telomere elongation, the question remains: how else could TERT contribute to cancer progression? TERT might therefore have non-telomeric functions in important cellular homeostasis pathways. In this section, we will first discuss the mechanisms by which TERT is reactivated in cancer, after which we will visit the literature that focuses on the non-telomeric role of TERT in cancer.
Re-activation mechanisms of TERT in cancer
Telomerase is expressed in germ cells, stem cells and some of the immune cells [34]. During the differentiation of stem cells to somatic cells, telomerase activity is repressed by transcriptional inactivation of the TERT gene which encodes the catalytic subunit of telomerase holoenzyme. However, TERT expression is re-activated in 90% of cancers, granting them potential to proliferate indefinitely. The mechanisms that re-activate TERT gene expression in cancer vary in different types of cancers. These mechanisms include copy number increase, activation of oncogenic pathways and cancer-specific TERT promoter mutations.
Copy number increase
TERT gene is located at the 5p15.33 chromosome band and consists of 16 exons. Deletions and insertions of chromosomes or chromosome arms are observed in many human tumors [35]. Chromosome 5p is mostly amplified in neuroblastoma, medulloblastoma, osteosarcoma, head and neck, lung, and cervical cancers [36,37,38]. Correlation of copy number gain with TERT gene expression or telomerase activity in a variety of cancer types have been extensively reviewed [33]. TERT expression or telomerase activity is highly correlated with copy number gain in neuroblastomas, cervical cancer and lung cancer, whereas no correlation has been found in melanoma, colorectal and hepatocellular carcinomas [33].
Oncogene activation
The TERT promoter contains various transcription factor binding motifs and epigenetic regulatory sequences that allow for a dynamic regulation of TERT expression. The proximal TERT promoter harbors E-boxes and GC boxes to host respective transcription factors, which include SP1, Myc and other associated epigenetic modifiers such as histone modifiers [34]. The distal TERT promoter on the other hand, contains binding motifs for AP1, p53, HIF1 and p21, of which the binding of these factors are influenced by cellular homeostasis and oncogenic pathways [34].
Cancer-specific TERT promoter mutations
Cancer-specific TERT promoter mutations, which are associated with enhanced TERT mRNA expression [39,40,41,42,43,44], are seen in ~ 19% of cancers [42], with higher prevalence in melanoma, bladder, glioblastoma, urothelial, thyroid and hepatocellular carcinoma. These mutations occur particularly in the immediate upstream of the ATG start codon and lead to C to T conversion, which in turn creates de novo Erythroblast Transformation Specific (ETS) transcription factor binding motifs [42]. Importantly, these de novo sites are very near to the GC-boxes in the core TERT promoter, which provide a unique regulatory mechanism for the reactivation of the TERT gene in cancers harboring TERT promoter mutations. Several groups have studied the reactivation of the mutant TERT promoter using recent genome editing tools and identified cancer-type-specific co-regulators that drive TERT expression in cells harboring the mutant TERT promoter [45, 46]. Bell et al. identified that the depletion of GA-binding protein (GABP) transcription factor using siRNAs dramatically reduces TERT expression as compared to the other ETS family proteins, suggesting that GABP interacts with the de novo site stronger than the other ETS proteins [47]. Detailed chromatin analysis of mutant TERT promoter revealed that the de novo GABPA site mediates a long-range chromatin interaction between the mutant TERT promoter and a long-distance region named T-INT1 (TERT-interacting region 1). This interaction stabilizes the GABPA protein on the TERT promoter by promoting GABPA-GABPA dimerization between GABPA sites located on the mutant TERT promoter and the T-INT1 region. Subsequently, chromatin modifiers like BRD4 are recruited to the proximal TERT promoter to re-activate TERT expression in the cell [48].
Non-canonical role of TERT in cancer
The impact of TERT overexpression on gene expression was first identified using immortalized fibroblasts [49]. TERT-mediated immortalization resulted in increased expression of epiregulin and Pol2-associated genes, epigenetic co-activators CBP/p300, protein biosynthesis-associated genes, growth factors and growth factor receptors [49]. Telomerase is recruited to telomere ends during S-phase to elongate telomeres [50]. Apart from telomere ends, telomerase has been detected in cajal bodies, mitochondria, cytoplasm, nucleus and nucleolus [51,52,53]. Overexpression of TERT and catalytically inactive TERT (DN-TERT) in ALT cells has been shown to promote cell adhesion and migration by upregulating the expression of extracellular matrix- and matrix metalloproteinase-specific genes [54]. Additionally, enhanced tumorigenesis by TERT overexpression occurred by activating various pathways including cell proliferation, anti-apoptosis and energy metabolism [55,56,57]. Mitochondrial TERT has been shown to reduce intracellular ROS production and hence, reduced ROS-induced cell death [57,58,59]. Furthermore, TERT inhibits cytosolic acidification, translocation of Bax, and the release of cytochrome C to the cytosol, which eventually inhibits apoptosis [60].
Telomerase overexpression has been shown to enhance cell proliferation rapidly, but in order for cells to become immortal, it requires additional events such as the activation of oncogenic pathways and the bypassing of multiple tumor suppressing mechanisms like senescence [49]. In TERT overexpressing cells, c-MYC oncogene and E2F pathways are activated [61, 62], whereas p16, a tumor suppressor gene that drives cells toward growth arrest and senescence, is transcriptionally silenced by increased promoter methylation [63].
In cancer, TERT has been shown to bind TCF binding elements (TBEs) to drive c-MYC expression [64]. In the presence of active Wnt signaling, TERT binds to TBEs associated with β-catenin and Brg1 to drive the expression of Wnt-dependent genes including cyclinD1, MYC and axin2 [64]. Indeed, the correlation between TERT and MYC is well-documented in various cancers. Koh et al. reported that TERT enhances MYC stability in vitro and in vivo which leads to increase of MYC occupancy on the promoters of its target genes [65, 66]. This regulation does not require the presence of TERC, suggesting that TERT–MYC interaction on MYC-regulated promoters are independent of telomerase activity and could also explain why TERT and MYC levels are highly correlated in cancer cells. On the other hand, in vivo studies revealed that overexpression of catalytically inactive TERT (DN-TERT) in the crypts of mouse intestines led to increased expression of a β-catenin driven gene, CD44, suggesting that reverse transcriptase activity is not required for TERT–TBE interaction [64]. Though the impact of TERT in Wnt pathway has been demonstrated both in vitro and in vivo, there are unaddressed issues that still remain. There are thousands of TCF binding sites in the genome and between 2000 and 3000 genes have been shown to be differentially expressed upon β-catenin depletion [67]. However, considering the limited number of TERT molecules in a cell [68], it is highly improbable that TERT is capable of regulating all these genes at once. Therefore, the question remains: which set of genes, and under what physiological condition, does this small pool of TERT molecules influence? Furthermore, the adaptor molecules required for the interaction of TERT with TBE sites remain to be elucidated. Indeed, this question is partially addressed by a genome-wide study that showed TERT binds to various genomic regions in different cell types [69]. Not surprisingly, the primary binding sites of TERT were found to be the sub-telomeric regions. However, TERT also binds to intergenic regions, promoters and introns in a cell type-dependent manner. Interestingly, among all the various cell types analyzed, TERT ubiquitously binds to regions that encode for tRNAs along with the RNA Pol III subunit RPC32. This binding enhances tRNAs expression and promotes rapid protein synthesis and cell growth [69].
In addition to regulating cell proliferation and the expression of pro-cancer genes, TERT has also been implicated in modulating inflammatory signals in cancer cells. NF-κB, the master regulator of inflammation in cells, is regulated by various molecules including cytokines, phosphatases, kinases and lncRNAs [70,71,72]. Once NF-κB is activated, it binds to its target promoters to drive the expression of pro-inflammatory and pro-survival genes. In the presence of an inflammatory stimuli, it has been shown that telomerase, together with p65, binds to the promoters of NF-κB target genes to regulate the expression of genes involved in inflammation and growth. [73, 74]. The depletion of TERT in an ovarian cancer cell line rapidly reduced cell proliferation, and this growth inhibition was reversed by the overexpression of p65. However, TERT overexpression failed to rescue the growth of p65-depleted cells, suggesting that p65 is the essential regulator in cell proliferation while TERT enhances its function [73]. Furthermore, the suppression of telomerase activity by MST-1, a known telomerase inhibitor, led to dramatic reduction of p65 occupancy on the promoters of NF-κB target genes [73], suggesting that co-regulation of p65-dependent gene expression is dependent on telomerase activity and the presence of other telomerase molecules like TERC as well Table 1.
Role of TERC in cancer
The telomerase RNA component, TERC, maps to chromosome 3q26.2 and encodes a 451 nucleotide lncRNA. Together with TERT, it forms the core component of the telomerase holoenzyme. In this complex, TERC functions mainly as a template for telomere elongation that is necessary for normal physiological processes such as stem cell renewal. In cancer, TERC is dysregulated by various mechanisms such as mutations and copy number increase that is generally correlated with cell proliferation and disease progression [77, 78].
Amplification of TERC in cancer
It has been proposed that elevated TERC expression could serve as a biomarker for cancer as its amplification has been detected in lung squamous cell carcinoma [79,80,81] and esophageal squamous cell carcinoma [77, 81]. Relatedly, TERC overexpression has been observed in prostate cancer (50). Additionally, TERC amplification correlates with disease progression in cervical cancer lesions (51–54) and esophageal lesions [77, 81]. In preclinical experiments, the overexpression of chicken TERC (cTERC) via genetically engineered Marek’s disease virus (MDV) containing cTERC instead of its viral TERC (vTERC), resulted in increased tumor incidence and visceral organ tumor load in chickens [82]. When chickens are infected with vTERC−/− MDV, tumor incidence is reduced by more than 60% as compared to those inoculated with MDV containing either one intact copy or both copies of vTERC [83]. Similarly, knocking down TERC using siRNA leads to inhibition of cell proliferation and induction of apoptosis in cancer cells [84, 85], as well as inhibition of xenograft tumor growth in nude mice [84]. The same effect was achieved when HeLa cells were treated with anti-sense TERC, which caused the cells to go into crisis state and stop proliferating from 23 to 26 cycles [86].
Although the tightly regulated TERT molecules are widely considered to be the limiting factor in the formation of active telomerase complex [34], the absence of TERC, under the context of TERT overexpression, has an inhibitory effect on tumorigenesis instead [87, 88]. K5-TERT mice, which overexpress TERT in their skin, develop more papillomas when exposed to chemical carcinogen TPA as compared to wild-type controls. However, when the same TPA treatment was applied to K5-TERT/TERC−/− mice, the number of papillomas developed were lower than those observed in wild-type and TERC−/− mice. In a related study, TPA treated G5 TERC−/− mice, which have critically shorter telomeres, developed fewer papillomas when compared to G1 TERC−/− mice or wild-type mice [89]. It would be interesting to perform the same TERC knockout study on mice expressing a dominant negative form of TERT (DN-TERT) that is catalytically inactive for its reverse transcriptase activity. This would help decipher whether reverse transcriptase activity is required for the observed inhibitory effect on tumorigenesis. Should this effect prove to be independent of reverse transcriptase activity, it will provide further insight on the non-canonical functions of TERT molecules and how they contribute to malignancies.
The combined evidence from all the TERC studies reviewed suggests an oncogenic role for TERC whereby dysregulation of its expression could promote tumor development. At the same time, targeting its expression specifically in cancer, regardless of TERT status, could serve as an attractive anti-tumor strategy.
Mutation of TERC in diseases
Mutations in TERC are observed in the transcribed region and more commonly associated with telomere biology diseases such as Dyskeratosis Congenita (DC) [90]. TERC mutation frequency, at a rate of about 1.5%, was identified in 210 patients with bone marrow failure syndrome [91]. In addition, a single mutation at nucleotide 305 (n305 G > A) was identified in a patient with clinical characteristics and family history of DC. Other identified TERC mutations include n322 (G > A), n450 (G > A) and n467 (T > C). As mutations in the TERC gene that alter its expression are rare, the regulation of TERC expression has been found to occur post-transcriptionally instead. Maturation of TERC transcripts is regulated by poly(A)-specific ribonuclease (PARN) which de-adenylates 3′ oligo(A) tails from nascent TERC RNA transcripts, thereby preventing their degradation by exosomes. This process is necessary for the maturation and maintenance of steady state levels of TERC in cells [92]. Therefore, patients with biallelic mutation of PARN gene suffer from severe DC [93].
Unlike TERT, mutations in the TERC gene, or its promoter, are less prevalent in cancer. The major allele of SNP rs2293607 was associated with higher susceptibility to colorectal cancer [94]. When overexpressed in colorectal cancer cell line HCT116, cells containing the major allele have longer telomeres resulting from higher TERC expression as compared to cells overexpressing the minor allele. This finding goes against the common observation that cancer cells have shorter telomeres. A possible explanation for this paradox could be that telomere shortening occurs post cancer diagnosis, as evidenced by weaker association of telomere length to cancer risk in prospective studies as compared to retrospective ones [95]. Alternatively, it could have also resulted from poorer survival of cancer patients with longer telomeres.
Dyskerin 1 in cancer
Dyskerin 1 is a widely expressed multifunctional protein and one of the components of telomerase complex which associates with TERC to provide structural stability required for telomerase activity [96, 97]. Mutations in the DKC1 gene lead to loss of function and cause telomere-associated diseases such as aplastic anemia, bone marrow failure syndromes and pulmonary fibrosis [98]. Expression of DKC1 is controlled by c-MYC transcriptionally in MYC-dependent cancers and enhances cell proliferation and growth [99, 100]. DKC1 overexpression has been reported in many cancers, including neuroblastoma, lymphoma, melanoma, colorectal cancer, ovarian carcinoma, breast cancer and hepatocellular carcinoma [101,102,103,104,105,106,107,108,109], and often results in poor prognosis due to aggressive tumor growth and resistance to therapy [104, 106, 110].
Away from its role on telomeres, DKC1 has been shown to bind HIF1α promoter and increases the expression of HIF1α in colorectal cancer (CRC). This results in enhanced expression of VEGF which promotes CRC progression [98]. In MYC-amplified neuroblastoma cells, n-MYC and c-MYC bind to the proximal DKC1 promoter and drive the expression of DKC1 gene [111]. Depletion of DKC1 in neuroblastoma cells reduced cell growth. Importantly, depletion of DKC1 in cells with ALT mechanism, a homologous recombination-based telomere maintenance mechanism, showed the same effect, suggesting a telomerase independent function of DKC1 for regulating cell proliferation in cancer [111]. One of the main functions of DKC1 is to process the H/ACA small nucleolar ribonucleoprotein required for ribosome synthesis [112]. Depletion of DKC1 in neuroblastoma cells induces ribosomal stress by dispersal of ribosomal proteins, which leads to inhibition of cell proliferation via p53-dependent G1 cell cycle arrest [111]. The correlation of MYC with TERT and DKC1 in cancers suggests that oncogene activation is one of the important events that initiates cancer-specific non-telomeric functions of TERT and DKC1.
GAR1 and NOP10 in cancer
GAR1 and NOP10, together with DKC1 and NHP2, associate with H/ACA snoRNAs to form the snoRNPs complex. This snoRNPs complex functions mainly as the catalytic unit for post-transcriptional pseudouridylation of rRNAs. Additionally, these snoRNPs proteins also bind to the H/ACA domain present on TERC to form the telomerase complex. Mutations in NOP10 gene have been implicated in various forms of DC [113, 114] but their role in cancer is less established. Clinically, NOP10 expression was found to be significantly decreased in a cohort of patients with chronic lymphocytic leukemia [115], but in another study on gastric and colorectal cancers, no difference in NOP10 expression was observed between normal and cancer tissues [116]. Individuals with homozygous loss-of-function mutation in NOP10 suffer from significant telomere shortening and reduced TERC expression [113]. This link between TERC levels and functional NOP10 expression was confirmed by in vitro studies in HeLa cells. The studies demonstrated that siRNA knockdown of NOP10, or overexpression of mutant NOP10, leads to reduction in TERC expression. This result hints that TERC stability might be dependent on NOP10 expression level which suggests that activating mutations in NOP10 could possibly lead to oncogenic transformation through increase in TERC expression. Although GAR1 mutation has been identified in patients with aplastic anemia [117], the resulting amino acid substitution did not affect telomere length in affected patients. Unlike NOP10, siRNA knockdown of GAR1 in HeLa cells was not associated with reduction in TERC levels [118]. Therefore, the role of GAR1 in carcinogenesis, if any, remains to be determined.
TCAB1 in cancer
TCAB1 functions as a scaffold protein during telomere maintenance by recruiting and localizing the telomerase complex to Cajal bodies present in the nucleus. This process brings the telomerase complex into close proximity to telomeres in the nucleus, which facilitates telomere elongation. Various studies have indicated that TCAB1 is overexpressed in a variety of carcinomas, in both primary patient samples and cancer cell lines [119, 120]. Moreover, high levels of TCAB1 correlate with poor prognosis and resistance to radiotherapy in head and neck carcinoma patients [120]. The promotion of carcinogenic transformation in cells overexpressed with TCAB1 has prompted speculation that it could function as an oncogene. In line with this proposition, several studies have shown that knockdown of TCAB1 leads to decreased proliferation and invasion in cancer cells [119, 121] while promoting cell cycle arrest [121] and/or cellular apoptosis [119, 122], possibly through the mitochondrial pathway [120]. As trafficking of TERT into nucleus is disrupted in TCAB1 knockdown cancer cells [121], it suggests that the oncogenic potential of TCAB1 acts through enhancing telomerase activity in the nucleus.
However, contradictory reports from a separate clinical study suggest that higher nuclear expression of TCAB1 is associated with increased sensitization to radiotherapy, better disease-free progression and overall survival in both ovarian cancer [123], as well as in head and neck carcinoma [124, 125]. One possible explanation for the conflicting reports on whether higher or lower TCAB1 is linked with better prognosis lies in the subcellular compartmentalization of TCAB1 expression. It was shown that nuclear staining, but not total staining which includes cytoplasmic expression, predicts a favorable clinical outcome [125]. Mechanistically, nuclear TCAB1 expression facilitates recruitment of repair factors, such as RNF168, BRCA1 and RAD51, to DNA double strand breaks following ionizing radiation damage in ovarian cancer cells [123]. This results in rapid clearance of γH2AX and promotes DNA repair [126].
It is puzzling as to how a better DNA damage repair response conferred by nuclear TCAB1 expression would actually improve radio-sensitivity and predict a better prognostic outcome in cancer patients following radiotherapy. More research on TCAB1 needs to be performed to elucidate under which specific conditions TCAB1 function as an oncogene or tumor suppressor and whether telomerase activity is implicated in these roles.
Pontin and Reptin in cancer
Pontin and Reptin are members of ATPases associated superfamily that regulate diverse functions like chromatin remodeling, transcription regulation and telomerase assembly. Although Pontin and Reptin are fairly abundant in the cell, overexpression of Pontin and Reptin has been reported in cancers and has been correlated with poor prognosis [127]. In MYC-driven cancers, MYC regulates the expression of Pontin and Reptin, which interact with β-catenin and MYC. Although these proteins belong to the same superfamily, they have different functions under various conditions. Pontin forms a complex with Polymerase I at ribosomal transcription sites together with c-MYC and regulates ribosomal RNA synthesis [128]. Reptin regulates cyclin D1 gene expression by modulating chromatin structure. Reptin displaces the cyclin D1 enhancer bound histone variant H2A.Z, inhibiting the repressive chromatin loop, which allows binding of estrogen receptors to the cyclin D1 promoter [129]. Interestingly, while TIP60–Pontin interaction increases expression of the metastasis suppressor KAII in non-metastatic prostate cancer cells, β-catenin-Reptin complex reduces KAI1 expression in metastatic prostate cancer cells [130]. Similarly, c-MYC, Pontin and Reptin together, decondense the chromatin at the end of mitosis and increase cell proliferation in Xenopus laevis egg extracts and also in human cancer cells in an ATP-dependent manner [131, 132].
Reptin and Pontin increase cancer progression via interacting with tumor suppressor p53 gene [133]. Pontin has been shown to interact with mutant p53 to regulate genes responsible for tumor migration and invasion in colorectal and breast cancer lines [134]. Reptin on the other hand, interacts with wild-type p53 to inhibit its tumor suppressor activity. Reptin, together with anterior gradient-2, function as a p53 inhibitor complex to diminish the expression of p53-dependent genes, which leads to increase in cell proliferation and metastasis [135,136,137,138]. Reptin also inhibits p14ARF, a tumor suppressor that inactivates MDM2, which leads to the activation of MDM2 and destabilization of p53, resulting in enhanced proliferation of cancer cells [138, 139]. Increased cell proliferation requires adjustments in energy metabolism. To support energy requirements, cancer cells predominantly utilize glycolysis which increases the hypoxic conditions. Pontin and Reptin regulate HIF1 pathway through TIP60 interaction to modulate hypoxic gene expression. TIP60 binds to the HIF1-regulated gene promoters to recruit RNA Pol II to drive gene expression [140]. Post-translational modification of Pontin and Reptin proteins are crucial for their chromatin remodeling activities. Uncontrolled proliferation of cells in cancer leads to deprivation of oxygen and induces hypoxic response [141]. Methylation of Pontin by the hypoxia-induced G9a and GLP proteins recruits p300, whereas methylation of Reptin by G9a recruits HDAC1 to the hypoxia target gene promoters [142, 143]. Given that the majority of Pontin- and Reptin-regulated hypoxia genes do not overlap, Pontin and Reptin co-activate and repress the hypoxia pathway genes in response to different environmental stimuli in a context-dependent manner Tables 2 and 3 [144].
The shelterin complex
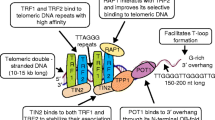
Telomeres are protected and maintained by a protein complex known as shelterin. Shelterin is made up of six specific proteins, TRF1, TRF2, RAP1, TIN2, TPP1 and POT1, that stably assemble along telomeres and confer protection to chromosome ends against unwarranted DNA damage repair [7]. Shelterin also functions to regulate telomerase activity and telomere length. The shelterin proteins are highly abundant and bind both double-stranded and single-stranded telomeric DNA. TRF1 and TRF2 form homodimers that bind double-stranded TTAGGG repeats [145] while POT1 specifically binds to single-stranded TTAGGG repeats in the 3′-overhang of telomeres [146]. The assembly of shelterin is controlled by TIN2 [147], which connects TRF1, TRF2 and TPP1 [148,149,150]. In addition, TPP1 also forms a heterodimer with POT1 and brings it into the proximity of telomeres, thus forming a bridge between the proteins bound to the duplex part of telomeric DNA to those bound to the 3′ overhang [151, 152]. Lastly, RAP1 is recruited to the shelterin complex by its binding to TRF2 [153].
TRF1 in cancer
Conventionally, TRF1 functions as a tumor suppressor by protecting telomeres from replication-dependent DNA loss and inhibiting telomerase activity [154,155,156]. Conditional TRF1 knockout mice experiments have shown that its deletion led to an accumulation of sister chromatid telomere fusions, chromosome end-to-end fusions and multi telomeric signals which result from telomere breakages [157]. The downregulation of TRF1 has also been reported in a number of human malignancies (Table 4). In particular, multiple studies have reported downregulation of TRF1 in breast cancer [158,159,160,161]. This is correlated with the overexpression of an oncomiR, miR-155, which targets a partially conserved site in the 3′UTR of TRF1. Overexpression of miR-155 has been reported in over 80% of breast cancers that were classified with TRF1 downregulation [161]. In a separate study, immunostaining revealed that TRF1 was far less abundant in almost all breast cancer tissues examined compared to normal tissue, and it was suggested that this enabled the maintenance of longer telomeres for prolonged proliferation of cancer cells [159]. Interestingly, in the conditional TRF1 knockout mouse experiment, it was observed that the downregulation of TRF1 was not the reason for the elongation of telomeres but was a result of increased fusion that occurred with the removal of TRF1 from telomeres [157]. These results suggest that in most cancers, downregulation of TRF1 contributes to cancer development by rendering telomeres fragile and susceptible to the accumulation of aberrant telomeric structures and chromosomal instability. In addition, the activity of telomerase in the absence of TRF1 allows for the maintenance of telomere length rather than for its continual elongation to ensure cell immortality.
Upregulation of TRF1 has been observed in many human malignancies (Table 4); and in some cancers, a progressive increase of TRF1 expression was observed during the transition of pre-malignant lesions to cancer. In hepatocellular carcinoma, increase in TRF1 expression corresponded to an increase in the neoplastic potential of different types of nodular lesions [162]. Benign large regenerative nodules had low TRF1 expression similar to normal liver tissues, whereas pre-malignant dysplastic nodules had significantly increased levels of TRF1 expression. Accordingly, the highest levels of TRF1 expression was observed in cancerous liver tissue. These corresponded to a decrease in telomere length, with high-grade dysplastic nodules displaying the shortest telomeres [162]. Interestingly, telomere lengths varied in cancerous liver tissue, though in most cases, it was still shorter than that of adjacent normal tissue [162]. Similar associations were also reported in development of gastric cancer [163]. On the other hand, in the development of lung cancers, despite a similar progressive upregulation of TRF1 from pre-malignant lesions to invasive carcinoma, telomere shortening and DNA damage response were suggested to precede this upregulation and that relative telomere length was shortest in benign squamous metaplasia as compared to low- and high-grade dysplasia and in-situ carcinomas [164]. Instead, telomeres increased in length from benign lesions to dysplasia, and finally stabilized in in-situ carcinomas with a slight decrease observed when progressing to invasive squamous cell carcinoma. Conversely, it was reported that in some cancers, after an initial upregulation of TRF1, a subsequent decrease in its levels was observed. Like other pre-malignant lesions, higher levels of TRF1 were detected in low-grade astrocytoma and were associated with short telomeres [165]. Upon progression to anaplastic astrocytoma and glioblastoma, TRF1 levels decrease as a result of its ADP-ribosylation mediated by the overexpression of PARP1 [165]. It has been proposed that this inhibition of TRF1 leads to its removal from telomeres and allows increased telomerase activity and prolonged proliferation of cancer cells [165].
In its contribution to cancer, the upregulation of TRF1, which occurs independent of telomerase activity and telomere length, has also been implicated in the dedifferentiation and maintenance of pluripotency of cells [166, 167]. Furthermore, higher TRF1 expression was also observed in less differentiated and more aggressive cancers with poorer prognosis [162, 163]. How the differential expression of TRF1 contributes to the development and progression of cancer appears to be time-dependent and could explain why in some cancers, both an upregulation or downregulation has been reported. With the decreasing cost of expression profiling technologies, prospective studies in which patients who present with pre-malignant lesions or early stage cancer could also be carried out to further investigate this. In addition, the effect of TRF1 seems to be influenced by other dysregulated components of the cell, and further investigation of these interactions could explain the disparity in mechanisms by which TRF1 is involved in different cancer types and could also reveal other pathways and molecular mechanisms by which TRF1 could contribute to cancer development.
TRF2 in cancer
TRF2 protects chromosome ends by inducing T-loop formation and preventing end-to-end fusion [190]. It was thought to be a negative regulator of telomere length [191]. This is consistent with early studies that observed that downregulation of TRF2 in cancers was usually accompanied by the downregulation of TRF1 [170, 172, 181, 192,193,194]. However, in the majority of human malignancies that reported TRF2 dysregulation, an upregulation of TRF2 is detected instead (Table 5). Similarly, the upregulation of TRF2 also coincided with upregulation of TRF1 and corresponded to shorter telomeres [175, 179]. In a study using telomerase active HT1080 human fibrosarcoma cell line, the overexpression of TRF2 led to stochastic telomere shortening whereby the loss of nearly the entire telomere tract was observed in some chromosomes which led to chromosome end-to-end fusion [195]. Sequencing of fusion products revealed the absence of telomeric repeats and large deletions were often found in the adjacent subtelomeric tracts. The upregulation of TRF2 in the presence of active telomerase caused telomeric replication stalling and the accumulation of ultrafine anaphase bridges and increased chromosomal instability. Despite the development of critically short telomeres with increased levels of TRF2, it has been observed that the accumulation of chromosomal aberrations was less than anticipated [196]. It was then proposed that TRF2 had a protective effect on these critically short telomeres and was able to prevent or delay cells from undergoing senescence. In addition, TRF2 has been shown to be a downstream target of the Wnt/β-catenin signaling pathway, which could imply that this upregulation of TRF2 in cancers could be mediated by the Wnt/β-catenin signaling pathway [197]. Though the upregulation of TRF1 and TRF2 usually go hand-in-hand, it also appears that dysregulation of either protein alone would be sufficient to drive the initiation of cancer formation together with activation of other cancer driving genes. It will be interesting to find out why they are dysregulated concurrently or if these observations are a result of the effect of one protein on the other.
Aside from its role as a telomeric protein, chromatin immunoprecipitation (ChIP)-sequencing has revealed that TRF2 binds to extratelomeric sites, of which some are located within the proximity of genes, implying that TRF2 might have non-telomeric roles [198, 199]. For instance, TRF2 has also been shown to modulate immune response to promote an immunosuppressive microenvironment conducive to the survival of cancer cells. In the development of colon cancer, the expression of TRF2 was found to be increased from pre-malignant low- and high-grade adenomas to intramucosal adenocarcinomas, and this corresponded to a decrease in NK cell numbers [200]. This suggests that TRF2 upregulation contributes to early cancer development by allowing cancer cells to escape innate immune responses. Further investigation using two different mouse models, one in which B16F10 murine melanoma cells were injected into immunocompetent mice and the other in which transformed human fibroblast BJcl2 cells were xenografted into nude mice, revealed that increased TRF2 levels promoted the recruitment of myeloid-derived suppressor cells (MDSCs) to sites of tumor formation. These MDSCs in turn express arginase 1, IL‐10, and TGF‐β, and inhibit the infiltration and activation of NK cells [201]. Furthermore, the recruitment of regulatory T cells with the suppression of CD8+ T cells was also observed, indicating that TRF2 is also capable of suppressing adaptive immune response at the tumor microenvironment via the recruitment of MDSCs. Patients with increased TRF2 expression and MDSC infiltration have been reported to have poorer prognosis. TRF2 has also been identified as a transcriptional activator of angiogenesis. Histopathological examination of different tumor types revealed that TRF2 is overexpressed in endothelial cells in tumor tissue but not in those of the adjacent healthy tissue [202]. These tumors include glioblastoma, liposarcoma, pancreas, colon, prostate and ovarian carcinomas. Primary endothelial cells isolated from mouse tumors have increased expression of TRF2 compared to that of normal lung endothelial cells, and this corresponded to an increase in angiogenic properties such as proliferation, migration and tube formation [202]. In addition, the upregulation of TRF2 in normal lung endothelial cells also led to similar increase in angiogenic properties whereas the knockdown of TRF2 in tumor endothelial cells led to a reversal of these observations. The promotion of angiogenesis by TRF2 has been attributed to its ability to bind the PDGFRβ promoter and activate its expression. Furthermore, the upregulation of TRF2 is also regulated by WT1, a protein known to regulate mediators of angiogenesis. In a more recent study, TRF2 was shown to indirectly regulate VEGF-A extracellular release by HCT116 colon cancer cells via the upregulation of SULF2 [203]. SULF2 is a sulfatase that carries out post-synthetic modification of heparan sulfate proteoglycans, which are thus inhibited from binding to VEGF-A and are released into the tumor microenvironment instead. In colorectal cancer patients, the upregulation of TRF2 was directly correlated with SULF2 upregulation, and tumors which had higher TRF2 levels also displayed greater angiogenesis. These oncogenic properties of TRF2 have been determined to be independent from its effects on telomeres and DNA damage response [202] and therapeutic strategies could be targeted at suppressing TRF2 in these cancers.
TIN2 in cancer
TIN2 as an adaptor is crucial for the recruitment, formation and stabilization of the shelterin complex [147] and its dysregulation can affect the functions of other shelterin proteins [213]. In vitro studies have demonstrated that mutations in TIN2, that disrupted either TRF1 or TRF2 binding, led to the uncapping of telomeres and activated DNA damage response [214]. Furthermore, the overexpression of TIN2 mutant protein significantly reduced both TRF1 and TRF2 levels [214], suggesting that TIN2 is critical for the stability and functions of TRF1 and TRF2. The downregulation of TIN2 in human malignancies are commonly observed as well, and many of these studies also reported concordant downregulation of TRF1 and TRF2 [170, 192, 193]. However, in chronic lymphocytic leukemia (CLL), the downregulation of mRNA was not accompanied by the significant downregulation of TRF1 and TRF2 mRNA, though the accumulation of telomere DNA damage was seen [215]. This suggests that in CLL in particular, loss of TIN2 did not affect the expression of these genes. Rather, the loss of TIN2 led to the failure of shelterin complex assembly on telomeres possibly resulting in observations similar to that seen in TRF1 or TRF2 downregulation. In a separate study, the downregulation of TIN2 in CLL was also associated with the presence of mutant p53 and significantly shorter telomeres [182]. Additionally, it has been observed in CLL that a spliced isoform of TIN2 with a deletion of exon 2 was upregulated while full-length TIN2 isoform was downregulated [213]. Co-immunoprecipitation experiments revealed that the spliced TIN2 isoform did not interact with TRF2. Moreover, TRF2 was also detected in the cytoplasm of lymphocytes which is suggestive of shelterin assembly dysfunction. It will be interesting to further investigate the role of this isoform and its contribution to the development of cancer, if any.
Similarly, in cancers that were found to overexpress TIN2, an upregulation of TRF1 and TRF2 was observed as well [162, 163, 175, 177, 179]. However, whether this upregulation of TIN2 is a direct consequence of the overexpression of TRF1 and/or TRF2, or vice versa, is not known. A study has shown that TIN2 was overexpressed in more than half of the breast cancer cell lines studied, and its silencing by shRNA caused a decrease in cell proliferation and migration [216]. Although the expression of TRF1 and TRF2 was not examined in this study, it is likely that the silencing of TIN2 would also cause the suppression of TRF1 and TRF2.
TIN2 has also been implicated in metabolism where it has also been found to be localized in the mitochondria [217, 218]. This localisation of TIN2 induces a morphological change causing the mitochondria to take on a more spherical shape in which ATP production capacity is decreased [218]. In addition, mitochondrial reactive oxygen species (ROS) production is increased along with HIF-1 activation which has been implicated in cancer [218, 219].
Given TIN2′s interaction with other components of the shelterin complex, subcomplexes containing TIN2 have been isolated from extracts obtained by nuclear extraction with differing salt concentrations [220]. It has been suggested that the two major TIN2-containing subcomplexes identified, TIN2-TRF1 and TIN2-TRF2/RAP1-TPP1/POT1, bind to and function at different locations along telomeres [220]. In vitro experiments have shown that the TIN2-TRF1 subcomplex regulates interactions between telomeric tracts [221], whereas the TIN2-TRF2/RAP1-TPP1/POT1 subcomplex protects telomere ends and prevents chromosome fusions by ensuring proper formation of the t-loop structure [220]. Interestingly, it has been observed that despite the absence of p53, the disruption of the TIN2-TRF2/RAP1-TPP1/POT1 subcomplex led to eventual cell death by causing severe genomic damage and mitotic catastrophe, making this subcomplex an attractive target of anticancer therapies [220]. In a more recent study, two other stable subcomplexes, TIN2-TRF2/RAP1 and TIN2-POT1/TPP1, were purified and structurally characterized [222]. However, whether these subcomplexes have distinct functions and roles in regulating telomeres remain to be elucidated Table 6.
RAP1 in cancer
The upregulation of RAP1 has been detected in a few cancers with short telomeres [224, 225]. However, the role of RAP1 in directly regulating telomeres is debatable as contradictory results have been reported [226, 227]. RAP1 functions to protect telomeres by suppressing homology-directed repair which can lead to telomere recombination events and changes in telomere lengths [228]. In particular, RAP1 has been found to protect critically short telomeres, in which its downregulation in senescent cells led to increased telomeric instability [229, 230].
RAP1 has non-telomeric roles, such as in metabolism whereby RAP1-knockout mice were observed to have fatty livers and higher risks of developing hepatocellular carcinomas [231]. Upon the treatment of carcinogen, DEN, Rap1-/- mice developed malignant tumors and had reduced survival as compared to wild-type mice [231]. Unlike human RAP1, murine RAP1 is not essential for the protection and maintenance of telomeres [231, 232], further implying its function as a non-telomeric protein. Chromatin immunoprecipitation experiments have also revealed that RAP1 binds to extratelomeric sites and is capable of regulating gene expression [199, 232]. In the cytoplasm, RAP1 complexes with IKKs to activate NF-κB signaling pathways that is crucial in cancer [233]. The presence of two conserved NF-κB binding sites in the RAP1 promoter could also further promote the development of cancer via a feed-forward mechanism [233,234,235,236,237]. In breast cancer, levels of RAP1 and NF-κB were highly correlated and associated with higher cancer grades [233], making RAP1 a good marker of prognosis Table 7.
TPP1 in cancer
The binding of TPP1 to POT1 is essential in the role of chromosome ends protection by POT1. A study has demonstrated that in the absence of TPP1, POT1 does not bind telomeres and loses its protective function [243, 244]. The downregulation of TPP1, as observed in some human malignancies (Table 8), could therefore indirectly elicit chromosomal instability and promote the development of cancer. TPP1 is also fundamental to the recruitment of telomerase to telomeres, in which mutations in its oligonucleotide/oligosaccharide-binding (OB)-fold domain prevents it interaction with TERC and reduces telomerase localization to telomeres [245, 246]. Cancers that were found to overexpress TPP1 (Table 8) were reported to have increased telomere length and were associated with more aggressive disease with poorer prognosis. Unlike the other shelterin proteins discussed earlier, rare deleterious mutations have been detected in TPP1 that were linked to a predisposition to cancer [247,248,249,250]. Site-directed mutagenesis in leukemia cell lines found that a mutation in TPP1 led to telomere elongation, prolonged proliferation and protection against apoptosis [247]. On the contrary, a mutation found in the TIN2-binding domain of TPP1 was found to hinder telomerase activity and cause shortened telomeres instead, but it similarly led to cell proliferation [250]. Other mutations in TIN2 were mostly found in its POT1-binding domain and were predicted to disrupt the formation of functional shelterin complexes [248]. Furthermore, these loss-of-function mutations were detected in patients presented with early onset melanoma, suggesting that dysregulation in shelterin could accelerate the development of cancer.
POT1 in cancer
Similar to TRF1 and TRF2, POT1 acts as a negative regulator of telomere length by binding strongly to the single-stranded 3′-overhang of telomeres and hindering the access of telomerase. This binding is regulated by TRF1 and its inhibition has led to telomere elongation [253, 254]. Similar to that of RAP1, POT1 also protects chromosome ends by preventing homologous recombination. Loss of POT1 leads to accumulation of anaphase bridges and chromosomal fusions which promotes cancer development in mice [255]. POT1 downregulation in human malignancies was associated with telomere dysfunction and formation of anaphase bridges [256, 257]. Patients diagnosed with these cancers also had poorer prognoses. Nonetheless, as with the other shelterin proteins, upregulation of POT1 has also been detected in cancer (Table 9). It has been suggested that the upregulation of POT1 was necessary in restoring and maintaining 3′-overhang lengths in telomerase reactivated cancers, so as to prevent DNA damage response activation and to prolong survival and proliferation of cancer cells [258]. Numerous deleterious mutations in POT1 have been identified in different human cancers [259], many of which exist in the DNA-binding domain and disrupt POT1′s binding to 3′-overhangs, promoting telomere elongation and chromosomal instability. A handful of mutations occur in the TPP1-binding domain. In particular, POT1 has been found to be commonly mutated in CLL [260, 261]. This suggests that POT1 may play a crucial role in the disease causing mechanisms of CLL and POT1-related therapeutic strategies can be a potential new angle in treating this disease. In familial melanoma, mutations in POT1 have also been identified and carriers of these mutations are more susceptible to cancer development [90, 262, 263]. As such, POT1 has been identified as a high penetrant gene in these cancers and has been suggested to be included in gene panel testing for families that come in for screening.
Conclusion
Increasing evidence suggests various non-canonical roles of telomeric proteins in cancer development and cellular homeostasis. These roles are hard to differentiate from their telomeric functions as anomalies associated with telomere length, such as genomic instability, can lead to cancer development as well. This is further complicated by the absence of good reagents and methods to distinguish between its telomeric and non-telomeric functions. To resolve this dilemma, comparison between wild-type TERT and its dominant-negative form named DN-TERT (catalytically inactive for its reverse transcriptase activity) has been proposed to discern the non-canonical role in cancer development and aging-related diseases. Indeed, deciphering of the canonical function of TERT has been investigated by many groups. Over-expression of WT-TERT in aged mice reverses the telomere shortening effects of aging by elongating their telomeres and extending their life span, whereas overexpression of DN-TERT is incapable of doing the same. This study clearly proves that TERT is critical in extending longevity and delaying physiological aging, and that its reverse transcriptase activity is absolutely required for this function [270]. However, the non-telomeric functions of telomerase still remain unaddressed and further experiments are necessary to understand how the non-canonical roles of TERT regulate gene expression and cancer progression. As telomerase influences gene expression in collaboration with other oncogenic proteins in cancer, it is hard to identify the exact molecular mechanisms involved in its gene regulatory function as a co-transcription factor. Hence, DN-TERT is an excellent tool for studying TERT’s role in gene expression regulation by assessing occupancy of TERT on specific genomic regions under various physiological conditions. This is fundamental in deepening our understanding of the telomeric vs. non-telomeric functions of TERT in cancer development and other diseases.
Similar to the telomerase complex molecules, shelterin proteins exhibit non-telomeric roles by regulating cancer-specific gene expression directly or indirectly in tumors. Canonically, expression of shelterin proteins is expected to be mainly downregulated in the course of cancer development and progression. Loss of these proteins make chromosome ends vulnerable to telomere loss, which leads to genetic instability. Interestingly, expression levels among the different shelterin proteins are highly correlated and loss of a particular shelterin protein generally leads to concurrent decrease of its associated protein level. These findings suggest a common transcriptional regulatory mechanism for the expression of these genes. Indeed, genes that encode the shelterin proteins have been shown to be regulated epigenetically in breast cancer. Cells treated with 5-Aza-CdR, an epigenetic drug that targets DNA methylation, enhances expression of shelterin and shelterin-associated genes and increases telomere length [271], supporting the notion that shelterin genes are transcriptionally regulated by a common mechanism.
All these findings indicate that most of these telomeric proteins play a significant role in cancer progression apart from their telomeric functions. Although the general outcome is enhanced cell proliferation, increased malignancy and genomic instability, the exact molecular mechanisms are yet to be thoroughly understood.
As discussed earlier, beta-catenin and c-MYC promote the expression of TERT, which in turn interacts back with these oncogenic drivers, to regulate the expression of cancer-specific genes in what appears to be a feed-forward loop. Given that the expression of shelterin and some of the telomerase complex molecules are significantly correlated during the course of cancer progression, it is highly likely that other such interactions exist between telomeric proteins and telomere-associated molecules. Therefore, it would be critical to perform biochemical assays to identify novel binding partners using both telomerase-dependent and independent cells under various cell growth conditions or oncogenic stimuli. To understand the underlying molecular mechanisms and investigate the dependencies on other telomeric molecules for its function, overexpression and knockdown of each telomeric protein individually and in combination will be beneficial in deciphering the sequence of events and in identifying context-dependent associated functions in the course of carcinogenesis. The downstream targets identified from these preliminary biochemical analyses will serve as a good starting point to further elucidate any non-telomeric roles that each telomeric protein may play in cancer progression. Additionally, genome-wide correlation analyses of these telomeric proteins and their targets may be useful in identifying specific gene expression signatures and molecular mediators that could serve as unique targets under specific biochemical processes or disease conditions. Furthermore, these multidimensional correlation analyses would deepen our understanding of the molecular mechanisms underlying telomere regulation in cancer, and allow us to design disease/pathway-associated reporter systems by utilizing the latest CRISPR-based high-throughput screening methods to identify novel therapeutic targets. This will also allow us to target the specific roles of these telomeric proteins in cancer without causing pro-aging effects.
Chromatin immunoprecipitation analyses revealed that in cancer, shelterin proteins (TRFs and RAP1) and TERT, apart from telomeric regions, also bind to intronic and distal promoter regions [198, 199]. Although the number of binding sites are limited for each protein, the genes located in these loci have important functions for cancer metabolism. Hence, it might prove intriguing to block or remove their binding through genome editing of these binding sites, and determine the functional outcome of such interactions in cancer progression.
Telomerase targeting approaches for cancer therapy have attracted the attention of many cancer researchers over the years. However, to date, all the telomerase inhibitors failed in clinical trials due to undesirable cytotoxic events as these molecules are crucial for telomere maintenance in stem cells and germ cells. Additionally, inhibiting telomerase has been shown to activate the ALT mechanism [272], a homologous recombination-based telomere maintenance mechanism observed in 15% of cancers [273,274,275], as a resistance strategy in cancer cells. Therefore, there is a pressing need to identify cancer-specific molecular interactors of telomeric molecules and their downstream targets to generate novel therapeutic strategies for cancer. While cancer-specific mutations often lead to malignancies, they also present as a great opportunity for cancer-specific growth inhibition. One of the recent examples was a study focusing on cancer-specific TERT promoter mutations which are observed in ~ 19% of the cancers but not in healthy tissues [48]. These promoter mutations mediate a unique regulatory mechanism that is specific to activation of mutant TERT promoter, hence targeting this particular mechanism would eliminate any potential cytotoxic effects on the stem cell compartment.
In conclusion, emerging evidence suggests that telomerase and shelterin components have key roles in cancer progression, independent of their telomere-associated functions (Fig. 1). It is important to extend our understanding of these molecular mechanisms and dynamics in physiological conditions so as to identify more efficient therapeutic strategies for cancer.

Summary of the roles of telomerase and shelterin complex
References
Moyzis RK et al (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A 85(18):6622–6626
Blackburn EH (2000) The end of the (DNA) line. Nat Struct Biol 7(10):847–850
von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem Sci 27(7):339–344
Greider CW, Blackburn EH (1987) The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51(6):887–898
Huffman KE et al (2000) Telomere shortening is proportional to the size of the G-rich telomeric 3’-overhang. J Biol Chem 275(26):19719–19722
Greider CW (2012) Molecular biology. Wnt regulates TERT–putting the horse before the cart. Science 336(6088):1519–1520
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19(18):2100–2110
Rha SY et al (2000) Effect of telomere and telomerase interactive agents on human tumor and normal cell lines. Clin Cancer Res 6(3):987–993
Teixeira MT et al (2004) Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell 117(3):323–335
Schmidt JC, Zaug AJ (2016) Cech, TR live cell imaging reveals the dynamics of telomerase recruitment to telomeres. Cell 166(5):1188–1197
Armstrong CA, Tomita K (2017) Fundamental mechanisms of telomerase action in yeasts and mammals: understanding telomeres and telomerase in cancer cells. Open Biol 7(3):160338
Guterres AN, Villanueva J (2020) Targeting telomerase for cancer therapy. Oncogene 39(36):5811–5824
Rousseau P, Autexier C (2015) Telomere biology: Rationale for diagnostics and therapeutics in cancer. RNA Biol 12(10):1078–1082
Shay JW (2016) Role of telomeres and telomerase in aging and cancer. Cancer Discov 6(6):584–593
Fernandes SG et al (2020) Role of telomeres and telomeric proteins in human malignancies and their therapeutic potential. Cancers (Basel) 12(7):1901
Greider CW, Blackburn EH (1989) A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337(6205):331–337
Yu GL et al (1990) In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature 344(6262):126–132
Chew CL et al (2018) Noncoding RNAs: master regulators of inflammatory signaling. Trends Mol Med 24(1):66–84
Weinrich SL et al (1997) Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet 17(4):498–502
Cohen SB et al (2007) Protein composition of catalytically active human telomerase from immortal cells. Science 315(5820):1850–1853
Nguyen THD et al (2018) Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557(7704):190–195
Jády BTE, Bertrand E (2004) Kiss TS Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body–specific localization signal. J Cell Biol 164(5):647–652
Venteicher AS et al (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323(5914):644–648
Mitchell JR, Cheng J, Collins K (1999) A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3’ end. Mol Cell Biol 19(1):567–576
Collins K (2006) The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol 7(7):484–494
Darzacq X et al (2006) Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J Cell Biol 173(2):207–218
Mason PJ, Bessler M (2011) The genetics of dyskeratosis congenita. Cancer Genet 204(12):635–645
Grozdanov PN et al (2009) SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 15(6):1188–1197
Venteicher AS et al (2008) Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132(6):945–957
Machado-Pinilla R et al (2012) Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18(10):1833–1845
Cristofari G et al (2007) Human telomerase RNA accumulation in cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol Cell 27(6):882–889
Bodnar AG et al (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279(5349):349–352
Cao Y, Bryan TM, Reddel RR (2008) Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci 99(6):1092–1099
Akincilar SC, Unal B, Tergaonkar V (2016) Reactivation of telomerase in cancer. Cell Mol Life Sci 73(8):1659–1670
Rooney PH et al (1999) Comparative genomic hybridization and chromosomal instability in solid tumours. Br J Cancer 80(5–6):862–873
Knuutila S et al (1998) DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol 152(5):1107–1123
Mosse YP et al (2005) High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer 43(4):390–403
Coe BP et al (2005) High-resolution chromosome arm 5p array CGH analysis of small cell lung carcinoma cell lines. Genes Chromosomes Cancer 42(3):308–313
Killela PJ et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 110(15):6021–6026
Griewank KG et al (2013) TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br J Cancer 109(2):497–501
Landa I et al (2013) Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab 98(9):E1562–E1566
Vinagre J et al (2013) Frequency of TERT promoter mutations in human cancers. Nat Commun 4:2185
Borah S et al (2015) Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 347(6225):1006–1010
Li Y et al (2016) Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci U S A 113(50):14402–14407
Li Y et al (2015) Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol 17(10):1327–1338
Xu X et al (2018) Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat Commun 9(1):3183
Bell RJ et al (2015) Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348(6238):1036–1039
Akincilar SC et al (2016) Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov 6(11):1276–1291
Lindvall C et al (2003) Molecular characterization of human telomerase reverse transcriptase-immortalized human fibroblasts by gene expression profiling: activation of the epiregulin gene. Cancer Res 63(8):1743–1747
Tomlinson RL et al (2006) Cell cycle-regulated trafficking of human telomerase to telomeres. Mol Biol Cell 17(2):955–965
Zhong F et al (2011) Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 25(1):11–16
Chiodi I, Mondello C (2012) Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front Oncol 2:133
Ozturk MB, Li Y, Tergaonkar V (2017) Current insights to regulation and role of telomerase in human diseases. Antioxidants (Basel) 6(1):17
Liu H et al (2016) hTERT promotes cell adhesion and migration independent of telomerase activity. Sci Rep 6:22886
Zhou L et al (2009) Telomerase reverse transcriptase activates the expression of vascular endothelial growth factor independent of telomerase activity. Biochem Biophys Res Commun 386(4):739–743
Rahman R, Latonen L, Wiman KG (2005) hTERT antagonizes p53-induced apoptosis independently of telomerase activity. Oncogene 24(8):1320–1327
Shin WH, Chung KC (2020) Human telomerase reverse transcriptase positively regulates mitophagy by inhibiting the processing and cytoplasmic release of mitochondrial PINK1. Cell Death Dis 11(6):425
Ahmed S et al (2008) Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci 121(Pt 7):1046–1053
Wu L et al (2020) Telomerase: Key regulator of inflammation and cancer. Pharmacol Res 155:104726
Indran IR, Hande MP, Pervaiz S (2011) hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res 71(1):266–276
Wang J, Hannon GJ, Beach DH (2000) Risky immortalization by telomerase. Nature 405(6788):755–756
Xiang H et al (2002) Human telomerase accelerates growth of lens epithelial cells through regulation of the genes mediating RB/E2F pathway. Oncogene 21(23):3784–3791
Farwell DG et al (2000) Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol 156(5):1537–1547
Park JI et al (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460(7251):66–72
Koh CM et al (2015) Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest 125(5):2109–2122
Khattar E, Tergaonkar V (2017) Transcriptional regulation of telomerase reverse transcriptase (TERT) by MYC. Front Cell Dev Biol 5:1
Herbst A et al (2014) Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genom 15:74
Akincilar SC et al (2015) Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett 589(9):974–984
Khattar E et al (2016) Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J Clin Invest 126(10):4045–4060
Yamamoto Y, Gaynor RB (2004) IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci 29(2):72–79
Akincilar SC et al (2020) NAIL: an evolutionarily conserved lncRNA essential for licensing coordinated activation of p38 and NFkappaB in colitis. Gut. https://doi.org/10.1136/gutjnl-2020-322980
Zhang Q, Lenardo MJ, Baltimore D (2017) 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168(1–2):37–57
Ghosh A et al (2012) Telomerase directly regulates NF-kappaB-dependent transcription. Nat Cell Biol 14(12):1270–1281
Puar YR et al (2018) Evidence for the involvement of the master transcription factor NF-kappaB in cancer initiation and progression. Biomedicines 6(3):82
Gonzalez OG et al (2014) Telomerase stimulates ribosomal DNA transcription under hyperproliferative conditions. Nat Commun 5:4599
Akiyama M et al (2003) Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res 63(1):18–21
Hu Y et al (2019) The clinicopathological correlations of hTERC amplification with esophageal squamous cell precursor lesions. Dig Dis Sci 64(1):68–75
Soder AI et al (1997) Amplification, increased dosage and in situ expression of the telomerase RNA gene in human cancer. Oncogene 14(9):1013–1021
Soder AI et al (1998) Tumour specific regulation of telomerase RNA gene expression visualized by in situ hybridization. Oncogene 16(8):979–983
Storti CB et al (2020) Telomere-associated genes and telomeric lncRNAs are biomarker candidates in lung squamous cell carcinoma (LUSC). Exp Mol Pathol 112:104354
Barthel FP et al (2017) Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 49(3):349–357
Kheimar A et al (2019) Overexpression of cellular telomerase RNA enhances virus-induced cancer formation. Oncogene 38(10):1778–1786
Trapp S et al (2006) A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J Exp Med 203(5):1307–1317
Li Y et al (2011) Telomerase inhibition strategies by siRNAs against either hTR or hTERT in oral squamous cell carcinoma. Cancer Gene Ther 18(5):318–325
Wen R et al (2006) Attenuation of telomerase activity by siRNA targeted telomerase RNA leads to apoptosis and inhibition of proliferation in human renal carcinoma cells. Chinese J Clin Oncol 3(5):326–331
Feng J et al (1995) The RNA component of human telomerase. Science 269(5228):1236–1241
Cayuela ML, Flores JM, Blasco MA (2005) The telomerase RNA component Terc is required for the tumour-promoting effects of Tert overexpression. EMBO Rep 6(3):268–274
Chiba K et al (2015) Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. https://doi.org/10.7554/eLife.07918
González-Suárez E et al (2000) Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 26(1):114–117
Shi J et al (2014) Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 46(5):482–486
Yamaguchi H et al (2003) Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 102(3):916–918
Boyraz B et al (2016) Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J Clin Invest 126(9):3377–3382
Tummala H et al (2015) Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest 125(5):2151–2160
Jones AM et al (2012) TERC polymorphisms are associated both with susceptibility to colorectal cancer and with longer telomeres. Gut 61(2):248–254
Pooley KA et al (2010) Telomere length in prospective and retrospective cancer case-control studies. Cancer Res 70(8):3170–3176
Meier UT, Blobel G (1994) NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J Cell Biol 127(6 Pt 1):1505–1514
Hamma T, Ferre-D’Amare AR (2006) Pseudouridine synthases. Chem Biol 13(11):1125–1135
Hou P et al (2020) DKC1 enhances angiogenesis by promoting HIF-1alpha transcription and facilitates metastasis in colorectal cancer. Br J Cancer 122(5):668–679
Alawi F, Lee MN (2007) DKC1 is a direct and conserved transcriptional target of c-MYC. Biochem Biophys Res Commun 362(4):893–898
Alawi F et al (2011) Correlation of dyskerin expression with active proliferation independent of telomerase. Head Neck 33(7):1041–1051
Westermann F et al (2007) High Skp2 expression characterizes high-risk neuroblastomas independent of MYCN status. Clin Cancer Res 13(16):4695–4703
Piva R et al (2006) Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest 116(12):3171–3182
McDonald SL et al (2004) Expression analysis of genes identified by molecular profiling of VGP melanomas and MGP melanoma-positive lymph nodes. Cancer Biol Ther 3(1):110–120
Montanaro L et al (2006) Dyskerin expression influences the level of ribosomal RNA pseudo-uridylation and telomerase RNA component in human breast cancer. J Pathol 210(1):10–18
Montanaro L et al (2008) Relationship between dyskerin expression and telomerase activity in human breast cancer. Cell Oncol 30(6):483–490
Sieron P et al (2009) DKC1 overexpression associated with prostate cancer progression. Br J Cancer 101(8):1410–1416
Witkowska A et al (2010) Expression profile of significant immortalization genes in colon cancer. Int J Mol Med 25(3):321–329
Schaner ME et al (2003) Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14(11):4376–4386
Liu B et al (2012) Dyskerin overexpression in human hepatocellular carcinoma is associated with advanced clinical stage and poor patient prognosis. PLoS ONE 7(8):e43147
Sbarrato T et al (2016) A ribosome-related signature in peripheral blood CLL B cells is linked to reduced survival following treatment. Cell Death Dis 7(6):e2249
O’Brien R et al (2016) MYC-Driven Neuroblastomas Are Addicted to a Telomerase-Independent Function of Dyskerin. Cancer Res 76(12):3604–3617
McCaul JA et al (2002) Telomerase inhibition and the future management of head-and-neck cancer. Lancet Oncol 3(5):280–288
Walne AJ et al (2007) Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet 16(13):1619–1629
Kannengiesser C et al (2020) First heterozygous <em>NOP10</em> mutation in familial pulmonary fibrosis. Eur Respir J 55(6):1902465
Dos Santos PC et al (2017) Dysregulation of H/ACA ribonucleoprotein components in chronic lymphocytic leukemia. PLoS ONE 12(6):e0179883
Kim MS et al (2012) Expressional analysis of NOLA1, NOLA2, NOLA3 and DKC1, the core proteins in H/ACA riboproteins, in gastric and colorectal cancers. Pathology 44(6):576–577
Pigullo S et al (2009) NOLA1 gene mutations in acquired aplastic anemia. Pediatr Blood Cancer 52(3):376–378
Vulliamy T et al (2008) Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci 105(23):8073–8078
Sun CK et al (2014) TCAB1: a potential target for diagnosis and therapy of head and neck carcinomas. Mol Cancer 13:180
Mahmoudi S et al (2011) WRAP53 promotes cancer cell survival and is a potential target for cancer therapy. Cell Death Dis 2(1):e114–e114
Yuan P et al (2014) Telomerase Cajal body protein 1 depletion inhibits telomerase trafficking to telomeres and induces G(1) cell cycle arrest in A549 cells. Oncol Lett 8(3):1009–1016
Wang K et al (2017) Epstein-Barr virus-induced up-regulation of TCAB1 is involved in the DNA damage response in nasopharyngeal carcinoma. Sci Rep 7(1):3218
Hedström E et al (2015) Downregulation of the cancer susceptibility protein WRAP53β in epithelial ovarian cancer leads to defective DNA repair and poor clinical outcome. Cell Death Dis 6(10):e1892–e1892
Garvin S et al (2015) Nuclear expression of WRAP53β is associated with a positive response to radiotherapy and improved overall survival in patients with head and neck squamous cell carcinoma. Oral Oncol 51(1):24–30
Tiefenböck-Hansson K et al (2017) WRAP53β, survivin and p16INK4a expression as potential predictors of radiotherapy/chemoradiotherapy response in T2N0-T3N0 glottic laryngeal cancer. Oncol Rep 38(4):2062–2068
Rassoolzadeh H et al (2016) Overexpression of the scaffold WD40 protein WRAP53β enhances the repair of and cell survival from DNA double-strand breaks. Cell Death Dis 7(6):e2267
Huber O et al (2008) Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res 68(17):6873–6876
Cvackova Z et al (2008) Pontin is localized in nucleolar fibrillar centers. Chromosoma 117(5):487–497
Dalvai M et al (2013) H2A.Z-dependent crosstalk between enhancer and promoter regulates cyclin D1 expression. Oncogene 32(36):4243–4251
Kim JH et al (2005) Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature 434(7035):921–926
Etard C et al (2005) Pontin and Reptin regulate cell proliferation in early Xenopus embryos in collaboration with c-Myc and Miz-1. Mech Dev 122(4):545–556
Magalska A et al (2014) RuvB-like ATPases function in chromatin decondensation at the end of mitosis. Dev Cell 31(3):305–318
Muller PA, Vousden KH (2013) p53 mutations in cancer. Nat Cell Biol 15(1):2–8
Zhao Y et al (2015) Pontin, a new mutant p53-binding protein, promotes gain-of-function of mutant p53. Cell Death Differ 22(11):1824–1836
Maslon MM et al (2010) A divergent substrate-binding loop within the pro-oncogenic protein anterior gradient-2 forms a docking site for Reptin. J Mol Biol 404(3):418–438
Gray TA et al (2013) Development of a fluorescent monoclonal antibody-based assay to measure the allosteric effects of synthetic peptides on self-oligomerization of AGR2 protein. Protein Sci 22(9):1266–1278
Clarke DJ et al (2016) Mass spectrometry analysis of the oxidation states of the pro-oncogenic protein anterior gradient-2 reveals covalent dimerization via an intermolecular disulphide bond. Biochim Biophys Acta 1864(5):551–561
Mao YQ, Houry WA (2017) The role of pontin and reptin in cellular physiology and cancer etiology. Front Mol Biosci 4:58
Xie C et al (2012) RUVBL2 is a novel repressor of ARF transcription. FEBS Lett 586(4):435–441
Perez-Perri JI et al (2016) The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep 16(1):37–47
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11(6):393–410
Lee JS et al (2010) Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell 39(1):71–85
Lee JS et al (2011) Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci U S A 108(33):13510–13515
Matias PM et al (2015) The AAA+ proteins Pontin and Reptin enter adult age: from understanding their basic biology to the identification of selective inhibitors. Front Mol Biosci 2:17
Fairall L et al (2001) Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell 8(2):351–361
Baumann P, Cech TR (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292(5519):1171–1175
O’Connor MS et al (2006) A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc Natl Acad Sci 103(32):11874–11879
Kim SH, Kaminker P, Campisi J (1999) TIN2, a new regulator of telomere length in human cells. Nat Genet 23(4):405–412
Houghtaling BR et al (2004) A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol 14(18):1621–1631
Ye JZ et al (2004) TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem 279(45):47264–47271
Liu D et al (2004) PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 6(7):673–680
Ye JZ-S et al (2004) POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev 18(14):1649–1654
Li B, Oestreich S, de Lange T (2000) Identification of human Rap1: implications for telomere evolution. Cell 101(5):471–483
van Steensel B, de Lange T (1997) Control of telomere length by the human telomeric protein TRF1. Nature 385(6618):740–743
Broccoli D et al (1997) Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet 17(2):231–235
Sfeir A et al (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138(1):90–103
Martínez P et al (2009) Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23(17):2060–2075
Poonepalli A et al (2008) Telomere-mediated genomic instability and the clinico-pathological parameters in breast cancer. Genes Chromosomes Cancer 47(12):1098–1109
Kishi S et al (2001) Telomeric protein Pin2/TRF1 induces mitotic entry and apoptosis in cells with short telomeres and is down-regulated in human breast tumors. Oncogene 20(12):1497–1508
Heng J et al (2017) Integrated analysis of promoter methylation and expression of telomere related genes in breast cancer. Oncotarget 8(15):25442–25454
Dinami R et al (2014) miR-155 Drives Telomere Fragility in Human Breast Cancer by Targeting TRF1. Can Res 74(15):4145–4156
Oh B-K et al (2005) Up-regulation of telomere-binding proteins, TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am J Pathol 166(1):73–80
Hu H et al (2010) Expression of TRF1, TRF2, TIN2, TERT, KU70, and BRCA1 proteins is associated with telomere shortening and may contribute to multistage carcinogenesis of gastric cancer. J Cancer Res Clin Oncol 136(9):1407–1414
Lantuejoul S et al (2010) Telomere maintenance and DNA damage responses during lung carcinogenesis. Clin Cancer Res 16(11):2979–2988
La Torre D et al (2013) Telomere length modulation in human astroglial brain tumors. PLoS ONE 8(5):e64296
Marión RM et al (2017) Common telomere changes during in vivo reprogramming and early stages of tumorigenesis. Stem Cell Rep 8(2):460–475
Schneider RP et al (2013) TRF1 is a stem cell marker and is essential for the generation of induced pluripotent stem cells. Nature Commun 4(1):1946
Kanauchi H et al (2003) Diagnostic and prognostic value of fas and telomeric-repeat binding factor-1 genes in adrenal tumors. J Clin Endocrinol Metabol 88(8):3690–3693
Garcia-Aranda C et al (2006) Correlations of telomere length, telomerase activity, and telomeric-repeat binding factor 1 expression in colorectal carcinoma. Cancer 106(3):541–551
Kojima K et al (2011) Telomerase activation without shortening of telomeric 3’-overhang is a poor prognostic factor in human colorectal cancer. Cancer Sci 102(2):330–335
Raynaud CM et al (2008) Telomere shortening is correlated with the DNA damage response and telomeric protein down-regulation in colorectal preneoplastic lesions. Ann Oncol 19(11):1875–1881
Miyachi K et al (2002) Correlation between telomerase activity and telomeric-repeat binding factors in gastric cancer. J Exp Clin Cancer Res 21(2):269–275
Matsutani N et al (2001) Expression of telomeric repeat binding factor 1 and 2 and TRF1-interacting nuclear protein 2 in human gastric carcinomas. Int J Oncol 19(3):507–512
Bejarano L et al (2017) Inhibition of TRF1 telomere protein impairs tumor initiation and progression in glioblastoma mouse models and patient-derived xenografts. Cancer Cell 32(5):590-607.e4
Lee JE et al (2008) Telomeric 3’ overhangs in chronic HBV-related hepatitis and hepatocellular carcinoma. Int J Cancer 123(2):264–272
Igarashi M et al (2003) Interferon can block telomere erosion and in rare cases result in hepatocellular carcinoma development with telomeric repeat binding factor 1 overexpression in chronic hepatitis C. Clin Cancer Res 9(14):5264–5270
Yokota T et al (2003) Telomere length variation and maintenance in hepatocarcinogenesis. Cancer 98(1):110–118
Ohyashiki JH et al (2001) Impaired telomere regulation mechanism by TRF1 (telomere-binding protein), but not TRF2 expression, in acute leukemia cells. Int J Oncol 18(3):593–598
Bellon M et al (2006) Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int J Cancer 119(9):2090–2097
Poncet D et al (2008) Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B-chronic lymphocytic leukemia. Blood 111(4):2388–2391
Véronèse L et al (2013) Telomeres and chromosomal instability in chronic lymphocytic leukemia. Leukemia 27(2):490–493
Guièze R et al (2016) Telomere status in chronic lymphocytic leukemia with TP53 disruption. Oncotarget 7(35):56976–56985
Campbell LJ et al (2006) hTERT, the catalytic component of telomerase, is downregulated in the haematopoietic stem cells of patients with chronic myeloid leukaemia. Leukemia 20(4):671–679
Nakanishi K et al (2003) Expression of mRNAs for telomeric repeat binding factor (TRF)-1 and TRF2 in atypical adenomatous hyperplasia and adenocarcinoma of the lung. Clin Cancer Res 9(3):1105–1111
Hu J et al (2006) Expression of telomeric repeat binding factor 1 in non-small cell lung cancer. J Surg Oncol 93(1):62–67
Lin X et al (2006) Expression of telomere-associated genes as prognostic markers for overall survival in patients with non-small cell lung cancer. Clin Cancer Res 12(19):5720–5725
Yajima T et al (2001) Telomerase reverse transcriptase and telomeric-repeat binding factor protein 1 as regulators of telomerase activity in pancreatic cancer cells. Br J Cancer 85(5):752–757
Chen C et al (2017) Structural insights into POT1-TPP1 interaction and POT1 C-terminal mutations in human cancer. Nature Commun 8:14929–14929
Pal D et al (2015) Over-expression of telomere binding factors (TRF1 & TRF2) in renal cell carcinoma and their inhibition by using SiRNA induce apoptosis, reduce cell proliferation and migration invitro. PLoS ONE 10(3):e0115651–e0115651
van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92(3):401–413
Smogorzewska A et al (2000) Control of human telomere length by TRF1 and TRF2. Mol Cell Biol 20(5):1659–1668
Yamada K et al (2002) Decreased gene expression for telomeric-repeat binding factors and TIN2 in malignant hematopoietic cells. Anticancer Res 22(2b):1315–1320
Yamada M et al (2002) Down-regulation of TRF1, TRF2 and TIN2 genes is important to maintain telomeric DNA for gastric cancers. Anticancer Res 22(6a):3303–3307
Chuang H-C et al (2011) Reduced expression of TRF1 is associated with tumor progression and poor prognosis in oral squamous cell carcinoma. Experim Therapeutic Med 2(1):63–67
Nera B et al (2015) Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun 6:10132
Karlseder J, Smogorzewska A, de Lange T (2002) Senescence induced by altered telomere state, not telomere loss. Science 295(5564):2446–2449
Diala I et al (2013) Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep 14(4):356–363
Simonet T et al (2011) The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 21(7):1028–1038
Yang D et al (2011) Human telomeric proteins occupy selective interstitial sites. Cell Res 21(7):1013–1027
Biroccio A et al (2013) TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat Cell Biol 15(7):818–828
Cherfils-Vicini J et al (2019) Cancer cells induce immune escape via glycocalyx changes controlled by the telomeric protein TRF2. EMBO J 38(11):e100012
El Maï M et al (2014) The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep 9(3):1047–1060
Zizza P et al (2019) TRF2 positively regulates SULF2 expression increasing VEGF-A release and activity in tumor microenvironment. Nucleic Acids Res 47(7):3365–3382
Dong W et al (2009) Sp1 upregulates expression of TRF2 and TRF2 inhibition reduces tumorigenesis in human colorectal carcinoma cells. Cancer Biol Ther 8(22):2166–2174
Diehl MC et al (2011) Elevated TRF2 in advanced breast cancers with short telomeres. Breast Cancer Res Treat 127(3):623–630
Bai Y et al (2014) Molecular targeting of TRF2 suppresses the growth and tumorigenesis of glioblastoma stem cells. Glia 62(10):1687–1698
Roy S et al (2018) p38 MAPK pathway and its interaction with TRF2 in cisplatin induced chemotherapeutic response in head and neck cancer. Oncogenesis 7(7):53
Klapper W et al (2003) DNA damage transiently increases TRF2 mRNA expression and telomerase activity. Leukemia 17(10):2007–2015
Knecht H, Mai S (2017) LMP1 and dynamic progressive telomere dysfunction: a major culprit in EBV-associated hodgkin’s lymphoma. Viruses 9(7):164
Lajoie V et al (2015) LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 125(13):2101–2110
Frías C et al (2008) Telomere shortening is associated with poor prognosis and telomerase activity correlates with DNA repair impairment in non-small cell lung cancer. Lung Cancer 60(3):416–425
Muñoz P et al (2005) XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat Genet 37(10):1063–1071
Ishdorj G et al (2017) A novel spliced variant of the TIN2 shelterin is present in chronic lymphocytic leukemia. Leuk Res 59:66–74
Kim SH et al (2004) TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem 279(42):43799–43804
Augereau A et al (2011) Telomeric damage in early stage of chronic lymphocytic leukemia correlates with shelterin dysregulation. Blood 118(5):1316–1322
Gao R et al (2015) Targeting of DNA damage signaling pathway induced senescence and reduced migration of cancer cells. J Gerontol A Biol Sci Med Sci 70(6):701–713
Billard P, Poncet DA (2019) Replication stress at telomeric and mitochondrial DNA: common origins and consequences on ageing. Int J Mol Sci 20(19):4959
Chen LY et al (2012) Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol Cell 47(6):839–850
Lee JH et al (2018) Loss of RNA-binding protein HuR facilitates cellular senescence through posttranscriptional regulation of TIN2 mRNA. Nucleic Acids Res 46(8):4271–4285
Kim S-H et al (2008) Telomere dysfunction and cell survival: roles for distinct TIN2-containing complexes. J Cell Biol 181(3):447–460
Kim S-H et al (2003) The human telomere-associated protein TIN2 stimulates interactions between telomeric DNA tracts in vitro. EMBO Rep 4(7):685–691
Oviya I (2018) Biochemical and structural analysis of shelterin subcomplexes. Nanyang Technological University, Nanyang
Kim S-H et al (2010) Androgen receptor interacts with telomeric proteins in prostate cancer cells. J Biolog Chem 285(14):10472–10476
Hsu CP et al (2007) Modulation of telomere shelterin by TRF1 [corrected] and TRF2 interacts with telomerase to maintain the telomere length in non-small cell lung cancer. Lung Cancer 58(3):310–316
Menendez JA et al (2015) Heregulin, a new regulator of telomere length in human cells. Oncotarget 6(37):39422–39436
O’Connor MS et al (2004) The human Rap1 protein complex and modulation of telomere length. J Biol Chem 279(27):28585–28591
Kabir S, Hockemeyer D, de Lange T (2014) TALEN gene knockouts reveal no requirement for the conserved human shelterin protein Rap1 in telomere protection and length regulation. Cell Rep 9(4):1273–1280
Sfeir A et al (2010) Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 327(5973):1657–1661
Lototska L et al (2020) Human RAP1 specifically protects telomeres of senescent cells from DNA damage. EMBO Rep 21(4):e49076
Benarroch-Popivker D et al (2016) TRF2-mediated control of telomere DNA topology as a mechanism for chromosome-end protection. Mol Cell 61(2):274–286
Ferrara-Romeo I, Martínez P, Blasco MA (2018) Mice lacking RAP1 show early onset and higher rates of DEN-induced hepatocellular carcinomas in female mice. PLoS ONE 13(10):e0204909
Martinez P et al (2010) Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol 12(8):768–780
Teo H et al (2010) Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol 12(8):758–767
Zhang Y et al (2015) Rap1-mediated nuclear factor-kappaB (NF-kappaB) activity regulates the paracrine capacity of mesenchymal stem cells in heart repair following infarction. Cell Death Discov 1:15007
Cai Y et al (2015) Rap1 induces cytokine production in pro-inflammatory macrophages through NFkappaB signaling and is highly expressed in human atherosclerotic lesions. Cell Cycle 14(22):3580–3592
Ding Y et al (2018) Rap1 deficiency-provoked paracrine dysfunction impairs immunosuppressive potency of mesenchymal stem cells in allograft rejection of heart transplantation. Cell Death Dis 9(3):386
Poon MW et al (2015) Inhibition of RAP1 enhances corneal recovery following alkali injury. Invest Ophthalmol Vis Sci 56(2):711–721
Khattar E et al (2019) Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nature Commun 10(1):5349–5349
Anuja K et al (2020) Role of telomeric RAP1 in radiation sensitivity modulation and its interaction with CSC marker KLF4 in colorectal cancer. Int J Radiat Biol 96(6):790–802
Cantara S et al (2012) Lack of mutations of the telomerase RNA component in familial papillary thyroid cancer with short telomeres. Thyroid 22(4):363–368
Kim H et al (2013) Telomere length, TERT and shelterin complex proteins in hepatocellular carcinomas expressing “stemness”-related markers. J Hepatol 59(4):746–752
Hoxha M et al (2014) Relevance of telomere/telomerase system impairment in early stage chronic lymphocytic leukemia. Genes Chromosomes Cancer 53(7):612–621
Hockemeyer D et al (2007) Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat Struct Mol Biol 14(8):754–761
Guo X et al (2007) Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J 26(22):4709–4719
Abreu E et al (2010) TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol 30(12):2971–2982
Zhong FL et al (2012) TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 150(3):481–494
Spinella J-F et al (2015) A novel somatic mutation in ACD induces telomere lengthening and apoptosis resistance in leukemia cells. BMC cancer 15:621–621
Aoude LG et al (2015) Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst. https://doi.org/10.1093/jnci/dju408
Potjer TP et al (2019) Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer 144(10):2453–2464
Li J et al (2018) A rare variant P507L in TPP1 interrupts TPP1-TIN2 interaction, influences telomere length, and confers colorectal cancer risk in chinese population. Cancer Epidemiol Biomarkers Prev 27(9):1029–1035
Yang L et al (2013) Telomere-binding protein TPP1 modulates telomere homeostasis and confers radioresistance to human colorectal cancer cells. PLoS ONE 8(11):e81034
Zhao Y et al (2017) The transcription factor RFX5 is a transcriptional activator of the TPP1 gene in hepatocellular carcinoma. Oncol Rep 37(1):289–296
Loayza D, de Lange T (2003) POT1 as a terminal transducer of TRF1 telomere length control. Nature 423(6943):1013–1018
Kelleher C, Kurth I, Lingner J (2005) Human protection of telomeres 1 (POT1) is a negative regulator of telomerase activity in vitro. Mol Cell Biol 25(2):808–818
Wu L et al (2006) Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126(1):49–62
Fujii K et al (2008) Protection of telomeres 1 protein levels are associated with telomere length in gastric cancer. Int J Mol Med 21(5):599–604
Panero J et al (2014) Expression profile of shelterin components in plasma cell disorders. Clinical significance of POT1 overexpression. Blood Cells Mol Dis 52(2–3):134–139
Hsu C-P et al (2005) Clinical significance of telomerase and its associate genes expression in the maintenance of telomere length in squamous cell carcinoma of the esophagus. World J Gastroenterol 11(44):6941–6947
Amir M et al (2020) Structural features of nucleoprotein CST/shelterin complex involved in the telomere maintenance and its association with disease mutations. Cells 9(2):359
Ramsay AJ et al (2013) POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet 45(5):526–530
Speedy HE et al (2016) Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 128(19):2319–2326
Robles-Espinoza CD et al (2014) POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 46(5):478–481
Wong K et al (2019) Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol 155(5):604–609
Salhab M et al (2008) The expression of gene transcripts of telomere-associated genes in human breast cancer: correlation with clinico-pathological parameters and clinical outcome. Breast Cancer Res Treat 109(1):35–46
Aljarbou F et al (2018) The expression of telomere-related proteins and DNA damage response and their association with telomere length in colorectal cancer in Saudi patients. PLoS ONE 13(6):e0197154
Kondo T et al (2004) Expression of POT1 is associated with tumor stage and telomere length in gastric carcinoma. Cancer Res 64(2):523–529
Ferrandon S et al (2013) Telomere profiling: toward glioblastoma personalized medicine. Mol Neurobiol 47(1):64–76
Vega F et al (2008) Splenic marginal zone lymphomas are characterized by loss of interstitial regions of chromosome 7q, 7q31.32 and 7q36.2 that include the protection of telomere 1 (POT1) and sonic hedgehog (SHH) genes. Br J Haematol 142(2):216–226
Panero J et al (2010) Altered mRNA expression of telomere-associated genes in monoclonal gammopathy of undetermined significance and multiple myeloma. Mol Med 16(11–12):471–478
Bernardes de Jesus B et al (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med 4(8): 691–704
Motevalli A et al (2014) The effect of chemotherapeutic agents on telomere length maintenance in breast cancer cell lines. Breast Cancer Res Treat 145(3):581–591
Recagni M et al (2020) The role of alternative lengthening of telomeres mechanism in cancer: translational and therapeutic implications. Cancers (Basel). https://doi.org/10.3390/cancers12040949
Bryan TM et al (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3(11):1271–1274
Bryan TM et al (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 14(17):4240–4248
Dunham MA et al (2000) Telomere maintenance by recombination in human cells. Nat Genet 26(4):447–450
Funding
We thank the Agency for Science Technology and Research, Singapore (A*STAR) for funding and support to the V.T. laboratory which is supported by grant NRF-CRP17-2017-02 from the National Research Foundation Singapore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akincilar, S.C., Chan, C.H.T., Ng, Q.F. et al. Non-canonical roles of canonical telomere binding proteins in cancers. Cell. Mol. Life Sci. 78, 4235–4257 (2021). https://doi.org/10.1007/s00018-021-03783-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03783-0